
Morphinan Alkaloids and Their Transformations: A Historical Perspective of a Century of Opioid Research in Hungary †

Abstract
Contents
- 1.
- Introduction
- 2.
- Chemistry
- 2.1.
- Poppy alkaloids
- 2.2.
- The stereochemistry of morphinans
- 2.3.
- Biosynthesis of morphinan alkaloids
- 2.4.
- The Makleit-Bognár nomenclature
- 2.5.
- Early syntheses of morphine derivatives with pharmaceutical importance
- 2.6.
- Reduction of thebaine
- 2.7.
- Synthesis of desomorphine
- 2.8.
- Nucleophilic substitution reactions in the morphine series
- 2.8.1.
- Reactions of 7,8-dihydro compounds
- 2.8.2.
- Reactions of Δ7,8-unsaturated derivatives
- 2.8.3.
- Reactions of pseudocodeine tosylate
- 2.8.4.
- Neopine derivatives
- 2.8.5.
- Alkyl mesylate and allyl halide structural units in the same molecule
- 2.8.6.
- Substrates containing double allylic system
- 2.8.6.1.
- Substrates with allyl halide and allyl tosylate sub-units
- 2.8.6.2.
- Substrates containing double allyl halide sub-structural units
- 2.9.
- Azidomorphinans
- 2.9.1.
- Azidomorphine analogues
- 2.9.2.
- 14-Hydroxy-8-azido-8-desoxyallopseudocodeine derivatives
- 2.9.3.
- Azido derivatives of 6,14-ethenomorphinans
- 2.9.4.
- 6-Azido-6-demethoxythebaine
- 2.10.
- Fluorinated morphinans
- 2.10.1.
- Ring-C fluorinated morphinans
- 2.10.2.
- 1-Fluoro-substituted morphinans
- 2.10.3.
- Fluorinated 6,14-ethenomorphinans
- 2.11.
- Application of the Mitsunobu reaction in the morphine series
- 2.11.1.
- The first application
- 2.11.2.
- Preparation of isomorphine and isocodeine derivatives
- 2.11.3.
- Reactions of codeine isomers and neopine
- 2.11.4.
- Synthesis of 6β-aminomorphinans
- 2.11.5.
- Synthesis of 6β-succinimido derivatives
- 2.11.6.
- Reaction of 14-halogenocodeines
- 2.11.7.
- Novel applications
- 2.12.
- Poppy alkaloids as starting materials for molecular imaging
- 2.13.
- Other semi-synthetic derivatives
- 3.
- Summary and conclusions
| „Az ember ezt, ha egykor ellesi, Vegykonyhájában szintén megteszi. – Te nagy konyhádba helyzéd embered, S elnézed néki, hogy kontárkodik, Kotyvaszt, s magát Istennek képzeli.” | “Man will certainly learn this by watching And will simulate it in his kitchen. – You put into your great kitchen your man And of his bungling you take no notice, He brews and fancies himself to be God.” |
| Madách Imre: Az ember tragédiája | Imre Madách: Tragedy of the man (translation: Tomschey, O.) |
1. Introduction
2. Chemistry
2.1. Poppy Alkaloids
2.2. The Stereochemistry of Morphinans
2.3. Biosynthesis of Morphinan Alkaloids
2.4. The Makleit–Bognár Nomenclature
2.5. Early Syntheses of Morphine Derivatives with Pharmaceutical Importance
2.6. Reduction of Thebaine
- (A)
- 1,2-Addition of hydrogen to the Δ7,8 double bond of thebaine (4). This results in dihydrothebaine (48), which is transformable to dihydrocodeinone (49) through hydrolysis;
- (B)
- 1,4-Addition of hydrogen to the conjugated system of thebaine (4) in ring-C. This leads to codeine methyl ether (44), which is reducible to tetrahydrothebaine (50);
- (C)
- 1,4-Addition of hydrogen to the oxygen atom of the E-ring and to C-7. This constitutes a hydrogenolysis of the allyl ether group, which results in the opening of the ether bridge. In the first step, Δ5,8-phenolic-dihydrothebaine (43, dihydrothebaine-Φ) is formed, which can be transformed to dihydrothebainone-Δ5-methylenolate (51). The enol ether hydrolysis of this compound results in dihydrothebainone (52). Subsequent hydrogenation leads to dihydrothebainol-6-methyl ether (53);
- (D)
- 1,6-Addition of hydrogen to the oxygen atom of the E-ring and to C-14. This process results in Δ5,7-phenolic-dihydrothebaine (45). The following/further hydrogenation of the latter compound forms dihydrothebainone-Δ5-methyl enolate (51) and dihydrothebainol-6-methyl ether (53).
2.7. Synthesis of Desomorphine
2.8. Nucleophilic Substitution Reactions in the Morphine Series
2.8.1. Reactions of 7,8-Dihydro Compounds
2.8.2. Reactions of Δ7,8-Unsaturated Derivatives
2.8.3. Reactions of Pseudocodeine Tosylate
2.8.4. Neopine Derivatives
2.8.5. Alkyl Mesylate and Allyl Halide Structural Units in the Same Molecule
2.8.6. Substrates Containing Double Allylic System
Substrates with Allyl Halide and Allyl Tosylate Subunits
Substrates Containing Double Allyl Halide Sub-Structural Units
2.9. Azidomorphinans
2.9.1. Azidomorphine Analogs
2.9.2. 14-Hydroxy-8-azido-8-desoxyallopseudocodeine Derivatives
2.9.3. Azido Derivatives of 6,14-Ethenomorphinans
2.9.4. 6-Azido-6-demethoxythebaine
2.10. Fluorinated Morphinans
2.10.1. Ring-C Fluorinated Morphinans
2.10.2. 1-Fluoro-Substituted Morphinans
2.10.3. Fluorinated 6,14-Ethenomorphinans
2.11. Application of the Mitsunobu Reaction in the Morphine Series
- (1)
- In the first elementary step of the process, a reactive betaine (XVII) is formed from the azodicarboxylic acid ester (244) and trialkyl or triarylphosphine (245) in an irreversible reaction;
- (2)
- In the second step, the reactive zwitterionic adduct (XVII) reacts with the acidic pronucleophile (H-Nu, 243) to produce the nucleophile (Nu⊖);
- (3)
- The alcoholic hydroxyl group (2a) attacks the protonated betaine (XVIII) to form an alkoxyphosphonium intermediate (XIX). During this process, 1,2-hydrazinecarboxylic acid dialkyl ester (247) arises;
- (4)
- The conjugated base form (Nu⊖) of the pronucleophile (H-Nu, 243) reacts with the alkylphosphonium intermediate (XIX), from which emerges the product (R-Nu, 246) as well as triaryl or trialkylphosphine oxide (248). When a chiral secondary alcohol is applied as the starting substrate, a change in the configuration of the chiral center is to be expected (Walden inversion).
2.11.1. The First Application
2.11.2. Preparation of Isomorphine and Isocodeine Derivatives
2.11.3. Reactions of Codeine Isomers and Neopine
2.11.4. Synthesis of 6β-aminomorphinans
2.11.5. Synthesis of 6β-Succinimido Derivatives
2.11.6. Reaction of 14-Halogenocodeines
2.11.7. Novel Applications
2.12. Poppy Alkaloids as Starting Materials for Molecular Imaging
2.13. Other Semisynthetic Derivatives
3. Summary and Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Sertürner, F.W. Über das Morphium, eine neue salzfähige Grundlage, und die Mekonsäure, als Hauptbestandteile des Opiums. Ann. Der Phys. 1817, 55, 56–89. [Google Scholar]
- Schmitz, R. Friedrich Wilhelm Sertürner and the Discovery of Morphine. Pharm. Hist. 1985, 27, 61–74. [Google Scholar]
- Van Zee, A. The promotion and marketing of oxycontin: Commercial triumph, public health tragedy. Am. J. Publ. Health 2009, 99, 221–227. [Google Scholar] [CrossRef]
- Kabay, J.J. János Kabay, The Life of an Inventor; National Library of Australia: Victoria, Australia, 1990; ISBN 0-646-01672-5.
- Kabay, J.J. Kabay János Magyar Feltaláló Élete; Alkaloida Vegyészeti Gyár Részvénytársaság: Tiszavasvári, Hungary, 1992; ISBN 9-630-29978-x. [Google Scholar]
- Hosztafi, S. Kabay János a magyar morfingyártás megalapítója. In Kabay Jánosra Emlékezünk; Évi Pályázat Díjnyertes Munkái; Galó, M., Ed.; Szabolcs-Szatmár-Bereg Megyei Tudományos Közalapítvány Kuratóriuma: Nyíregyháza, Hungary, 1996; Volume 8, pp. 27–53. [Google Scholar]
- Hosztafi, S. János Kabay, the founder of morphine manufacture in Hungary. Gyógyszerészet 1997, 41, 25–37. [Google Scholar]
- Móra, L.; Próder, I. A Magyar Kémia és Vegyipar Kronológiája 1800–1950, Magyar Tudománytörténeti Intézet Tudományos Közleményei; Gazda, I., Ed.; Magyar Tudománytörténeti Intézet: Budapest, Hungary, 2015; Volume 51, pp. 1–128. [Google Scholar]
- Próder, I. Kabay János morfinelőállítási eljárása. Magy. Kém. Lapja 1998, 53, 169–172. [Google Scholar]
- Kabay, J. Eljárás Ópiumalkaloidák Előállítására. Hungarian Patent 89,464, 1 May 1925. [Google Scholar]
- Kabay, J. Eljárás Ópiumalkaloidák Előállítására. Hungarian Patent 109,788, 30 November 1931. [Google Scholar]
- Bayer, I. Manufacture of alkaloids from the poppy plant in Hungary. Bull. Narc. 1961, 1, 21–28. [Google Scholar]
- Szántay, C.; Nemes, A. A magyar alkaloidkémia története. Magy. Kém. Foly. 1994, 100, 423–442. [Google Scholar]
- Bernáth, J. Poppy, The Genus Papaver; Harwood Academic Publishers: Amsterdam, The Netherlands, 1998; pp. 1–342. [Google Scholar]
- Herczegh, P. Bognár Rezső szerves kémikus egyetemi tanár, akadémikus, tudománypolitikus (1913–1990). Gerund. Egyetemtörténeti Közlemények 2016, 7, 235–240. [Google Scholar]
- Bognár, R.; Gaál, G.D.; Mészáros, Z.; Szabó, S.; Zeller, I. Eljárás Morphin Társalkaloidjainak Kinyerésére Máknövény Részeinek Kivonatolása Során Kapott Anyagból. Hungarian Patent 145,999, 3 July 1958. [Google Scholar]
- Szlávik, L.; Bognár, R.; Bartók, A.; Gaál, G.; Kaskötő, Z.; Nagy, S.; Gyöngy, I.; Maczkó, G.; Kerekes, P.; Selmeci, G. Eljárás Ópiumalkaloidák Előállítására. Hungarian Patent 155,407, 12 July 1967. [Google Scholar]
- Bognár, R.; Gaál, G.; Kerekes, P.; Szabó, S. On the accompanying alkaloids of morphine 1. Isolation of codeine, thebaine and narcotine. Pharmazie 1967, 22, 452–454. [Google Scholar]
- Bognár, R.; Gaál, G.; Górecki, P.; Szabó, S. On the companion alkaloids of morphine. 2. Isolation of narcotoline and papaverine. Pharmazie 1967, 22, 525–526. [Google Scholar]
- Górecki, P.; Bognár, R. On the accompanying alkaloids of morphine. 3. Modified preparation of codeine, thebaine and papaverine. Pharmazie 1968, 23, 590–593. [Google Scholar] [PubMed]
- Blaskó, G.; Gula, D.J.; Shama, M. The phthtalideisoquinoline alkaloids. J. Nat. Prod. 1982, 45, 105–122. [Google Scholar] [CrossRef]
- Bentley, K.W. The Chemistry of Morphine Alkaloids; Clarendon Press: Oxford, UK, 1954; pp. 1–453. [Google Scholar]
- Casy, A.F.; Parfitt, R.T. Diels-Alder adducts of thebaine. In Opioid Analgesics: Chemistry and Receptors; Plenum Press: New York, NY, USA, 1986; pp. 69–84. [Google Scholar]
- Szántay, C.; Dörnyei, G.; Blaskó, G. The morphine alkaloids. In The Alkaloids: Chemistry and Physiology; Academic Press Inc.: New York, NY, USA; London, UK, 1994; Volume 45, pp. 128–232. [Google Scholar]
- Hosztafi, S. III. Chemistry-Biochemistry of Poppy, 1. Chemical Structures of Alkaloids. In Poppy, The Genus Papaver; Bernáth, J., Ed.; Harwood Academic Publishers: Amsterdam, The Netherlands, 1998; pp. 105–159. [Google Scholar]
- Lee, S.; Park, N.I.; Park, Y.; Park, K.C.; Kim, E.S.; Son, Y.K.; Choi, B.S.; Kim, N.S.; Choi, I.Y. O- and N-Methyltransferases in benzylisoquinoline alkaloid producing plants. Genes Genom. 2023, 46, 367–378. [Google Scholar] [CrossRef]
- Marques, I.A.; Brodelius, P.E. Elicitor-Induced l-Tyrosine Decarboxylase from Plant Cell Suspension Cultures: II. Partial Characterization. Plant Physiol. 1988, 88, 52–55. [Google Scholar] [CrossRef]
- Lee, E.J.; Facchini, P.J. Tyrosine aminotransferase contributes to benzylisoquinoline alkaloid biosynthesis in opium poppy. Plant Physiol. 2011, 157, 1067–1078. [Google Scholar] [CrossRef] [PubMed]
- Luk, L.Y.; Bunn, S.; Liscombe, D.K.; Facchini, P.J.; Tanner, M.E. Mechanistic studies on norcoclaurine synthase of benzylisoquinoline alkaloid biosynthesis: An enzymatic Pictet-Spengler reaction. Biochemistry 2007, 46, 10153–10161. [Google Scholar] [CrossRef]
- Robin, A.Y.; Giustini, C.; Graindorge, M.; Matringe, M.; Dumas, R. Crystal structure of norcoclaurine-6-O-methyltransferase, a key rate-limiting step in the synthesis of benzylisoquinoline alkaloids. Plant J. 2016, 87, 641–653. [Google Scholar] [CrossRef]
- Yu, M.; Facchini, P.J. cDNA cloning and characterization of (S)-N-methylcoclaurine 3′-hydroxylase (CYP80B1) from elicitortreated opium poppy cell suspension cultures (accession no. AF191772). Plant Physiol. 2000, 122, 1457. [Google Scholar]
- Lannuzel, A.; Michel, P.P.; Caparros-Lefebvre, D.; Abaul, J.; Hocquemiller, R.; Ruberg, M. Toxicity of Annonaceae for dopaminergic neurons: Potential role in atypical parkinsonism in Guadeloupe. Mov. Disord. 2002, 17, 84–90. [Google Scholar] [CrossRef]
- Fürst, S.; Hosztafi, S. Pharmacology of poppy alkaloids. In Poppy—The Genus Papaver; Bernáth, J., Ed.; Harwood Academic Publisher: Amsterdam, The Netherlands, 1998; pp. 291–318. [Google Scholar]
- Aceto, M.D.; Harris, L.S.; Abood, M.E.; Rice, K.C. Stereoselective mu- and delta-opioid receptor-related antinociception and binding with (+)-thebaine. Eur. J. Pharm. 1999, 365, 143–147. [Google Scholar] [CrossRef]
- Cumming, P.; Marton, J.; Lilius, T.O.; Olberg, D.E.; Rominger, A. A Survey of Molecular Imaging of Opioid Receptors. Molecules 2019, 24, 4190. [Google Scholar] [CrossRef]
- Nyman, U.; Bruhn, J.G. Papaver bracteatum—A summary of current knowledge. Planta Med. 1979, 35, 97–117. [Google Scholar] [CrossRef] [PubMed]
- Milo, J.; Levy, A.; Palevitch, D. An alternative raw-the cultivation and breeding of Papaver bracteatum. In Poppy—The Genus Papaver; Bernáth, J., Ed.; Harwood Academic Press: Amsterdam, The Netherlands, 1998; pp. 279–290. [Google Scholar]
- Millgate, A.G.; Pogson, B.J.; Wilson, I.W.; Kutchan, T.M.; Zenk, M.H.; Gerlach, W.L.; Fist, A.; Larkin, P.J. Morphine-pathway block in top1 poppies. Nature 2004, 431, 413–414. [Google Scholar] [CrossRef] [PubMed]
- Dastmalchi, M.; Chang, L.; Torres, M.A.; Ng, K.K.S.; Facchini, P.J. Codeinone reductase isoforms with differential stability, efficiency and product selectivity in opium poppy. Plant J. 2018, 95, 631–647. [Google Scholar] [CrossRef] [PubMed]
- Bird, D.A.; Franceschi, V.R.; Facchini, P.J. A tale of three cell types: Alkaloid biosynthesis is localized to sieve elements in opium poppy. Plant Cell 2003, 15, 2626–2635. [Google Scholar] [CrossRef]
- Onoyovwe, A.; Hagel, J.M.; Chen, X.; Khan, M.F.; Schriemer, D.C.; Facchini, P.J. Morphine biosynthesis in opium poppy involves two cell types: Sieve elements and laticifers. Plant Cell 2013, 25, 4110–4122. [Google Scholar] [CrossRef]
- Lim, H.-Y.; Kwok, S.-F. Differentiation and Comparison of Raw, Prepared and Dross Opium; United Nations Office on Drugs and Crime: Vienna, Austria, 1981; Available online: https://www.unodc.org/unodc/en/data-and-analysis/bulletin/bulletin_1981-01-01_1_page006.html (accessed on 15 January 2025).
- Stranska, I.; Skalicky, M.; Novak, J.; Matyasova, E.; Hejnak, V. Analysis of selected poppy (Papaver somniferum L.) cultivars: Pharmaceutically important alkaloids. Ind. Crops Prod. 2013, 41, 120–126. [Google Scholar] [CrossRef]
- Carlin, M.G.; Dean, J.R.; Ames, J.M. Opium alkaloids in harvested and thermally processed poppy seeds. Front. Chem. 2020, 8, 737. [Google Scholar] [CrossRef]
- Haber, I.; Pergolizzi, J., Jr.; LeQuang, J.A. Poppy Seed Tea: A Short Review and Case Study. Pain Ther. 2019, 8, 151–155. [Google Scholar] [CrossRef]
- Bishop-Freeman, S.C.; Fox, L.; Winecker, R.E.; Hudson, J.S. Death from poppy tea consumption. J. Anal. Toxicol. 2020, 44, 734–740. [Google Scholar] [CrossRef]
- Steentoft, A.; Kaa, E.; Worm, K. Fatal intoxications in Denmark following intake of morphine from opium poppies. Z. Rechtsmed. 1988, 101, 197–204. [Google Scholar] [CrossRef] [PubMed]
- Penafiel, R.; Yoo, D.; Turner, C.; Brown, J.A.; McDonald, C.; Tran, J.; Shaw, V.; Roberts, D.M. Toxicokinetics of thebaine in those consuming non-food grade poppy seeds as a tea. Clin. Toxicol. 2023, 61, 644–648. [Google Scholar] [CrossRef] [PubMed]
- Benyhe, S.; Zádor, F.; Ötvös, F. Biochemistry of opioid (morphine) receptors: Binding, structure and molecular modelling. Acta Biol. 2015, 59 (Suppl. 1), 17–37. [Google Scholar]
- Tanguturi, P.; Streicher, J.M. The role of opioid receptors in modulating Alzheimer’s Disease. Front. Pharmacol. 2023, 14, 1056402. [Google Scholar] [CrossRef]
- Che, T.; Dwivedi-Agnihotri, H.; Shukla, A.K.; Roth, B.L. Biased ligands at opioid receptors: Current status and future directions. Sci. Signal. 2021, 14, 677. [Google Scholar] [CrossRef]
- De Neve, J.; Barlow, T.M.A.; Tourwé, D.; Bihel, F.; Simonin, F.; Ballet, S. Comprehensive overview of biased pharmacology at the opioid receptors: Biased ligands and bias factors. RSC Med. Chem. 2021, 12, 828–870. [Google Scholar] [CrossRef]
- DePriest, A.Z.; Puet, B.L.; Holt, A.C.; Roberts, A.; Cone, E.J. Metabolism and disposition of prescription opioids: A review. Forensic Sci. Rev. 2015, 27, 115–145. [Google Scholar]
- Makleit, S.; Bognár, R. Amino-morfidok és amino-kodidok előállításának és térkémiájának vizsgálatáról. Kém. Közl. 1968, 30, 289–295. [Google Scholar]
- Bognár, R.; Makleit, S. Conversions of tosyl and mesyl derivatives of the morphine group II. Aminomorphides and aminocodides. Acta Chim. Acad. Sci. Hung. 1968, 58, 203–205. [Google Scholar]
- Crabbendam, P.R.; Lie, T.S.; Linders, J.T.M.; Maat, L. Synthesis of 6, 14-ethenoisomorphinans and 6, 14-ethenomorphinans based on Diels–Alder adducts of 6-demethoxythebaine and 6-demethoxy-β-dihydrothebaine; pharmacology of the isomorphinans (Chemistry of Opium Alkaloids, Part XIX). Recl. Trav. Chim. Pays-Bas 1984, 103, 296–300. [Google Scholar] [CrossRef]
- Marton, J.; Fekete, A.; Cumming, P.; Hosztafi, S.; Mikecz, P.; Henriksen, G. Diels–Alder adducts of morphinan-6, 8-dienes and their transformations. Molecules 2022, 27, 2863. [Google Scholar] [CrossRef]
- Rodionov, V.M. L’importance des ethers alcoyliques des acides sulfoaromatiquea pour l’alcoylation des composes organiques. Bull. Soc. Chim. Fr. 1926, 39, 305–325. [Google Scholar]
- Heumann, W.R. The manufacture of codeine from morphine. Bull. Narcotics 1958, 10, 15–17. [Google Scholar]
- Gyöngy, I.; Makleit, S.; Mészáros, Z.; Szlávik, L. Eljárás Folcodin (Morfolinil-Etil-Morfin) Előállítására. Hungarian Patent 144,637, 5 April 1956. [Google Scholar]
- Mertes, P.M.; Petitpain, N.; Tacquard, C.; Delpuech, M.; Baumann, C.; Malinovsky, J.M.; Longrois, D.; Gouel-Cheron, A.; Quang, D.L.; Demoly, P.; et al. Pholcodine exposure increases the risk of perioperative anaphylaxis to neuromuscular blocking agents: The ALPHO case-control study. Br. J. Anaesth. 2023, 131, 150–158. [Google Scholar] [CrossRef] [PubMed]
- Mahase, E. UK withdraws pholcodine-containing cough and cold medicines over anaphylaxis risk in surgery. BMJ Brit. Med. J. 2023, 380, 609. [Google Scholar] [CrossRef]
- Szabó, S.; Bognár, R. Eljárás N-allil-normorfin tisztítására. Hungarian Patent 1483, 30 May 1956. [Google Scholar]
- McCawley, E.L.; Hart, E.R.; Marsh, D.F. The Preparation of N-Allylnormorphine. J. Am. Chem. Soc. 1941, 63, 314. [Google Scholar] [CrossRef]
- von Braun, J. Die Einwirkung von Bromcyan auf tertiäre Amine. Berichte 1900, 33, 1438–1452. [Google Scholar] [CrossRef]
- Berényi, S.; Hosztafi, S.; Makleit, S.; Szeifert, I. A New route for the preparation of N-substituted N-demethylapocodeine derivatives. Acta Chim. Acad. Sci. Hung. 1982, 110, 363–369. [Google Scholar]
- Berényi, S.; Hosztafi, S.; Makleit, S.; Molnár, I. Preparation of demethoxyoripavine and its conversion into N-substituted N-demethylapomorphine derivatives. Acta Chim. Hung. 1983, 113, 51–60. [Google Scholar]
- Bognár, R.; Makleit, S. A new preparation of dihydro-6-deoxymorphine. Chem. Ind. 1956, 1239. [Google Scholar]
- Bognár, R.; Makleit, S. Eine neue Darstellung des Dihydro-6-desoxymorphins. Arzneim. Forsch./Drug. Res. 1958, 8, 323–325. [Google Scholar]
- Bognár, R.; Makleit, S. A dihidro-6-desoxymorphin új előállítása. Acta Univ. Debreceniensis Ludovico Kossuth Nomin. 1958, 5, 225–229. [Google Scholar]
- Holmes, H.L. The Morphine Alkaloids I. In The Alkaloids, Chemistry and Physiology; Manske, R.H.F., Holmes, H.L., Eds.; Academic Press Inc.: New York, NY, USA, 1952; Volume 2, pp. 2–162. [Google Scholar]
- Holmes, H.L.; Stok, G. The Morphine Alkaloids II. In The Alkaloids, Chemistry and Physiology; Manske, R.H.F., Holmes, H.L., Eds.; Academic Press Inc.: New York, NY, USA, 1952; Volume 2, pp. 162–219. [Google Scholar]
- Small, L.; Browning, G.L. Reduction studies in the morphine series. VII. Thebaine. J. Org. Chem. 1939, 3, 618–637. [Google Scholar] [CrossRef]
- Stork, G. The reduction of thebaine. J. Am. Chem. Soc. 1952, 74, 768–773. [Google Scholar] [CrossRef]
- Bentley, K.W.; Robinson, R. Some notes on the reduction of thebaine and related topics. Experientia 1950, 65, 353–354. [Google Scholar] [CrossRef] [PubMed]
- Bentley, K.W.; Robinson, R.; Wain, A.E. The reduction of thebaine and dihydrothebaine by sodium and ammonia. J. Chem. Soc. 1952, 958–966. [Google Scholar] [CrossRef]
- Razdan, R.K.; Portlock, D.E.; Dalzell, H.C.; Malmberg, C. Synthesis of β-dihydrothebaine. J. Org. Chem. 1978, 43, 3604–3606. [Google Scholar] [CrossRef]
- Schmid, H.; Karrer, P. Über das β-Dihydro-thebain. Helv. Chim. Acta 1950, 33, 863–873. [Google Scholar] [CrossRef]
- Bentley, K.W.; Lewis, J.W.; Taylor, J.B. Reduction of thebaine with mixed hydride reducing agents. J. Chem. Soc. C 1969, 15, 1945–1946. [Google Scholar] [CrossRef]
- Linders, J.T.M.; Adriaansens, R.J.O.; Lie, T.S.; Maat, L. Scission of the epoxy ring in 4.5α-epoxymorphinans: A convenient synthesis of β-dihydrothebaine, 6-demethoxy-β-dihydrothebaine and desoxycodeine-A. (Chemistry of Opium Alkaloids, Part 21). Recl. Trav. Chim. Pays-Bas 1986, 105, 27–29. [Google Scholar] [CrossRef]
- Robinson, R. Address of the President Sir Robert Robinson, at the anniversary meeting, 1 December 1947. Proc. Roy. Soc. Lond. Ser. B Biol. Sci. 1947, 135, 5–19. [Google Scholar]
- Robinson, R. An essay in correlation arising from alkaloid chemistry. Nature 1947, 160, 815–817. [Google Scholar] [CrossRef] [PubMed]
- Small, L.F. The reduction of thebaine. Neopine methyl ether. J. Org. Chem. 1955, 20, 953–958. [Google Scholar] [CrossRef]
- Bognár, R.; Szabó, S. A tebain hidrogénezéséről. Magy. Kém. Foly. 1953, 59, 321–325. [Google Scholar]
- Szabó, S. Újabb Adatok a Tebain Redukciós Termékeinek Kémiájához. C.Sc. Dissertation, Kossuth Lajos Tudományegyetem (KLTE), Debrecen, Hungary, 1967. [Google Scholar]
- Schöpf, C. Die Konstitution der Morphiumalkaloide. Justus Liebigs Ann. Chem. 1927, 452, 211–267. [Google Scholar] [CrossRef]
- Florez, D.H.Â.; dos Santos Moreira, A.M.; da Silva, P.R.; Brandão, R.; Borges, M.M.C.; de Santana, F.J.M.; Borges, K.B. Desomorphine (Krokodil): An overview of its chemistry, pharmacology, metabolism, toxicology and analysis. Drug Alcohol Depend. 2017, 173, 59–68. [Google Scholar] [CrossRef] [PubMed]
- Alves, E.A. DARK classics in chemical neuroscience: Krokodil. ACS Chem. Neurosci. 2020, 11, 3968–3978. [Google Scholar] [CrossRef]
- Mella-Raipan, J.; Romero-Parra, J.; Recabarren-Gajardo, G. DARK classics in chemical neuroscience: Heroin and desomorphine. ACS Chem. Neurosci. 2020, 11, 3905–3927. [Google Scholar] [CrossRef]
- Rapoport, H.; Bonner, R.M. Δ7-Desoxymorphine. J. Am. Chem. Soc. 1951, 73, 5485. [Google Scholar] [CrossRef]
- Cunningham, C.W.; Mercer, S.L.; Hassan, H.E.; Traynor, J.R.; Eddington, N.D.; Coop, A. Opioids and efflux transporters. Part 2: P-glycoprotein substrate activity of 3-and 6-substituted morphine analogs. J. Med. Chem. 2008, 51, 2316–2320. [Google Scholar] [CrossRef]
- Eddy, N.B.; Halbach, H.; Braenden, O.J. Synthetic substances with morphine-like effect: Relationship between analgesic action and addiction liability, with a discussion of the chemical structure of addiction-producing substances. Bull. World Health Org. 1956, 14, 353–402. [Google Scholar] [PubMed]
- Braenden, O.J.; Eddy, N.B.; Halbach, H. Synthetic substances with morphine-like effect: Relationship between chemical structure and analgesic action. Bull. World Health Org. 1955, 13, 937–998. [Google Scholar] [PubMed]
- Reden, J.; Reich, M.F.; Rice, K.C.; Jacobson, A.E.; Brossi, A.; Streaty, R.A.; Klee, W.A. Deoxymorphines: Role of the phenolic hydroxyl in antinociception and opiate receptor interactions. J. Med. Chem. 1979, 22, 256–259. [Google Scholar] [CrossRef]
- Alves, E.A.; Grund, J.P.C.; Afonso, C.M.; Netto, A.D.P.; Carvalho, F.; Dinis-Oliveira, R.J. The harmful chemistry behind krokodil (desomorphine) synthesis and mechanisms of toxicity. Forensic Sci. Int. 2015, 249, 207–213. [Google Scholar] [CrossRef] [PubMed]
- Katselou, M.; Papoutsis, I.; Nikolaou, P.; Spiliopoulou, C.; Athanaselis, S. A “Krokodil” emerges from the murky waters of addiction. Abuse trends of an old drug. Life Sci. 2014, 102, 81–87. [Google Scholar] [CrossRef]
- Small, L.F.; Morris, D.E. The desoxymorphines. J. Am. Chem. Soc. 1933, 55, 2874–2885. [Google Scholar] [CrossRef]
- Small, L.F.; Yuen, K.C.; Eilers, L.K. The catalytic hydrogenation of the halogenomorphides: Dihydrodesoxymorphine-D. J. Am. Chem. Soc. 1933, 55, 3863–3870. [Google Scholar] [CrossRef]
- Welsh, L.H. O3-Monoacetylacetylmorphine. J. Org. Chem. 1954, 19, 1409–1415. [Google Scholar] [CrossRef]
- Schmid, H.; Karrer, P. Über die Umsetzungsprodukte von Toluolsulfonsäureestern mit Lithiumaluminiumhydrid. Helv. Chim. Acta 1949, 32, 1371–1378. [Google Scholar] [CrossRef]
- Makleit, S.; Berényi, S.; Bognár, R. Conversions of tosyl and mesyl derivatives of the morphine group, XIV. A new method for the preparation of “deoxymorphine E and dihydrodeoxymorphine D”. Acta Chim. Acad. Sci. Hung. 1976, 88, 409–411. [Google Scholar]
- Srimurugan, S.; Su, C.J.; Shu, H.C.; Murugan, K.; Chen, C. A facile and improved synthesis of desomorphine and its deuterium-labeled analogue. Monatsh. Chem. 2012, 143, 171–174. [Google Scholar] [CrossRef]
- Gahr, M.; Freudenmann, R.W.; Hiemke, C.; Gunst, I.M.; Connemann, B.J.; Schönfeldt-Lecuona, C. Desomorphine goes “crocodile”. J. Addict. Dis. 2012, 31, 407–412. [Google Scholar] [CrossRef] [PubMed]
- Neves, J.F.; Alves, E.A.; Soares, J.X.; Cravo, S.M.; Silva, A.M.; Pereira Netto, A.D.; Carvalho, F.; Dinis-Oliveira, R.J.; Afonso, C.M. Data analysis of “krokodil” samples obtained by street-like synthesis. Data Brief 2015, 6, 83–88. [Google Scholar] [CrossRef] [PubMed]
- Babapoor-Farrokhran, S.; Caldararo, M.D.; Rad, S.N.; Laborde, F.N.; Rehman, R.; Mejia, J. New case of krokodil (desomorphine) use. Int. J. Case Rep. Images 2018, 9, 1–4. [Google Scholar]
- Soares, J.X.; Alves, E.A.; Silva, A.M.N.; de Figueiredo, N.G.; Neves, J.F.; Cravo, S.M.; Rangel, M.; Netto, A.D.P.; Carvalho, F.; Dinis-Oliveira, R.J.; et al. Street-like synthesis of krokodil results in the formation of an enlarged cluster of known and new morphinans. Chem. Res. Toxicol. 2017, 30, 1609–1621. [Google Scholar] [CrossRef]
- Smith, M.B. Reactions, Mechanisms, and Structure, Chapter 10. Aliphatic substitution nucleophilic and organometallic. In March’s Advanced Organic Chemistry, 7th ed.; John Wiley & Sons Inc.: Hoboken, NJ, USA, 2013; pp. 373–568. ISBN 978-0-470-46259-1. [Google Scholar]
- Hamlin, T.A.; Swart, M.; Bickelhaupt, F.M. Nucleophilic substitution (SN2): Dependence on nucleophile, leaving group, central atom, substituents, and solvent. ChemPhysChem 2018, 19, 1315–1330. [Google Scholar] [CrossRef]
- Jacobson, O.; Kiesewetter, D.O.; Chen, X. Fluorine-18 radiochemistry, labeling strategies and synthetic routes. Bioconjugate Chem. 2015, 26, 1–18. [Google Scholar] [CrossRef]
- Stork, G.; Clarke, F.H. The SN2′ reaction. III. Structure and SN2′ reactions of the halocodides. J. Am. Chem. Soc. 1956, 78, 4619–4624. [Google Scholar] [CrossRef]
- Bognár, R.; Makleit, S.; Radics, L. Tozil-, ill. mezilszármazékok reakcióinak vizsgálata a morfinsorban, VIII. A 6-dezoxi-6-fluor-izo-kodein új előállítása és vizsgálata. Magy. Kém. Foly. 1970, 76, 461–464. [Google Scholar]
- Bognár, R.; Makleit, S.; Radics, L. Conversions of tosyl and mesyl derivatives of the morphine group, VIII. A new synthesis and investigations of 6-deoxy-6-fluoroisocodeine. Acta Chim. Acad. Sci. Hung. 1971, 67, 63–69. [Google Scholar]
- Makleit, S.; Mile, T.; Bognár, R. Tozil-, ill. mezilszármazékok vizsgálata a morfinsorban, XVI. Újabb adatok az allilátrendeződés mechanizmusához. Magy. Kém. Foly. 1975, 81, 564–565. [Google Scholar]
- Makleit, S.; Mile, T.; Bognár, R. Conversions of tosyl and mesyl derivatives in the morphine group, XVI. New data to the mechanism of allylic rearrangement. Acta Chim. Acad. Sci. Hung. 1976, 89, 275–277. [Google Scholar] [CrossRef]
- Makleit, S. Az un Morfin-Sor (Morfin, Kodein, Dihidromorfin, Dihidrokodein, 14-Hidroxi-Kodein és 14-Hidroxi-Dihidrokodein) 6-O-Tozil, ill, 6-O-Mezil-Származékai Előállítása és Nukleofil Szubsztitúciós Reakcióinak Tanulmányozása, Part I (1971), Part 2 (1972). D.Sc. Dissertation, Kossuth Lajos University, Debrecen, Hungary, 1972. [Google Scholar]
- Gaál, G.; Makleit, S. 2.1. Mákalkaloidok izolálása és kémiai átalakítása. In Kutatási eredmények 1950–1973, Jubileumi Kötet Bognár Rezső Hatvanadik Születésnapjára; Gaál, G., Ed.; Kossuth Lajos Tudományegyetem Szerves Kémiai Tanszéke; Alföldi Nyomda: Debrecen, Hungary, 1973; pp. 57–118. [Google Scholar]
- Bognár, R.; Makleit, S. Tozil-, ill. mezilszármazékok reakcióinak vizsgálata a morfinsorban, III. Azido- és amino-kodidok előállítása. Magy. Kém. Foly. 1968, 74, 523–526. [Google Scholar]
- Bognár, R.; Makleit, S. Conversions of tosyl and mesyl derivatives of the morphine group, III. Preparation of azido- and aminocodides. Acta Chim. Acad. Sci. Hung. 1969, 59, 373–378. [Google Scholar]
- Bognár, R.; Makleit, S.; Mile, T. Tozil-, ill. mezilszármazékok reakciónak vizsgálata a morfinsorban, IV. Azido-és amino-morfidok előállítása. Magy. Kém. Foly. 1968, 74, 526–530. [Google Scholar]
- Bognár, R.; Makleit, S.; Mile, T. Conversions of tosyl and mesyl derivatives of the morphine group, IV. Preparation of “azido- and aminomorphides”. Acta Chim. Acad. Sci. Hung. 1969, 59, 379–385. [Google Scholar]
- Makleit, S.; Radics, L.; Bognár, R.; Mile, T.; Oláh, É. Tozil-, illetve mezilszármazékok reakcióinak vizsgálata a morfinsorban, X. 14-Hidroxi-morfin-származékok, I. Azido- és aminovegyületek. Magy. Kém. Foly. 1972, 78, 223–228. [Google Scholar]
- Makleit, S.; Radics, L.; Bognár, R.; Mile, T.; Oláh, É. Conversions of tosyl and mesyl derivatives of the morphine group, X. 14-Hydroxymorpine derivatives, I. Azido and amino derivatives. Acta Chim. Acad. Sci. Hung. 1972, 74, 99–113. [Google Scholar]
- Makleit, S.; Bognár, R. Tozil-, ill. mezilszármazékok reakcióinak vizsgálata a morfinsorban, V. Izo-kodein és dihidro-izo-kodein új előállítása. Magy. Kém. Foly. 1969, 75, 235. [Google Scholar]
- Makleit, S.; Bognár, R. Conversions of tosyl and mesyl derivatives of the morphine group, V. A new method for the preparation of isocodeine and dihydroisocodeine. Acta Chim. Acad. Sci. Hung. 1969, 59, 387–388. [Google Scholar]
- Bognár, R.; Makleit, S.; Mile, T. Tozil-, ill. mezil-származékok reakcióinak vizsgálata a morfin-sorban. Acetil-merkapto és merkaptoszármazékok előállítása. Magy. Kém. Foly. 1969, 75, 330–332. [Google Scholar]
- Bognár, R.; Makleit, S.; Mile, T. Conversions of tosyl and mesyl derivatives of the morphine group. Synthesis of acetylmercapto and mercapto derivatives. Acta Chim. Acad. Sci. Hung. 1969, 59, 161–164. [Google Scholar]
- Bognár, R.; Makleit, S.; Mile, T.; Radics, L. Tozil ill. mezilszármazékok reakcióinak vizsgálata a morfinsorban, VI. Acetilmerkapto és merkaptoszármazékok előállítása. Magy. Kém. Foly. 1970, 76, 102–105. [Google Scholar]
- Bognár, R.; Makleit, S.; Mile, T.; Radics, L. Conversions of tosyl and mesyl derivatives of the morphine group, VI. Synthesis of acetylmercapto and mercapto derivatives. Acta Chim. Acad. Sci. Hung. 1970, 64, 273–279. [Google Scholar]
- Makleit, S.; Somogyi, G.; Bognár, R. Tozil, illetve mezilszármazékok vizsgálata a morfinsorban, XV.: A pszeudokodein-tozilát nukleofil szubsztitúciós reakcióinak tanulmányozása. Magy. Kém. Foly. 1975, 81, 517–519. [Google Scholar]
- Makleit, S.; Somogyi, G.; Bognár, R. Conversions of tosyl and mesyl derivatives in the morphine group XV. Nucleophilic substitution reactions of pseudocodeine tosylate. Acta Chim. Acad. Sci. Hung. 1976, 82, 173–179. [Google Scholar] [CrossRef]
- Makleit, S.; Bognár, R. Tozil-, illetve mezilszármazékok vizsgálata a morfinsorban, VII. Izo-morfin és dihidro-izo-morfin új előállítása. Magy. Kém. Foly. 1970, 76, 112–113. [Google Scholar]
- Makleit, S.; Bognár, R. Conversions of tosyl and mesyl derivatives of the morphine group, VII. A new method for the preparation of isomorphine and dihydroisomorphine. Acta Chim. Acad. Sci. Hung. 1970, 64, 281–283. [Google Scholar]
- Bognár, R.; Makleit, S.; Mile, T.; Radics, L. Tozil-, ill. mezilszármazékok reakcióinak vizsgálata a morfinsorban, IX. Magy. Kém. Foly. 1972, 78, 163–166. [Google Scholar]
- Bognár, R.; Makleit, S.; Mile, T.; Radics, L. Reaktionen von Tosyl- bzw. Mesyl-Derivaten in der Morphin-Reihe, 9. Mitt. Monatsh. Chem. 1972, 103, 143–149. [Google Scholar] [CrossRef]
- Small, L.F.; Lutz, R.E. Chemistry of the Opium Alkaloids; US Treasury Department Public Health Reports; US Treasury Department: Washington, DC, USA, 1932; Volume 103, pp. 1–374.
- Inoue, H.; Takeda, M.; Kugita, H. Synthesis of B/C trans-fused morphine structures. IV. Synthesis of B/C trans-isomorphine. Chem. Pharm. Bull. 1970, 18, 1569–1575. [Google Scholar] [CrossRef]
- Kugita, H.; Takeda, M.; Inoue, H. Synthesis of B/C trans-fused morphine structures. V. Pharmacological summary of trans-morphine derivatives and an improved synthesis of trans-codeine. J. Med. Chem. 1970, 13, 973–975. [Google Scholar] [CrossRef] [PubMed]
- Makleit, S.; Berényi, S.; Bognár, R. Új, hatékony módszer neopin előállítására. Magy. Kém. Foly. 1977, 83, 478–479. [Google Scholar]
- Makleit, S.; Berényi, S.; Bognár, R. A new and efficient method for the preparation of neopine. Acta Chim. Acad. Sci. Hung. 1977, 94, 165–168. [Google Scholar]
- Berényi, S.; Makleit, S. Az izoneopin előállítása. Magy. Kém. Foly. 1980, 86, 335–336. [Google Scholar]
- Berényi, S.; Makleit, S. Synthesis of isoneopine. Acta Chim. Acad. Sci. Hung. 1980, 104, 97–99. [Google Scholar]
- Berényi, S.; Makleit, S.; Dobány, Z. Morphine alkaloids Part 105. Isolation of neopine from poppy head. Herba Hung. 1986, 25, 87–89. [Google Scholar]
- Berényi, S.; Makleit, S.; Bognár, R.; Tegdes, A. Tozil-, illetve mezilszármazékok vizsgálata a morfinsorban, XXII. A 6-O-mezil-neopin néhány nukleofil szubsztitúciós reakciója. Magy. Kém. Foly. 1979, 85, 499–501. [Google Scholar]
- Berényi, S.; Makleit, S.; Bognár, R.; Tegdes, A. Conversions of tosyl and mesyl derivatives of the morphine group, XXII. Some nucleophilic substitution reactions of 6-O-mesylneopine. Acta Chim. Acad. Sci. Hung. 1980, 103, 365–369. [Google Scholar] [CrossRef]
- Crabbendam, P.R.; Maat, L.; Beyerman, H.C. Chemistry of opium alkaloids. Part XV. Preparation of 6-demethoxythebaine from neopine and its Diels-Alder reaction with ethyl acrylate. Recl. Trav. Chim. Pays-Bas 1981, 100, 293–294. [Google Scholar] [CrossRef]
- Hutchins, C.W.; Cooper, G.K.; Puerro, S.; Rapoport, H. 6-Demethoxythebaine and its conversion to analgesics of the 6,14-ethenomorphinan type. J. Med. Chem. 1981, 24, 773–777. [Google Scholar] [CrossRef] [PubMed]
- Beyerman, H.C.; Crabbendam, P.R.; Lie, T.S.; Maat, L. Convenient conversions of codeine into 6-demethoxythebaine via bond rearrangement (Chemistry of Opium Alkaloids, Part XVIII). Recl. Trav. Chim. Pays-Bas 1984, 103, 112–114. [Google Scholar] [CrossRef]
- Hauser, F.M.; Chen, T.-K.; Carroll, F.I. 14-Hydroxycodeinone. Improved synthesis. J. Med. Chem. 1974, 17, 1117. [Google Scholar] [CrossRef]
- Iijima, I.; Minamikawa, J.; Jacobson, A.E.; Brossi, A.; Rice, K.C.; Klee, W.A. Studies in the (+)-morphinan series. 5. Synthesis and biological properties of (+)-naloxone. J. Med. Chem. 1978, 21, 398–400. [Google Scholar] [CrossRef] [PubMed]
- Currie, A.C.; Gillon, J.; Newbold, G.T.; Spring, F.S. Some reactions of 14-hydroxycodeine. J. Chem. Soc. 1960, 773–781. [Google Scholar] [CrossRef]
- Seki, I. Studies on the morphine alkaloids and its related compounds. XIII. On the nucleophilic substitution reactions of the 14-hydroxylated morphine alkaloids. Chem. Pharm. Bull. 1966, 14, 445–453. [Google Scholar] [CrossRef] [PubMed]
- Simon, C.; Berényi, S.; Makleit, S.; Fekete, V. Tozil és mezilszármazékok vizsgálata a morfinsorban, XXV. A 6-O-mezil-7α-klór(bróm)-neopin nukleofil szubsztitúciós reakcióinak vizsgálata. Magy. Kém. Foly. 1986, 92, 402–406. [Google Scholar]
- Simon, C.; Berényi, S.; Makleit, S.; Fekete, V. Conversions of tosyl and mesyl derivatives of the morphine group, XXV. Studies of the nucleophilic substitution reactions of 6-O-mesyl-7α-chloro(bromo)neopine. Acta Chim. Hung. 1987, 124, 497–503. [Google Scholar]
- Abe, K.; Nakamura, Y.; Onda, M.; Okuda, S. Studies on morphine alkaloids—VIII. Some reactions of 14β-bromocodeine and related compounds. Tetrahedron 1971, 27, 4495–4509. [Google Scholar] [CrossRef]
- Conroy, H. Neopinone. J. Am. Chem. Soc. 1955, 77, 5960–5966. [Google Scholar] [CrossRef]
- Berényi, S. A 8,14-es Helyzetben Kettős Kötést és Kettősen Allil-Rendszert Tartalmazó Morfinszármazékok SN Típusú Reakciói; C.Sc. Dissertation, Kossuth Lajos Tudományegyetem Debrecen, Debrecen, Hungary, 1984. [Google Scholar]
- Berényi, S.; Makleit, S.; Szilágyi, L. Tozil- és mezilszármazékok vizsgálata a morfinsorban, XXIII. Új 6-szubsztituált-6-demetoxi-tebain származékok előállítása. Magy. Kém. Foly. 1984, 90, 154–157. [Google Scholar]
- Berényi, S.; Makleit, S.; Szilágyi, L. Conversions of tosyl and mesyl derivatives of the morphine group, XXIII. Preparation of new 6-substtituted-6-demethoxythebaine derivatives. Acta Chim. Hung. 1984, 117, 307–312. [Google Scholar]
- Berényi, S.; Makleit, S.; Sepsi, Á. Morphine Alkaloids. Part 106. Reactions of morphine derivatives containing double allylic system. Part 2. Studies of nucleophilic substitution reactions of dihalo derivatives. Acta. Chim. Hung. 1989, 126, 275–279. [Google Scholar] [CrossRef]
- Okuda, S.; Yamaguchi, S.; Tsuda, K. Studies on morphine alkaloids. II. Indolinocodeine. I. A new skeletal rearrangement of 14-bromocodeine. Chem. Pharm. Bull. 1965, 13, 1092–1103. [Google Scholar]
- Patai, S. The chemistry of the azido group. In The Chemistry of Functional Groups; Interscience Publishers a division of John Wiley & Sons Ltd.: London, UK; New York, NY, USA; Sydney, Australia; Toronto, ON, Canada, 1971; pp. 1–626. ISBN 10: 0 471 66925 3. [Google Scholar]
- Braese, S.; Gil, C.; Knepper, K.; Zimmermann, V. Organic azides: An exploding diversity of a unique class of compounds. Angew. Chem. Int. Ed. 2005, 44, 5188–5240. [Google Scholar] [CrossRef]
- Pinho e Melo, T.M.V.D. Part 1. Synthesis and safety, Chapter 3. Synthesis of azides. In Organic Azides. Syntheses and Applications; Braese, S., Banert, K., Eds.; John Wiley & Sons, Ltd.: Hoboken, NJ, USA, 2010; pp. 53–94. [Google Scholar]
- Xie, S.; Sundhoro, M.; Houk, K.N.; Yan, M. Electrophilic azides for materials synthesis and chemical biology. Acc. Chem. Res. 2020, 53, 937–948. [Google Scholar] [CrossRef]
- Aimi, T.; Meguro, T.; Kobayashi, A.; Hosoya, T.; Yoshida, S. Nucleophilic transformations of azido-containing carbonyl compounds via protection of the azido group. Chem. Commun. 2021, 57, 6062–6065. [Google Scholar] [CrossRef] [PubMed]
- Griffin, R.J. Chapter 3. The medicinal chemistry of the azido group. In Progress in Medicinal Chemistry; Ellis, G.P., Luscombe, D.K., Eds.; Elsevier Science: Amsterdam, The Netherlands, 1994; Volume 31, pp. 121–232. [Google Scholar]
- Cocker, J.D.; Cowley, B.R.; Cox, J.S.G.; Eardley, S.; Gregory, G.I.; Lazenby, J.K.; Long, A.G.; Sly, J.C.P.; Somerfield, G.A. 922. Cephalosporanic acids. Part II. Displacement of the acetoxy-group by nucleophiles. J. Chem. Soc. 1965, 5015–5031. [Google Scholar] [CrossRef]
- Willner, D.; Holdrege, C.T.; Baker, S.R.; Cheney, L.C. 7-(α-aminophenylacetamido)-3-azidomethyl-3-cephem-4-carboxylic acid. J. Antibiot. 1972, 25, 64–67. [Google Scholar] [CrossRef]
- Lister, R.G.; Nutt, D.J. Is Ro 15-4513 a specific alcohol antagonist? Trends Neurosci. 1987, 10, 223–225. [Google Scholar] [CrossRef]
- Bach, P.; de Timary, P.; Gründer, G.; Cumming, P. Molecular imaging studies of alcohol use disorder. In Current Topics in Behavioral Neurosciences; Springer: Berlin/Heidelberg, Germany, 2023. [Google Scholar] [CrossRef]
- Halldin, C.; Farde, L.; Litton, J.E.; Hall, H.; Sedvall, G. [11C]Ro 15-4513, a ligand for visualization of benzodiazepine receptor binding. Preparation, autoradiography and positron emission tomography. Psychopharmacology 1992, 108, 16–22. [Google Scholar] [CrossRef] [PubMed]
- Winter, B.A.; Goldstein, A. A photochemical affinity-labelling reagent for the opiate receptor (s). Mol. Pharmacol. 1972, 8, 601–611. [Google Scholar] [CrossRef] [PubMed]
- Fürst, S.; Hosztafi, S.; Friedmann, T. Structure-activity relationships of synthetic and semisynthetic opioid agonists and antagonists. Curr. Med. Chem. 1995, 1, 423–440. [Google Scholar] [CrossRef]
- Fürst, S.; Hosztafi, S. The chemical and pharmacological importance of morphine analogues. Acta Physiol. Hung. 2008, 95, 3–44. [Google Scholar] [CrossRef]
- Knoll, J. Azidomorphine and rymazolium an approach to the ideal analgesic. Pharmacol. Res. Commun. 1973, 5, 175–191. [Google Scholar] [CrossRef]
- Knoll, J. Azidomorphines: A new family of potent analgesics with low dependence capacity. Prog. Neuropsychopharmacol. 1979, 3, 95–108. [Google Scholar] [CrossRef]
- Knoll, J.; Fürst, S.; Mészáros, Z.; Nagy, G.; Dávid, Á.; Bognár, R.; Makleit, S.; Valovics, G. Pharmaceutical Composition Having Synergistic Analgesic Activity and Containing Azidomorphine or Azidocodeine, Chinoin Pharmaceutical and Chemical Works Ltd.: Budapest, Hungary. U.S. Patent 4,035,491, 29 June 1976. [Google Scholar]
- Knoll, J.; Fürst, S.; Kelemen, K. The pharmacology of azidomorphine and azidocodeine. J. Pharm. Pharmacol. 1973, 25, 929–939. [Google Scholar] [CrossRef] [PubMed]
- Bulaev, V.M.; Chichenkov, O.N. Effect of morphine and azidomorphine on cortical unit activity. Bull. Exp. Biol. Med. 1977, 83, 828–830. [Google Scholar] [CrossRef]
- Bulaev, V.M.; Zakusov, V.V.; Knoll, J.; Chichenkov, O.N. Pharmacology of azidomorphine. Bull. Exp. Biol. Med. 1978, 85, 749–751. [Google Scholar] [CrossRef]
- Knoll, J.; Zsilla, G.; Makleit, S. The metabolism of azidomorphine in the rat. Med. Biol. 1975, 53, 501–504. [Google Scholar]
- Knoll, J.; Fürst, S.; Vizi, E.S. Kinetic parameters of a new narcotic agonist, azidomorphine. Pharmacology 1973, 10, 354–362. [Google Scholar] [CrossRef] [PubMed]
- Horváth, K.; Wollemann, M. Azidomorphine is an agonist of high-affinity opioid receptor binding sites. Neurochem. Res. 1986, 11, 1565–1569. [Google Scholar] [CrossRef]
- Knoll, J.; Fürst, S.; Makleit, S. The pharmacology of 14-hydroxy-azidomorphine. J. Pharm. Pharmacol. 1974, 27, 99–105. [Google Scholar] [CrossRef] [PubMed]
- Cone, E.J. The metabolism of azidomorphine in a human subject. Xenobiotica 1978, 8, 301–304. [Google Scholar] [CrossRef]
- Sasvári, K.; Simon, K.; Bognár, R.; Makleit, S. The crystal and molecular structure of 6-deoxy-6-azidodihydroisomorphine, C17H20N4O2. Acta Cryst. B 1974, 30, 634–641. [Google Scholar] [CrossRef]
- Kálmán, A.; Ignáth, Z.; Simon, K.; Bognár, R.; Makleit, S. The crystal and molecular structure of 6-deoxy-6-azido-14-hydroxydihydroisomorphine, C17H20N4O3. Acta Cryst. B 1976, 32, 2667–2670. [Google Scholar] [CrossRef]
- Tamás, J.; Mák, M.; Makleit, S. Mass spectrometric studies on azidomorphine derivatives. Org. Mass Spectrom. 1974, 9, 847–853. [Google Scholar] [CrossRef]
- Dinya, Z.; Makleit, S.; Szabó, S.; Mile, T.; Bognár, R. A morfinszármazékok C-gyűrűjének térszerkezeti adottságai és az infravörös spektrumaik közötti összefüggések vizsgálata. Magy. Kém. Foly. 1971, 77, 265–274. [Google Scholar]
- Dinya, Z.; Makleit, S.; Bognár, R. A morfinszármazékok elektron és térszerkezetének vizsgálata és összefüggésük a farmakológiai hatással. Kém. Közl. 1973, 39, 141–172. [Google Scholar]
- Dinya, Z.; Makleit, S.; Bognár, R.; Jékel, P. Quantum-chemical calculations for morphine alkaloids. Acta Chim. Acad. Sci. Hung. 1972, 71, 125–126. [Google Scholar]
- Makleit, S.; Knoll, J.; Bognár, R.; Berényi, S.; Kiss, G. Tozil-, ill. mezilszármazékok vizsgálata a morfinsorban, XVII. “Azidomorfin” -származékok, I. Magy. Kém. Foly. 1976, 82, 430–431. [Google Scholar]
- Makleit, S.; Knoll, J.; Bognár, R.; Berényi, S.; Kiss, G. Conversions of tosyl and mesyl derivatives of the morphine group, XVII. “Azidomorphine” derivatives I. Acta Chim. Acad. Sci. Hung. 1977, 93, 165–168. [Google Scholar]
- Bognár, R.; Makleit, S.; Kiss, G.; Berényi, S.; Mile, T.; Knoll, J.; Elek, S.; Gyökér, I.; Zoltai, A.; Tóth, G.; et al. 6-Deoxy-6-Azido-14-Hydroxy-7,8-Dihydro Isomorphine or a Pharmacetuically Acceptable Salt Thereof. US Patent 4,144,236, 10 May 1977. [Google Scholar]
- Bognár, R.; Makleit, S.; Knoll, J.; Berényi, S.; Horváth, G. Tozil-, illetve mezilszármazékok reakcióinak vizsgálata a morfinsorban, XIII. Morfin-alkaloidok azidoszármazékai. Kém. Közl. 1975, 44, 1–10. [Google Scholar]
- Bognár, R.; Makleit, S.; Knoll, J.; Berényi, S.; Horváth, G. Conversions of tosyl and mesyl derivatives of the morphine group. XIII. Azido derivatives of morphine alkaloids. Bulg. Chem. Commun. 1975, 8, 203–215. [Google Scholar]
- Makleit, S.; Knoll, J.; Bognár, R.; Berényi, S.; Somogyi, G.; Kiss, G. Tozil-, ill. mezilszármazékok vizsgálata a morfinsorban, XVIII. “Azidomorfin”-származékok II. Magy. Kém. Foly. 1976, 82, 432–437. [Google Scholar]
- Makleit, S.; Knoll, J.; Bognár, R.; Berényi, S.; Somogyi, G.; Kiss, G. Conversions of tosyl and mesyl derivatives of the morphine group, XVIII. “Azidomorphine” derivatives, II. Acta Chim. Acad. Sci. Hung. 1977, 93, 169–174. [Google Scholar]
- Bognár, R.; Gaál, G.; Kerekes, P.; Horváth, G.; Kovács, M.T. Hydroxyl group elimination in the morphine series. Org. Prep. Proced. Int. 1974, 6, 305–311. [Google Scholar] [CrossRef]
- Makleit, S.; Berényi, S.; Bognár, R.; Elek, S. Tozil-, illetve mezilszármazékok vizsgálata a morfinsorban, XX. A mezilészterek előállításáról. Magy. Kém. Foly. 1977, 83, 190–191. [Google Scholar]
- Makleit, S.; Berényi, S.; Bognár, R.; Elek, S. Conversions of tosyl and mesyl derivatives of the morphine group, XX. Synthesis of mesyl esters. Acta Chim. Acad. Sci. Hung. 1977, 94, 161–163. [Google Scholar]
- Knoll, J.; Makleit, S.; Friedmann, T.; Hársing, L.G.; Hadházy, P. Circulatory, respiratory and antitussive effects of azidomorphine and related substances. Arch. Int. Pharmacodyn. Ther. 1974, 210, 241–249. [Google Scholar]
- Bognár, R.; Makleit, S.; Kiss, G.; Berényi, S.; Mile, T.; Knoll, J.; Elek, S.; Gyökér, I.; Zoltai, A.; Tóth, G.; et al. 7,8-Dihydroisomorphine derivative, namely azidoethylmorphine. U.S. Patent 4,167,636, 29 March 1978. [Google Scholar]
- Makleit, S.; Knoll, J.; Bognár, R.; Berényi, S.; Somogyi, G.; Kiss, G. Tozil-, ill. mezilszármazékok vizsgálata a morfinsorban, XIX. “Azidomorfin”-származékok. Magy. Kém. Foly. 1976, 82, 434–437. [Google Scholar]
- Makleit, S.; Knoll, J.; Bognár, R.; Berényi, S.; Somogyi, G.; Kiss, G. Conversions of tosyl and mesyl derivatives of the morphine group, XIX. “Azidomorphine” derivatives III. Acta Chim. Acad. Sci. Hung. 1977, 93, 175–181. [Google Scholar]
- Knoll, J.; Fürst, S.; Makleit, S. The pharmacology of N-substituted azidomorphines. Arch. Int. Pharmacodyn. Ther. 1977, 228, 268–292. [Google Scholar] [PubMed]
- Fürst, S.; Wagner, T.A.; Knoll, J. Azidomorphines and heterogenous opiate receptors. Pol. J. Pharmacol. Pharm. 1988, 40, 627–634. [Google Scholar]
- Fürst, S.; Friedmann, T.; Kovács, A.; Wagner, T. Comparison of the opioid activity of CAM and κ agonists. In NIDA Research Monograph 75, Proceedings of the 1986 INRC Progress in Opioid Research, International Narcotics Research Conference, San Francisco, CA, USA, 6–11 July 1986; Holaday, J.W., Law, P.-Y., Herz, A., Eds.; National Institute on Drug Abuse: Rockville, MD, USA, 1986; pp. 224–227. [Google Scholar]
- Knoll, J.; Makleit, S.; Berényi, S.; Hosztafi, S.; Fürst, S.; Knoll, B.; Kiss, G.; Gyulai, Z. Eljárás N-Szubsztituált-N-Demetil-Azidoetilmorfin-Származékok Előállítására. Hungarian Patent 199,843, 27 December 1985. [Google Scholar]
- Gulyás, G.; Berényi, S.; Makleit, S. Morfinvázas alkaloidok új epoxiszármazékainak előállítása és átalakításai. Magy. Kém. Foly. 1986, 92, 508–513. [Google Scholar]
- Gulyás, G.; Berényi, S.; Makleit, S. Morphine alkaloids, 104. Synthesis and conversions of new epoxy derivatives. Acta. Chim. Hung. 1988, 125, 255–265. [Google Scholar]
- Gulyás, G.; Makleit, S. A 8β,14β-epoxi-kodid reakcióinak tanulmányozása. Magy. Kém. Foly. 1988, 94, 517–519. [Google Scholar]
- Gulyás, G.; Makleit, S. Investigation of the reactions of 8,14β-epoxycodide. Acta Chim. Hung. 1990, 127, 99–102. [Google Scholar]
- Seki, I. Studies on the morphine alkaloids and its related compounds. XV. Isolations of 8β,14β-epoxy-codide and 14-hydroxy-dihydropseudocodeine from some nucleophilic substitution reaction products of tosylate of 14-hydroxy-codeine or its isomers. Chem. Pharm. Bull. 1969, 17, 1549–1554. [Google Scholar] [CrossRef]
- Seki, I.; Takagi, H. Studies on the morphine alkaloids and its related compounds. XVI. Synthesis of 14-hydroxy-allopseudocodeine 8-ethers and its derivatives. Chem. Pharm. Bull. 1969, 17, 1555–1559. [Google Scholar] [CrossRef]
- Bognár, R.; Makleit, S.; Radics, L.; Mile, T. Tozil, illetve mezilszármazékok reakcióinak vizsgálata a morfinsorban, XI.: 14-Hidroxi-morfin-származékok, II. Halogénvegyületek. Magy. Kém. Foly. 1972, 78, 228–229. [Google Scholar]
- Makleit, S.; Radics, L.; Mile, T.; Bognár, R. Conversions of tosyl and mesyl derivatives of morphine group. XI.: 14-Hydroxymorphine derivatives. II. Halogen compounds. Acta Chim. Acad. Sci. Hung. 1972, 74, 111–113. [Google Scholar]
- Bentley, K.W.; Hardy, D.G. Novel analgesics and molecular rearrangements in the morphine-thebaine group I. Ketones derived from 6,14-endo-ethenotetrahydrtothebaine. J. Am. Chem. Soc. 1967, 89, 3267–3273. [Google Scholar] [CrossRef]
- Linders, J.T.M.; Maat, L. Face selectivity of the Diels-Alder reaction of thebaine-like morphinandienes, A computational approach (Chemistry of opium alkaloids, Part XXXI). Bull. Soc. Chim. Belg. 1989, 98, 265–276. [Google Scholar] [CrossRef]
- Maat, L. Novel thebainelike morphinan-dienes and their Diels-Alder adducts. In NIDA Research Monograph Series, Drugs of Abuse: Chemistry, Pharmacology, Immunology, and AIDS; Pham, P.T.K., Rice, K.C., Eds.; National Institutes of Health: Washington, DC, USA, 1990; pp. 35–49. [Google Scholar]
- Lewis, J.W.; Husbands, S.M. The orvinols and related opioids—High affinity ligands with diverse efficacy profiles. Curr. Pharm. Des. 2004, 10, 717–732. [Google Scholar] [CrossRef]
- Berényi, S.; Gulyás, G.; Batta, G.; Gunda, T.; Makleit, S. Steric factors in the azidolysis–thermolysis of some 5-tosyloxymethylbicyclo [2.2.2] oct-2-enes to yield 4-azatetracyclo [4.4.0.02.4.03.8]decanes. J. Chem. Soc. Perkin Trans. 1991, 1, 1139–1142. [Google Scholar] [CrossRef]
- Berényi, S.; Gulyás, G.; Batta, G.; Makleit, S. Synthesis of a new azatetracyclodecane ring system via intramolecular addition of azide on double bond. Org. Prep. Proced. Int. 1991, 23, 111–113. [Google Scholar] [CrossRef]
- Batta, G.; Gunda, T.E.; Szabó, Z.; Berényi, S.; Gulyás, G.; Makleit, S. C-19 Configurational assignments in some morphine derivatives by homonuclear NOE. Magn. Reson. Chem. 1992, 30, 96–104. [Google Scholar] [CrossRef]
- Sepsi, Á.; Berényi, S.; Makleit, S.; Tóth, Z. Morphine Alkaloids, CXVII: Investigation of the azidolysis of tertiary alcohols of thebaine derivatives with bridged ring C. Arch. Pharm. 1993, 326, 313–317. [Google Scholar] [CrossRef]
- Pancrazi, A.; Khuong-Huu, Q. Alcaloides steroidiques—CLXVII: Synthese d’azides tertiaires en serie alicyclique—III. Tetrahedron 1974, 30, 2337–2343. [Google Scholar] [CrossRef]
- Cone, E.J.; Gorodetzky, C.W.; Darwin, W.D.; Buchwald, W.F. Stability of the 6,14-endo-ethanotetrahydrooripavine analgesics: Acid-catalyzed rearrangement of buprenorphine. J. Pharm. Sci. 1984, 73, 243–246. [Google Scholar] [CrossRef]
- Husbands, S.M.; Breeden, S.W.; Grivas, K.; Lewis, J.W. Ring constrained analogues of the orvinols: The furanomorphides. Bioorg. Med. Chem. Lett. 1999, 9, 831–834. [Google Scholar] [CrossRef]
- Csutorás, C.; Berényi, S.; Czakó, B.; Makleit, S. Syntheses and transformations of novel nitrogen and sulfur containing morphinandienes. Monatsh. Chem. 1997, 128, 1267–1273. [Google Scholar] [CrossRef]
- Gates, M.; Tschudi, G. The synthesis of morphine. J. Am. Chem. Soc. 1956, 78, 1380–1393. [Google Scholar] [CrossRef]
- Gates, M.; Shepard, M.S. The closure of the oxide bridge in the morphine series. J. Am. Chem. Soc. 1962, 84, 4125–4130. [Google Scholar] [CrossRef]
- Weller, D.D.; Rapoport, H. Practical synthesis of codeine from dihydrothebainone. J. Med. Chem. 1976, 19, 1171–1175. [Google Scholar] [CrossRef]
- Rice, K.C. Synthetic opium alkaloids and derivatives. A short total synthesis of (±)-dihydrothebainone, (±)-dihydrocodeinone, and (±)-nordihydrocodeinone as an approach to a practical synthesis of morphine, codeine and congeners. J. Org. Chem. 1980, 45, 3135–3137. [Google Scholar] [CrossRef]
- Tóth, G.; Mallareddy, J.R. Tritiated opioid receptor ligands as radiotracers. Curr. Pharm. Design 2013, 19, 7461–7472. [Google Scholar] [CrossRef]
- Filer, C.N. Morphinan alkaloids labeled with tritium: Synthesis and applications. J. Label. Compd. Radiopharm. 2013, 56, 639–648. [Google Scholar] [CrossRef]
- Gillis, E.P.; Eastman, K.J.; Hill, M.D.; Donnelly, D.J.; Meanwell, N.A. Applications of fluorine in medicinal chemistry. J. Med. Chem. 2015, 58, 8315–8359. [Google Scholar] [CrossRef]
- Sandulenko, I.V.; Ambartsumyan, A.A.; Moiseev, S.K. Fluorinated and [18F]fluorinated morphinan based opioid ligands. Org. Biomol. Chem. 2020, 18, 5533–5557. [Google Scholar] [CrossRef]
- Makleit, S.; Berényi, S.; Hoszafi, S.; Simon, C. Synthesis and chemical transformation of halogen-containing morphine derivatives. Magy. Kém. Lapja 1997, 52, 282–289. [Google Scholar]
- Somogyi, G.; Makleit, S.; Bognár, R. Tozil-, illetve mezilszármazékok vizsgálata a morfinsorban, XXI. A dihidrokodein C-6-halogénszármazékai. Magy. Kém. Foly. 1977, 83, 327–330. [Google Scholar]
- Somogyi, G.; Makleit, S.; Bognár, R. Conversions of tosyl and mesyl derivatives of the morphine group, XXI. C-6 Halogen derivatives of dihydrocodeine. Acta Chim. Acad. Sci. Hung. 1978, 97, 339–344. [Google Scholar]
- Boswell, G.A.; Henderson, R.M. Fluoro Analogs of Hydrocodone and Oxycodone Useful as Analgesics, Narcotic Antagonists or Both. U.S. Patent 4,241,065, 2 July 1979. [Google Scholar]
- Ganti, V. Analgesic, Antagonist, and/or Anorectic 14-Fluoromorphinans. US Patent 4,451,470, 6 July 1982. [Google Scholar]
- Rice, K.C.; Pert, C.B.; Burke, T.R.; Larson, S.M.; Eckelman, W.C.; Channing, M.A. Synthesis and Utilization of 17-methyl and 17-cyclopropylmethyl-3,14-dihydroxy-4,5α-epoxy-6β-fluoromorphinans (foxy and cyclofoxy) as 18F-Labeled Opioid Ligands for Positron Emission Transaxial Tomography (PETT). U.S. Patent 4,775,759, 27 November 1984. [Google Scholar]
- Burke, T.R.; Rice, K.C.; Pert, C.B. Probes for narcotic receptor mediated phenomena. 11. Synthesis of 17-methyl and 17-cycloporopylmethyl-3,14-dihydroxy-4,5α-epoxy-6β-fluoromorphinans (foxy and cyclofoxy) as a model of opioid ligands suitable for positron emission transaxial tomography. Heterocycles 1985, 23, 99–106. [Google Scholar]
- Pert, C.B.; Danks, J.A.; Channing, M.A.; Eckelman, W.C.; Larson, S.M.; Bennett, J.M.; Burke, T.R., Jr.; Rice, K.C. S-[18F]Acetylcyclofoxy: A useful probe for the visualization of opiate receptors in living animals. FEBS Letters 1984, 177, 281–286. [Google Scholar] [CrossRef]
- Channing, M.S.; Eckelman, W.C.; Bennett, J.M.; Burke, T.R.; Rice, K.C. Radiosynthesis of (S)[18F]F-3-acetylcyclofoxy: A high affinity opiate antagonist. Int. J. Appl. Radiat. Isot. 1985, 36, 429–433. [Google Scholar] [CrossRef]
- Newman, A.; Channing, M.; Finn, R.; Dunn, B.; Simpson, N.; Carson, R.; Ostrowski, N.; Cohen, R.; Burke, T.; Larson, S.; et al. Ligands for Imaging Opioid Receptors in Conscious Humans by Positron Emission Tomography (PET). In NIDA Research Monograph, Problems of Drug Dependence; National Institutes of Health: Washington, DC, USA, 1988; pp. 117–121. [Google Scholar]
- Kling, M.A.; Carson, R.E.; Borg, L.; Zametkin, A.; Matochik, J.A.; Schluger, J.; Herscovitch, P.; Rice, K.C.; Ho, A.; Eckelman, W.C.; et al. Opioid receptor imaging with positron emission tomography and [18F]cyclofoxy in long-term, methadone-treated former heroin addicts. J. Pharm. Exp. Ther. 2000, 295, 1070–1076. [Google Scholar] [CrossRef]
- Woudenberg, R.H.; Maat, L. Synthesis and biological activity of new etorphine analogues from 7-chloro-6-demethoxythebaine and 7-chloro-5β-methyl-6-demethoxythebaine (Chemistry of Opium Alkaloids, part XXXVII). Recl. Trav. Chim. Pays-Bas 1993, 112, 113–122. [Google Scholar] [CrossRef]
- Maat, L.; Woudenberg, R.H.; Meuzelaar, G.J.; Linders, J.T.M. Chemistry of opium alkaloids. Part 44: Synthesis and opioid receptor binding profile of substituted ethenoisomorphinans and ethenomorphinans. Bioorg. Med. Chem. 1999, 7, 529–541. [Google Scholar] [CrossRef]
- Marton, J.; Szabó, Z.; Csorvássy, I.; Simon, C.; Hosztafi, S.; Makleit, S. Reaction of morphinan-6, 8-dienes with azadienophiles. Tetrahedron 1996, 52, 2449–2464. [Google Scholar] [CrossRef]
- Jeong, I.H.; Kim, Y.S.; Cho, K.Y.; Kim, K.J. Unexpected effect of fluorine in Diels-Alder reaction of 2-fluoroacrolein with thebaine. Bull. Korean Chem. Soc. 1990, 11, 178–179. [Google Scholar]
- Jeong, I.H.; Kim, Y.S.; Cho, K.Y.; Kim, K.J. Reaction of thebaine with trifluoromethyl substituted acetylenic dienophiles. Bull. Korean Chem. Soc. 1990, 11, 258–260. [Google Scholar]
- Kim, K.J.; Kim, S.M. Synthesis of fluorinated new thebaine derivatives as analgesics. Yakhak Hoechi 1998, 42, 257–264. [Google Scholar]
- Berényi, S.; Hosztafi, S.; Makleit, S. A new efficient method for the preparation of 2-fluoro-N-propyl-norapomorphine. J. Chem. Soc. Perkin Trans. 1992, 1, 2693–2694. [Google Scholar] [CrossRef]
- Csutorás, C.; Berényi, S.; Neumeyer, J.L. Investigation of the acid-catalyzed rearrangement of morphinans. Lett. Org. Chem. 2007, 4, 409–413. [Google Scholar] [CrossRef]
- Berényi, S.; Tóth, Z.; Sepsi, Á.; Zékány, A.; Gyulai, S.; Makleit, S. Synthesis and biological evaluation of some halogenated 6,14-ethenomorphinan derivatives. Med. Chem. Res. 1995, 5, 26–32. [Google Scholar]
- Savić-Vujović, K.R.; Vučković, S.; Srebro, D.; Ivanović, M.; Došen-Mićović, L.; Vučetić, Č.; Džolijić, E.; Prostran, M. A comparison of the antinociceptive and temperature responses to morphine and fentanyl derivatives in rats. Arch. Pharm. Res. 2013, 36, 501–508. [Google Scholar] [CrossRef]
- Magnus, P.; Ghavimi-Alagha, B. Chemical Transformations of (-)-Codeine to Afford Derivatives Codeine and Morphine Thereof. WO 2013/184,902, 7 June 2012. [Google Scholar]
- Ramsby, S.; Neumeyer, J.L.; Grigoriadis, D.; Seeman, P. 2-Haloaporphines as potent dopamine agonists. J. Med. Chem. 1989, 32, 1198–1201. [Google Scholar] [CrossRef]
- Baldessarini, R.J.; Kula, N.S.; Gao, Y.; Campbell, A.; Neumeyer, J.L. R-(-)-2-Fluoro-N-n-propylnorapomorphine: A very potent and D2-selective dopamine agonist. Neuropharmacology 1991, 30, 97–99. [Google Scholar] [CrossRef]
- Hosztafi, S.; Makleit, S. Synthesis of new morphine derivatives containing halogen in the aromatic ring. Synth. Commun. 1994, 24, 3031–3045. [Google Scholar] [CrossRef]
- Hosztafi, S.; Makleit, S. Synthesis of new apomorphine derivatives containing halogen (CI and Br) in ring-D. Synth. Commun. 1996, 26, 3909–3918. [Google Scholar] [CrossRef]
- Filer, C.N.; Ahern, D.G. Preparation of (-)-[8,9-3H]apomorphine at high specific activity. J. Org. Chem. 1980, 45, 3918–3919. [Google Scholar] [CrossRef]
- Filer, C.N. Radiopharmaceutical and Methods of Synthesis and Use Thereof. US. Patent 7,323,566, 20 April 2005. [Google Scholar]
- Lousberg, R.J.J.C.; Weiss, U. The analgesic action of 1-fluorocodeine. Experientia 1974, 30, 1440–1441. [Google Scholar] [CrossRef]
- Makleit, S.; Dubina, V. A gram scale preparation of 1-fluorocodeine and 1-fluorodihydrocodeine. ACH-Models Chem. 2000, 137, 447–449. [Google Scholar]
- Hosztafi, S.; Marton, J. Synthesis of 1-fluoro-substituted codeine derivatives. Chem. Pap. 2016, 70, 973–982. [Google Scholar] [CrossRef]
- Hosztafi, S.; Kraszni, M.; Tóth, G.; Marton, J. Synthesis of 1-Iodo-substituted codeine derivatives. Lett. Org. Chem. 2018, 15, 1012–1020. [Google Scholar] [CrossRef]
- Sandulenko, I.V.; Kovaleva, E.S.; Peregudov, A.S.; Kalinin, V.N.; Moiseev, S.K. 21,21,21-Trifluorothevinone: The straightest way to fluorinated thevinols and orvinols. ChemistrySelect 2016, 1, 1004–1005. [Google Scholar] [CrossRef]
- Zelentsova, M.; Sandulenko, I.V.; Ambartsumyan, A.; Danshina, A.; Moiseev, S.K. C (21)-Di-and monofluorinated scaffold for thevinol/orvinol-based opioid receptor ligands. Org. Biomol. Chem. 2023, 21, 9091–9100. [Google Scholar] [CrossRef]
- Zelentsova, M.; Moiseev, S.K. Synthesis of aryl-21,21,21-triflurothevinols. Adv. Chem. Chemical Technol. (Uspekhi V Khimii I Khimicheskoi Tekhnologii) 2018, 32, 32–34. [Google Scholar]
- Zelentsova, M.V.; Sandulenko, I.V.; Melnikova, E.K.; Moiseev, S.K. 19F NMR determination of the C20 absolute configuration of C21-fluorinated arylthevinols. Mendeleev Commun. 2022, 32, 97–99. [Google Scholar] [CrossRef]
- Sandulenko, I.V.; Belozertseva, I.V.; Zvartau, E.E.; Zelentsova, M.V.; Ambartsumyan, A.A.; Smol’yakov, A.F.; Moiseev, S.K. C (21)-fluorinated thevinol scaffold for opioid ligands. 21, 21, 21-Trifluoro-6-O-nororvinols: Design, synthesis and analgesic activity. Eur. J. Med. Chem. 2023, 252, 115296. [Google Scholar] [CrossRef]
- Szűcs, E.; Marton, J.; Szabó, Z.; Hosztafi, S.; Kékesi, G.; Tuboly, G.; Bánki, L.; Horváth, G.; Szabó, P.T.; Tömböly, C.; et al. Synthesis, biochemical, pharmacological characterization and in silico profile modelling of highly potent opioid orvinol and thevinol derivatives. Eur. J. Med. Chem. 2020, 191, 112145. [Google Scholar] [CrossRef] [PubMed]
- Marton, J.; Hosztafi, S.; Berényi, S.; Simon, C.; Makleit, S. Herstellung von 6, 14-Ethenomorphinan-derivaten. Monatsh. Chem. 1994, 125, 1229–1239. [Google Scholar] [CrossRef]
- Sandulenko, I.V.; Kovaleva, E.S.; Zelentsova, M.V.; Ambartsumyan, A.A.; Gorlov, S.N.; Danshina, A.A.; Aysin, R.R.; Moiseev, S.K. Control of the diastereoselectivity at C (20) in the formation of C (21)-fluorinated thevinols. Org. Biomol. Chem. 2023, 21, 1440–1449. [Google Scholar] [CrossRef]
- Bentley, K.W.; Hardy, D.G.; Meek, B. Novel analgesics and molecular rearrangements in the morphine-thebaine group. II. Alcohols derived from 6, 14-endo-etheno-and 6, 14-endo-ethanotetrahydrothebaine. J. Am. Chem. Soc. 1967, 89, 3273–3280. [Google Scholar] [CrossRef] [PubMed]
- Kumar, V.; Ridzwan, I.E.; Grivas, K.; Lewis, J.W.; Clark, M.J.; Meurice, C.; Jimenez-Gomez, C.; Pogozheva, I.; Mosberg, H.; Traynor, J.R.; et al. Selectively promiscuous opioid ligands: Discovery of high affinity/low efficacy opioid ligands with substantial nociceptin opioid peptide receptor affinity. J. Med. Chem. 2014, 57, 4049–4057. [Google Scholar] [CrossRef]
- Husbands, S.M. Buprenorphine and related orvinols. In Research and Development of Opioid-Related Ligands; ACS Symposium Series; American Chemical Society: Washington, DC, USA, 2013; pp. 127–144. [Google Scholar]
- Sandulenko, I.V.; Zelentsova, M.V.; Tyutyunov, A.A.; Moiseev, S.K. Thevinoic acid fluoroalkyl esters. Russ Chem. Bull. 2022, 71, 2754–2756. [Google Scholar] [CrossRef]
- Finke, A.O.; Krasnov, V.I.; Rybalova, T.V.; Chirkova, V.Y.; Belenkaya, S.V.; Volosnikova, E.A.; Shcherbakov, D.N.; Shults, E.E. A straightforward trifluoromethylation at the C6 position of morphinane alkaloids, their modification and evaluation of inhibition of the SARS-CoV-2 main protease. J. Fluor. Chem. 2023, 271, 110189. [Google Scholar] [CrossRef]
- Mitsunobu, O.; Yamada, M. Preparation of esters of carboxylic and phosphoric acid via quaternary phosphonium salts. Bull. Chem. Soc. Jpn. 1967, 40, 2380–2382. [Google Scholar] [CrossRef]
- Mitsunobu, O. The use of diethyl azodicarboxylate and triphenylphosphine in synthesis and transformation of natural products. Synthesis 1981, 1, 1–28. [Google Scholar] [CrossRef]
- Fletcher, S. The Mitsunobu reaction in the 21st century. Org. Chem. Front. 2015, 2, 739–752. [Google Scholar] [CrossRef]
- Simon, C.; Hosztafi, S.; Makleit, S. Application of the Mitsunobu reaction in the field of alkaloids. J. Heterocycl. Chem. 1997, 34, 349–365. [Google Scholar] [CrossRef]
- Robson, L.E.; Kosterlitz, H.W. Specific protection of the binding sites of D-Ala2-D-Leu5-enkephalin (δ-receptors) and dihydromorphine (μ-receptors). Proc. Royal Soc. Lond. B. Biol. Sci. 1979, 205, 425–432. [Google Scholar]
- Smith, J.R.; Simon, E.J. Selective protection of stereospecific enkephalin and opiate binding against inactivation by N-ethylmaleimide: Evidence for two classes of opiate receptors. Proc. Natl. Acad. Sci. USA 1980, 71, 281–284. [Google Scholar] [CrossRef]
- Bowen, W.D.; Hellewell, S.B.; Kelemen, M.; Huey, R.; Stewart, D. Affinity labeling of δ-opiate receptors using [D-Ala2, Leu5, Cys6] enkephalin. Covalent attachment via thiol-disulfide exchange. J. Biol. Chem. 1987, 262, 13434–13439. [Google Scholar] [CrossRef]
- Fujii, I.; Togame, H.; Yamamoto, M.; Kanematsu, K.; Takayanagi, I.; Konno, F. Design and synthesis of sulfur-containing morphine and an opioid receptor probe. Chem. Pharm. Bull. 1988, 36, 2282–2285. [Google Scholar] [CrossRef] [PubMed]
- Okuda, S.; Yamaguchi, S.; Kawazoe, Y.; Tsuda, K. Studies on Morphine Alkaloids. I. Nuclear magnetic resonance spectral studies on morphine alkaloids. I. Chem. Pharm. Bull. 1964, 12, 104–112. [Google Scholar] [CrossRef]
- Kanematsu, K.; Naito, R.; Shimohigashi, Y.; Ohno, M.; Ogasawara, T.; Kurono, M.; Yagi, K. Design synthesis of an opioid receptor probe: Mode of binding of S-Activated (-)-6-β-sulfhydryldihydromorphine with the SH group in the mu-opioid receptor. Chem. Pharm. Bull. 1990, 38, 1438–1440. [Google Scholar] [CrossRef]
- Kanematsu, K.; Yoshiyasu, T.; Yoshida, M. Molecular srtucture of (-)-3-acetyl-6-beta-(acetylthio)-N-(cyclopropylmethyl) normorphine and its 14-hydroxy congeners. Chem. Pharm. Bull. 1990, 38, 1441–1443. [Google Scholar] [CrossRef]
- Fleischhacker, W.; Richter, B. Claisen-Reaktionen an Codeinen. 8-Alkyl-8, 14-dihydro-6-desmethoxythebaine und-oripavine. Chem. Ber. 1980, 113, 3866–3880. [Google Scholar] [CrossRef]
- Kirby, G.W.; Massey, S.R. An efficient preparation of isocodeine from codeine. J. Chem. Soc. C 1971, 3047–3048. [Google Scholar] [CrossRef] [PubMed]
- Simon, C. A Mitsunobu-Reakció Alkalmazása a Morfin-Alkaloidok Körében. C.Sc. Dissertation, Kossuth Lajos Tudományegyetem Debrecen, Debrecen, Hungary, 1993. [Google Scholar]
- Simon, C.; Hosztafi, S.; Makleit, S. Application of the Mitsunobu reaction for the preparation of isomorphine and isocodeine derivatives. Synth. Commun. 1991, 21, 407–412. [Google Scholar] [CrossRef]
- Friedmann, T.; Tóth, Z.; Hosztafi, S.; Simon, C.; Makleit, S.; Fürst, S. Influence of spatial orientation of the C-6-OH group in ring C of morphine derivatives on opioid activity. Arch. Int. Pharmacodyn. Thér. 1994, 328, 16–25. [Google Scholar]
- Maat, L.; De Groot, J.A.; Beyerman, H.C. Synthesis of 6β-Halogenodeoxyneopine from Neopine. Synth. Commun. 1979, 9, 713–718. [Google Scholar] [CrossRef]
- Jiang, J.B.; Hanson, R.N.; Portoghese, P.S.; Takemori, A.E. Stereochemical studies on medicinal agents. 23. Synthesis and biological evaluation of 6-amino derivatives of naloxone and naltrexone. J. Med. Chem. 1977, 20, 1100–1102. [Google Scholar] [CrossRef]
- Sayre, L.M.; Portoghese, P.S. Stereospecific synthesis of the 6α- and 6β-amino derivatives of naltrexone and oxymorphone. J. Org. Chem. 1980, 45, 3366–3368. [Google Scholar] [CrossRef]
- Mohamed, M.S.; Portoghese, P.S. Stereoselectivity of the reduction of naltrexone oxime with borane. J. Org. Chem. 1986, 51, 105–106. [Google Scholar] [CrossRef]
- Simon, C.; Hosztafi, S.; Makleit, S. Application of the Mitsunobu reaction for morphine compounds. Preparation of 6β-aminomorphine and codeine derivatives. Morphine Alkaloids Part 115. Synth. Commun. 1992, 22, 913–921. [Google Scholar] [CrossRef]
- Simon, C.; Hosztafi, S.; Makleit, S. Applicaltion of the Mitsunobu reaction in the morphine series. Preparation of 6β-amino-14β-hydroxymorphine and 14-hydroxycodeine derivatives. Heterocycles 1994, 38, 1347–1354. [Google Scholar]
- Simon, C.; Hosztafi, S.; Makleit, S. Stereoselective synthesis of β-naltrexol, β-naloxol β-naloxamine, β-naltrexamine and related compounds by the application of the Mitsunobu reaction. Tetrahedron 1994, 50, 9757–9768. [Google Scholar] [CrossRef]
- Szilágyi, L.; Makleit, S.; Hosztafi, S.; Simon, C. Substituent-dependent conformational changes in 6β-substituted codeine derivatives. Magn. Reson. Chem. 1992, 30, 552–557. [Google Scholar] [CrossRef]
- Riba, P.; Tóth, Z.; Hosztafi, S.; Friedmann, T.; Fürst, S. Structure-activity relationships of N-cyclopropylmethyl morphine derivatives in isolated organs. Analgesia 1995, 1, 785–788. [Google Scholar] [CrossRef]
- Abe, K.; Onda, M.; Okuda, S. Studies on Morphine Alkaloids. V. Reaction Mechanism of the Reduction of 14β-Bromocodeinone with Sodium Borohydride. Chem. Pharm. Bull. 1969, 17, 1847–1853. [Google Scholar] [CrossRef]
- Simon, C.; Hosztafi, S.; Makleit, S. Application of the Mitsunobu reaction in the morphine series: The reaction of 14β-chloro and 14β-bromocodeine with phthalimide and diphenylphosphoryl azide. Tetrahedron Lett. 1993, 34, 6475–6478. [Google Scholar] [CrossRef]
- Simon, C.; Hosztafi, S.; Makleit, S. The first preparation of 6β-bromo codeine and morphine derivatives. Kinetic vs. thermodynamic control. J. Chem. Res. 1997, 12, 2734–2742. [Google Scholar] [CrossRef]
- Fujimura, M.; Izumimoto, N.; Momen, S.; Yoshikawa, S.; Kobayashi, R.; Kanie, S.; Hirakata, M.; Komagata, T.; Okanishi, S.; Hashimoto, T.; et al. Characteristics of TRK-130 (Naltalimide), a novel opioid ligand, as a new therapeutic agent for overactive bladder. J. Pharmacol. Exp. Ther. 2014, 350, 543–551. [Google Scholar] [CrossRef]
- Izumimoto, N.; Kawai, K.; Kawamura, K.; Fujimura, M.; Komagata, T. Remedies or Preventives for Urinary Frequency or Urinary Incontinence and Morphinan Derivatives Having Nitrogen-Containing Heterocyclic Group. U.S. Patent 7,320,984, 8 October 2003. [Google Scholar]
- Matyas, G.R.; Rice, K.C.; Cheng, K.; Li, F.; Antoline, J.F.; Iyer, M.R.; Jacobson, A.E.; Mayorov, A.V.; Beck, Z.; Torres, O.B.; et al. Facial recognition of heroin vaccine opiates: Type 1 cross-reactivities of antibodies induced by hydrolytically stable haptenic surrogates of heroin, 6-acetylmorphine, and morphine. Vaccine 2014, 32, 1473–1479. [Google Scholar] [CrossRef] [PubMed]
- Jalah, R.; Torres, O.B.; Mayorov, A.V.; Li, F.; Antoline, J.F.; Jacobson, A.E.; Rice, K.C.; Deschamps, J.R.; Beck, Z.; Alving, C.R.; et al. Efficacy, but not antibody titer or affinity, of a heroin hapten conjugate vaccine correlates with increasing hapten densities on tetanus toxoid, but not on CRM197 carriers. Bioconjugate Chem. 2015, 26, 1041–1053. [Google Scholar] [CrossRef]
- Lam, R.; Gondin, A.B.; Canals, M.; Kellam, B.; Briddon, S.J.; Graham, B.; Scammells, P.J. Fluorescently labeled morphine derivatives for bioimaging studies. J. Med. Chem. 2018, 61, 1316–1329. [Google Scholar] [CrossRef]
- Nagase, H.; Hayakawa, J.; Kawamura, K.; Kawai, K.; Takezawa, Y.; Matsuura, H.; Tajima, C.; Endo, T. Discovery of a structurally novel opioid κ-agonist derived from 4,5-epoxymorphinan. Chem. Pharm. Bull. 1998, 46, 366–369. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Tang, K.; Inan, S.; Siebert, D.; Holzgrabe, U.; Lee, D.Y.; Huang, P.; Li, J.G.; Cowan, A.; Liu-Chen, L.Y. Comparison of pharmacological activities of three distinct κ ligands (Salvinorin A, TRK-820 and 3FLB) on κ opioid receptors in vitro and their antipruritic and antinociceptive activities in vivo. J. Pharm. Exp. Ther. 2005, 312, 220–230. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, S.; Sugawara, Y.; Tsuji, R.; Tanimura, R.; Kaneko, C.; Yuzawa, N.; Yagi, M.; Kawai, K. Discovery of highly selective κ-opioid receptor agonists: 10α-Hydroxy TRK-820 derivatives. Bioorg. Med. Chem. Lett. 2017, 27, 3920–3924. [Google Scholar] [CrossRef]
- Váradi, A.; Hosztafi, S.; Le Rouzic, V.; Tóth, G.; Urai, Á.; Noszál, B.; Pasternak, G.W.; Grinnell, S.G.; Majumdar, S. Novel 6β-acylaminomorphinans with analgesic activity. Eur. J. Med. Chem. 2013, 69, 786–789. [Google Scholar] [CrossRef]
- Urai, A.; Váradi, A.; Szőcs, L.; Komjáti, B.; Le Rouzic, V.; Hunkele, A.; Pasternak, G.W.; Majumdar, S.; Hosztafi, S. Synthesis and pharmacological evaluation of novel selective MOR agonist 6β-pyridinyl amidomorphines exhibiting long-lasting antinociception. MedChemComm 2017, 8, 152–157. [Google Scholar] [CrossRef]
- Lever, J.R. PET and SPECT imaging of the opioid system: Receptors, radioligands and avenues for drug discovery and development. Curr. Pharm. Design 2007, 13, 33–49. [Google Scholar] [CrossRef]
- Henriksen, G.; Willoch, F. Imaging of opioid receptors in the central nervous system. Brain 2008, 131, 1171–1196. [Google Scholar] [CrossRef] [PubMed]
- Dannals, R.F. Positron emission tomography radioligands for the opioid system. J. Label. Compd. Radiopharm. 2013, 56, 187–195. [Google Scholar] [CrossRef]
- Dannals, R.F.; Ravert, H.T.; Frost, J.J.; Wilson, A.A.; Burns, H.D.; Wagner, H.N. Radiosynthesis of an opiate receptor binding radiotracer: [11C]carfentanil. Int. J. Appl. Radiat. Isot. 1985, 36, 303–306. [Google Scholar] [CrossRef] [PubMed]
- Berényi, S.; Csutorás, C.; Sipos, A. Recent developments in the chemistry of thebaine and its transformation products as pharmacological targets. Curr. Med. Chem. 2009, 16, 3215–3242. [Google Scholar] [CrossRef]
- Lever, J.R.; Kinter, C.M.; Ravert, H.T.; Musachio, J.L.; Mathews, W.B.; Dannals, R.F. Synthesis of N1′-([11C]methyl)naltrindole ([11C]MeNTI): A radioligand for positron emission tomographic studies of delta opioid receptors. J. Label. Compd. Radiopharm. 1995, 36, 137–145. [Google Scholar] [CrossRef]
- Mathews, W.B.; Kinter, C.M.; Palma, J.; Daniels, R.V.; Ravert, H.T.; Dannals, R.F.; Lever, J.R. Synthesis of N1′-([18F]fluoroethyl)naltrindole ([18F]FEtNTI): A radioligand for positron emission tomographic studies of delta opioid receptors. J. Label. Compd. Radiopharm. 1999, 42, 43–54. [Google Scholar] [CrossRef]
- Majumdar, S.; Burgman, M.; Haselton, N.; Grinnell, S.; Ocampo, J.; Pasternak, A.R.; Pasternak, G.W. Generation of novel radiolabeled opiates through siete-selective iodination. Bioorg. Med. Chem. Lett. 2011, 21, 4001–4004. [Google Scholar] [CrossRef]
- Bentley, K.W. The morphine alkaloids. In The Alkaloids: Chemistry and Physiology; Manske, R.H.F., Ed.; Academic Press Inc.: New York, NY, USA; London, UK, 1971; Volume 13. [Google Scholar]
- Coop, A.; Lewis, J.W.; Rice, K.C. Direct and simple O-demethylation of thebaine to oripavine. J. Org. Chem. 1996, 61, 6774. [Google Scholar] [CrossRef]
- Coop, A.; Janetka, J.W.; Lewis, J.W.; Rice, K.C. L-Selectride as a general reagent for the O-demethylation and N-decarbomethoxylation of opium alkaloids and derivatives. J. Org. Chem. 1998, 63, 4392–4396. [Google Scholar] [CrossRef]
- Hosztafi, S. Recent Advances in the Chemistry of Oripavine and Its Derivatives. Adv. Biosci. Biotechnol. 2014, 5, 704–717. [Google Scholar] [CrossRef]
- Lever, J.R.; Dannals, R.F.; Wilson, A.A.; Ravert, H.T.; Wagner, H.N., Jr. Synthesis of carbon-11 labeled diprenorphine: A radioligand for positron emission tomographic studies of opiate receptors. Tetrahedron Lett. 1987, 28, 4015–4018. [Google Scholar] [CrossRef]
- Luthra, S.K.; Brady, F.; Turton, D.R.; Brown, D.J.; Dowsett, K.; Waters, S.L.; Jones, A.K.P.; Matthews, R.W.; Crowder, J.C. Automated radiosyntheses of [6-O-methyl-11C]diprenorphine and [6-O-methyl-11C]buprenorphine from 3-O-trityl protected precursors. Appl. Radiat. Isot. 1994, 45, 857–873. [Google Scholar] [CrossRef]
- Lever, J.R.; Mazza, S.M.; Dannals, R.F.; Ravert, H.T.; Wilson, A.A.; Wagner, J.H.N. Facile synthesis of [11C]buprenorphine for positron emission tomographic studies of opioid receptors. Int. J. Radiat. Appl. Instrum. Part A. Appl. Radiat. Isot. 1990, 41, 745–752. [Google Scholar] [CrossRef]
- Marton, J.; Schoultz, B.W.; Hjørnevik, T.; Drzezga, A.; Yousefi, B.H.; Wester, H.-J.; Willoch, F.; Henriksen, G. Synthesis and evaluation of a full-agonist orvinol for PET-Imaging of opioid receptors: [11C]PEO. J. Med. Chem. 2009, 52, 5586–5589. [Google Scholar] [CrossRef]
- Marton, J.; Henriksen, G. Design and synthesis of an 18F-labeled version of phenylethyl orvinol ([18F]FE-PEO) for PET-imaging of opioid receptors. Molecules 2012, 17, 11554–11569. [Google Scholar] [CrossRef] [PubMed]
- Riss, P.J.; Hong, Y.T.; Marton, J.; Caprioli, D.; Williamson, D.J.; Ferrari, V.; Saigal, N.; Roth, B.L.; Henriksen, G.; Fryer, T.D.; et al. Synthesis and evaluation of 18F-FE-PEO in rodents: An 18F-labeled full agonist for opioid receptor imaging. J. Nucl. Med. 2013, 54, 299–305. [Google Scholar] [CrossRef] [PubMed]
- Schoultz, B.W.; Hjørnevik, T.; Reed, B.J.; Marton, J.; Coello, C.S.; Willoch, F.; Henriksen, G. Synthesis and evaluation of three structurally related 18F-labeled orvinols of different intrinsic activities: 6-O-[18F]Fluoroethyl-diprenorphine ([18F]FDPN), 6-O-[18F]fluoroethyl-buprenorphine ([18F]FBPN), and 6-O-[18F]fluoroethyl-phenethyl-orvinol ([18F]FPEO). J. Med. Chem. 2014, 57, 5464–5469. [Google Scholar] [PubMed]
- Wester, H.-J.; Willoch, F.; Tölle, T.R.; Munz, F.; Herz, M.; Øye, I.; Schadrack, J.; Schwaiger, M.; Bartenstein, P. 6-O-(2-[18F]Fluoroethyl-6-O-desmethyldiprenorphine ([18F]DPN): Synthesis, biologic evaluation, and comparison with [11C]DPN in humans. J. Nucl. Med. 2000, 41, 1279–1286. [Google Scholar]
- Schoultz, B.W.; Reed, B.J.; Marton, J.; Willoch, F.; Henriksen, G. A fully automated radiosynthesis of [18F]fluoroethyl-diprenorphine on a single module by use of SPE cartridges for preparation of high quality 2-[18F]fluoroethyl tosylate. Molecules 2013, 18, 7271–7278. [Google Scholar] [CrossRef]
- Marton, J.; Cumming, P.; Bauer, B.; Henriksen, G. A new precursor for the radiosynthesis of 6-O-(2-[18F]fluoroethyl)-6-O-desmethyl-diprenorphine ([18F]FE-DPN) by nucleophilic radiofluorination. Lett. Org. Chem. 2021, 18, 344–352. [Google Scholar] [CrossRef]
- Németh, E.; Gyuricza, B.; Forgács, V.; Cumming, P.; Henriksen, G.; Marton, J.; Bauer, B.; Mikecz, P.; Fekete, A. Optimization of a Nucleophilic Two-Step Radiosynthesis of 6-O-(2-[18F]fluoroethyl)-6-O-desmethyl-diprenorphine ([18F] FE-DPN) for PET Imaging of Brain Opioid Receptors. Int. J. Mol. Sci. 2023, 24, 13152. [Google Scholar] [CrossRef]
- Levinstein, M.R.; Ventriglia, E.N.; Gomez, J.L.; Budinich, R.C.; Marton, J.; Henriksen, G.; Holt, D.P.; Dannals, R.F.; Pomper, M.G.; Zarate, C.A.; et al. 6-O-(2-[18F]Fluoroethyl)-6-O-desmethyl-diprenorphine ([18F]FE-DPN) preferentially binds to mu opioid receptors in vivo. Mol. Imag. Biol. 2023, 25, 384–390. [Google Scholar] [CrossRef]
- Horváth, G.; Makleit, S. Vizsgálatok a morfin szintézisével összefüggésben, II. Az O6-demetil-szalutaridin átalakítása szalutaridinné. Magy. Kém. Foly. 1980, 86, 260–263. [Google Scholar]
- Horváth, G.; Makleit, S. Studies related to the synthesis of morphine, II. Conversion of O6-demethylsalutaridine into salutaridine. Acta Chim. Acad. Sci. Hung. 1981, 106, 37–42. [Google Scholar] [CrossRef]
- Lewis, J.W.; Walter, D. Buprenorphine—Background to its development as a treatment for opioid dependence. In NIDA Research Monograph 121, Buprenorphine: An Alternative Treatment for Opioid Dependence; Blaine, J.D., Ed.; US Department of Health and Human Services, Public Health Service, Alcohol, Drug Abuse and Mental Health Administration, NIDA: Rockville, MD, USA, 1992; Volume 121, pp. 5–11. [Google Scholar]
- Berényi, S.; Hosztafi, S.; Makleit, S.; Simon, C.; Marton, J. New Process for Producing N-Substituted-6,14-endo-ethanotetrahydrothebaine and -Oripavine Derivatives and Their Salts. Hungarian Patent 210,158, 15 January 1991. [Google Scholar]
- Marton, J.; Simon, C.; Hosztafi, S.; Berényi, S.; Makleit, S.; Kabai, S.; Kolozsi, S.; Kovács, Z.; Nagy, I. New Process for Producing 6,14-ethenomorphinan Derivatives and Salts Thereof. Hungarian Patent 76,478, 10 July 1995. [Google Scholar]
- Marton, J.; Szabó, Z.; Hosztafi, S. Herstellung von 6,14 Ethenomorphinan-Derivaten. Liebigs Ann. Chem. 1993, 8, 915–919. [Google Scholar] [CrossRef]
- Antus, S. Invetsigation of biologically active compounds at the Department of Organic Chemistry of University of Debrecen between 1992–2009, I. Acta Pharm. Hung. 2009, 79, 95–103. [Google Scholar] [PubMed]
- Hosztafi, S.; Makleit, S.; Bognár, R. Morfin-alkaloidok N-demetilezése. A nor-neopin előállítása. Magy. Kém. Foly. 1980, 86, 15–17. [Google Scholar]
- Hosztafi, S.; Makleit, S.; Bognár, R. N-Demethylation of morphine alkaloids. Preparation of norneopine. Acta Chim. Acad. Sci. Hung. 1980, 103, 371–375. [Google Scholar]
- Hosztafi, S. Reactions of azodicarboxylic esters with amines. Sci. Pharm. 1987, 55, 61–75. [Google Scholar]
- Hosztafi, S. Morfinváz C Gyűrűjében Módosított Vegyületek Szintézise. D.Sc. Dissertation, Semmelweis University, Budapest, Hungary, 2020. [Google Scholar]
- Seller, S.; Szabó, M.; Palincza, J.; Imre, L.; Cselenyák, Z.; Tompa, J. Process for Separation of Opium Alkaloids. Hungarian Patent 225,038, 15 November 2002. [Google Scholar]
- Volkow, N.D.; Blanco, C. The changing opioid crisis: Development, challenges and opportunities. Mol. Psychiatry 2021, 26, 218–233. [Google Scholar] [CrossRef] [PubMed]
- Pieters, T. The imperative of regulation: The co-creation of a medical and non-medical US opioid crisis. Psychoactives 2023, 2, 317–336. [Google Scholar] [CrossRef]
- Dennen, A.C.; Blum, K.; Braverman, R.E.; Bowirrat, A.; Gold, M.; Elman, I.; Thanos, K.P.; Baron, D.; Gupta, A.; Edwards, D.; et al. How to Combat the Global Opioid Crisis. CPQ Neurol. Psychol. 2023, 5, 93. [Google Scholar]
- Engi, Z.; Benkő, R.; Soós, G.; Szok, D.; Csenki, M.; Csüllög, E.; Balog, A.; Csupor, D.; Viola, R.; Doró, P.; et al. Trends in opioid utilization in Hungary, 2006–2020: A nationwide retrospective study with multiple metrics. Eur. J. Pain 2022, 26, 1896–1909. [Google Scholar] [CrossRef]
- Berényi, S. A debreceni mákalkaloidkutatás 50 éve—50 years on the Research of Poppy Alkaloids in Debrecen. MKL–Magy. Kém. Lapja. 1999, 54, 548–549. [Google Scholar]


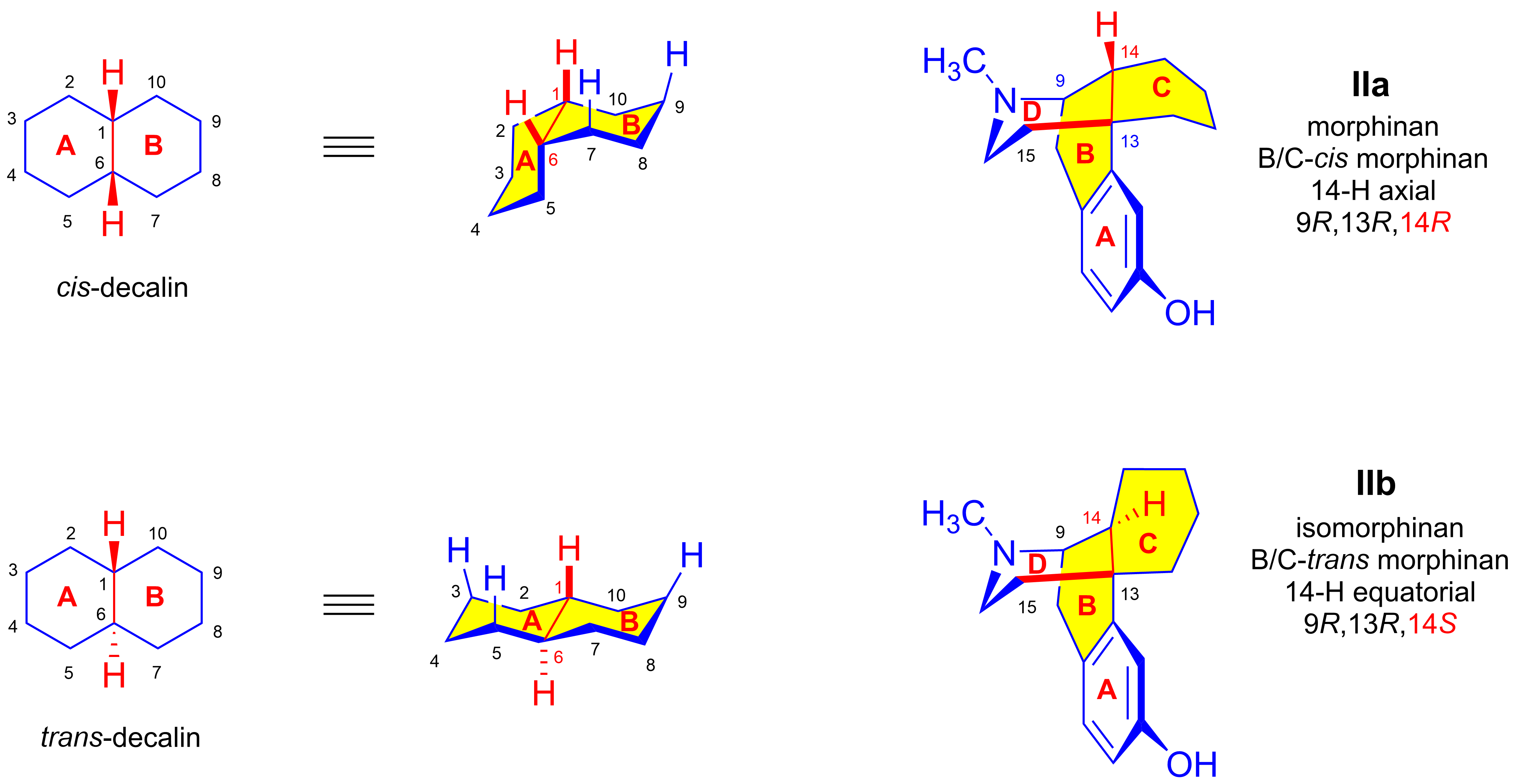
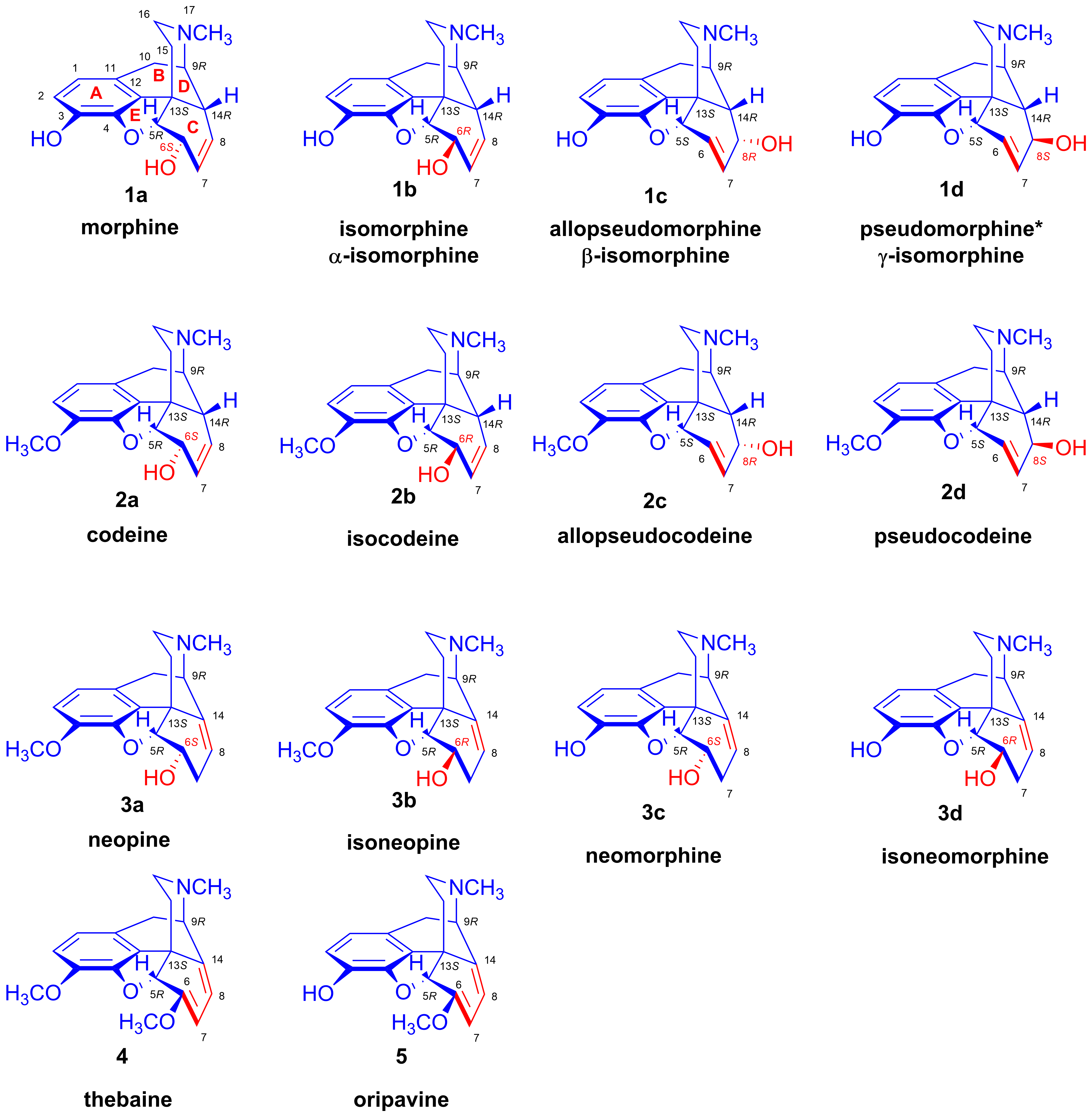
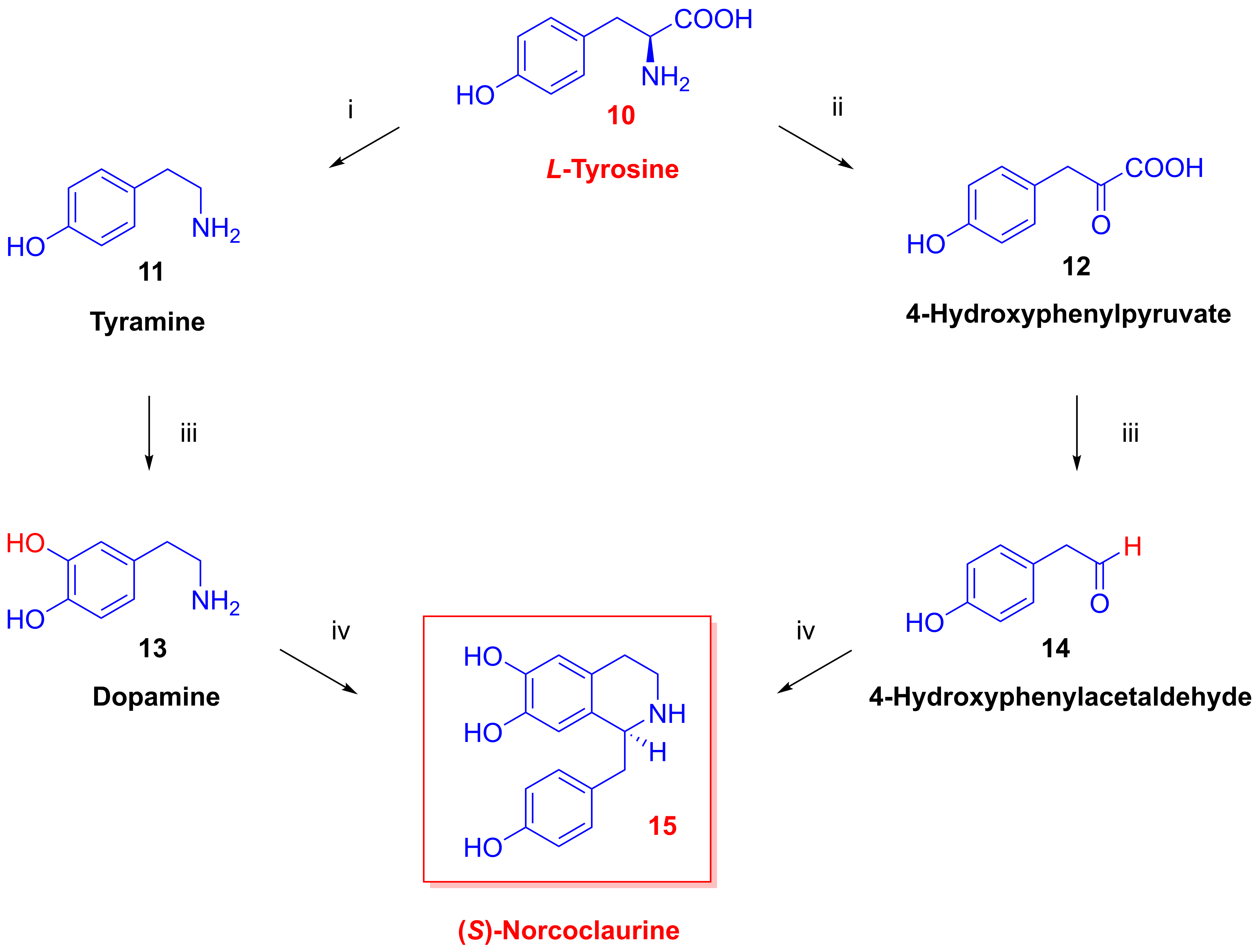
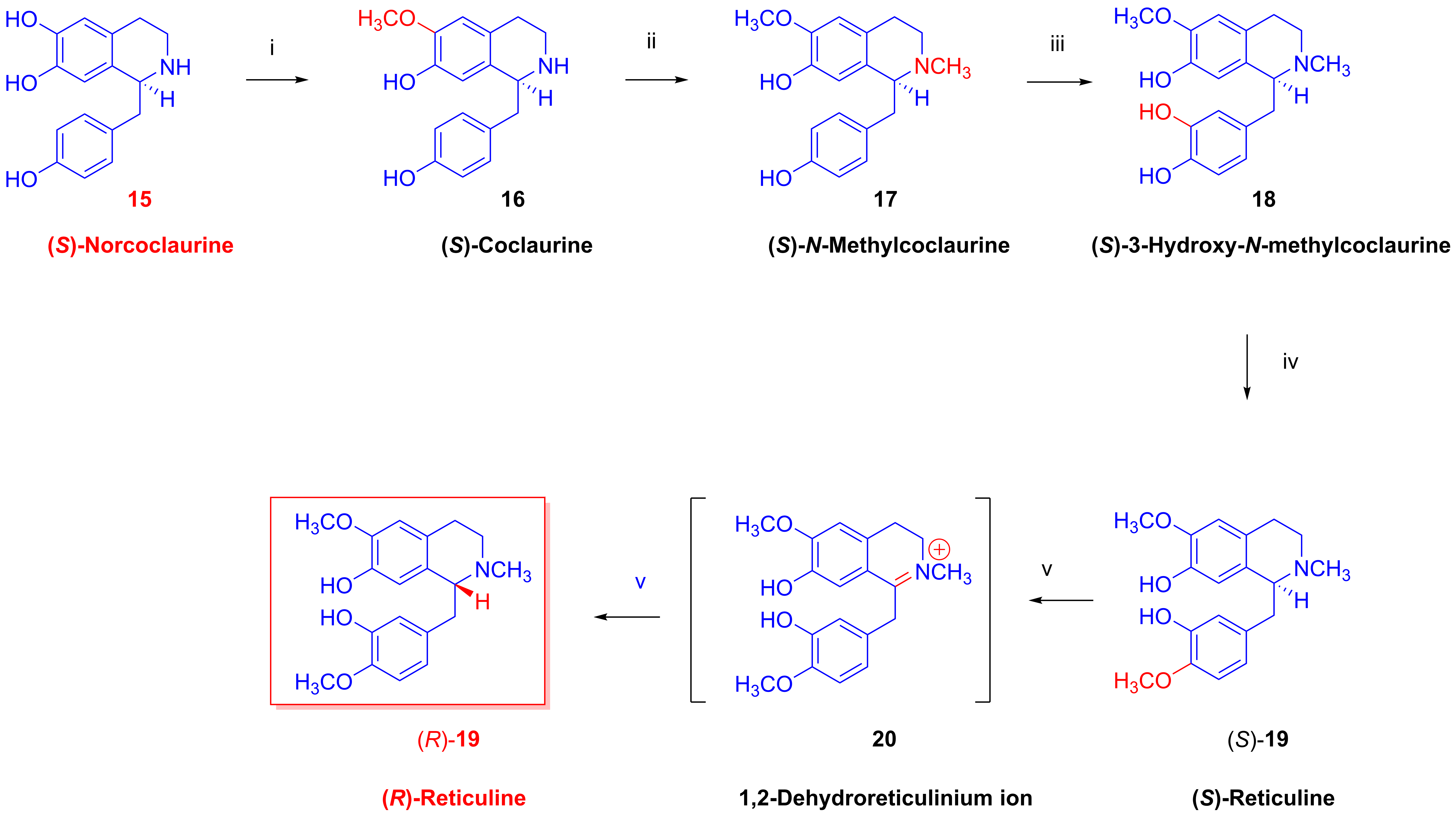



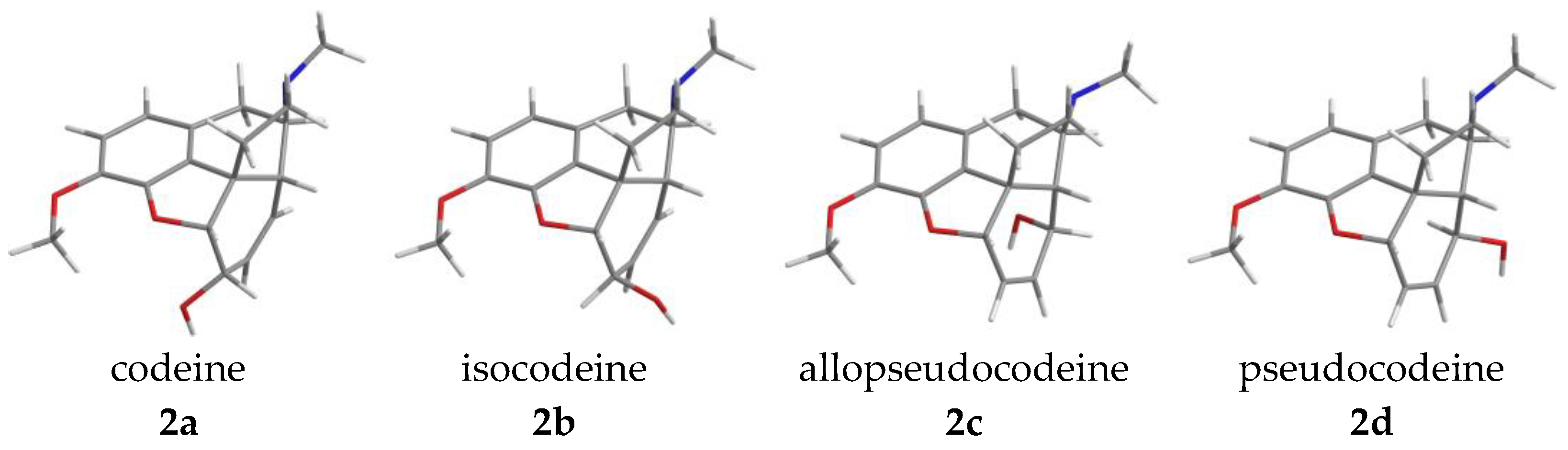
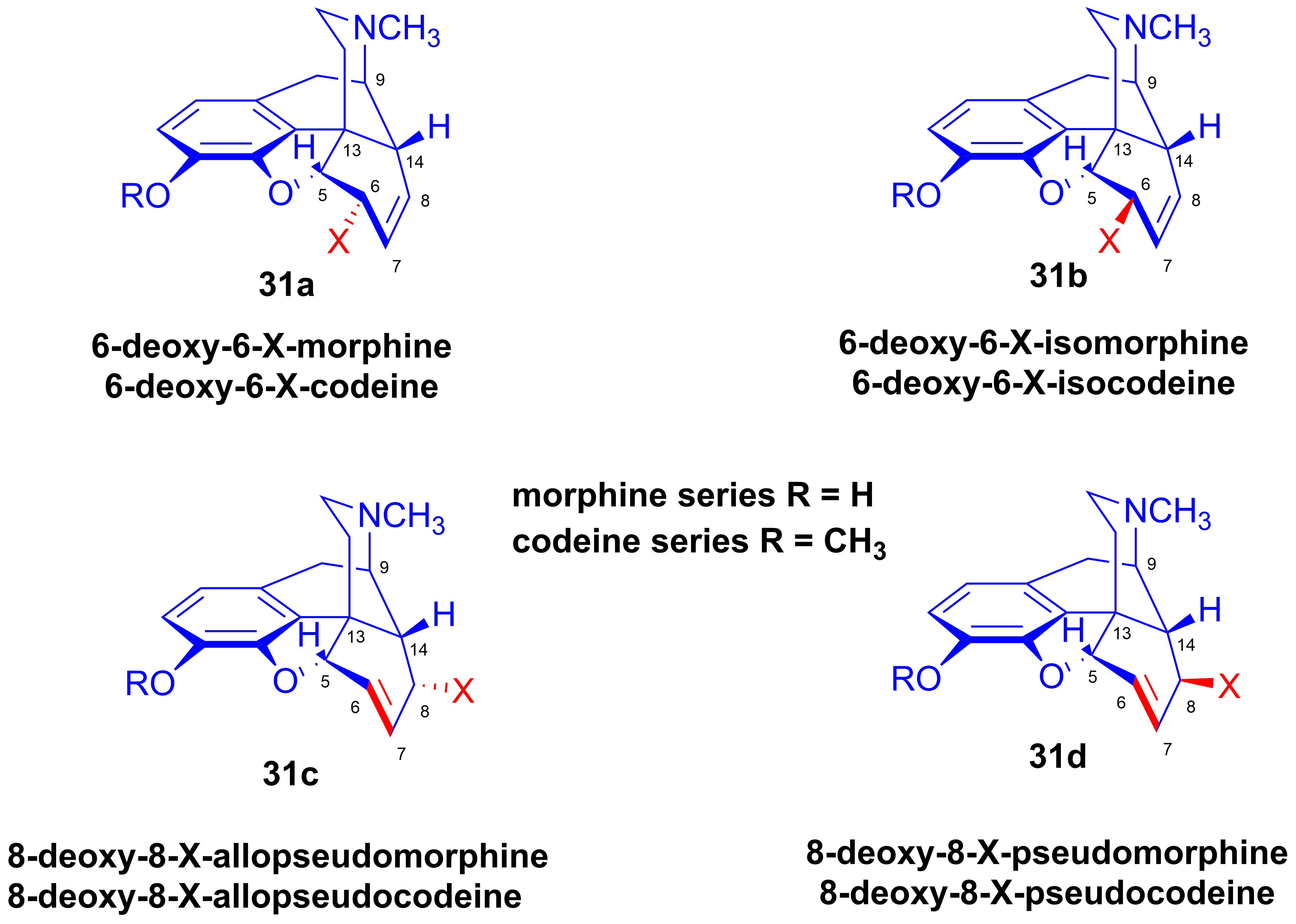
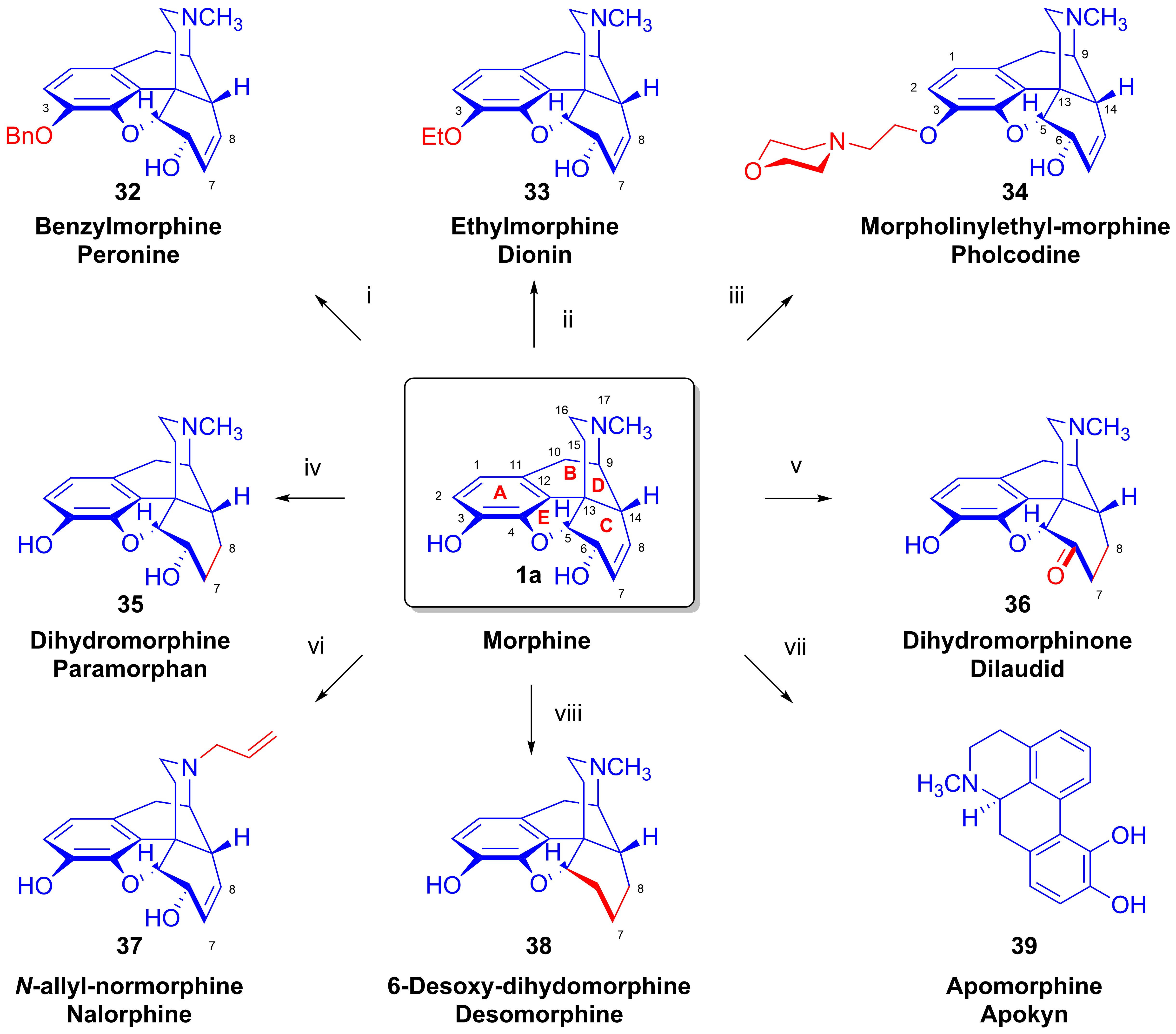
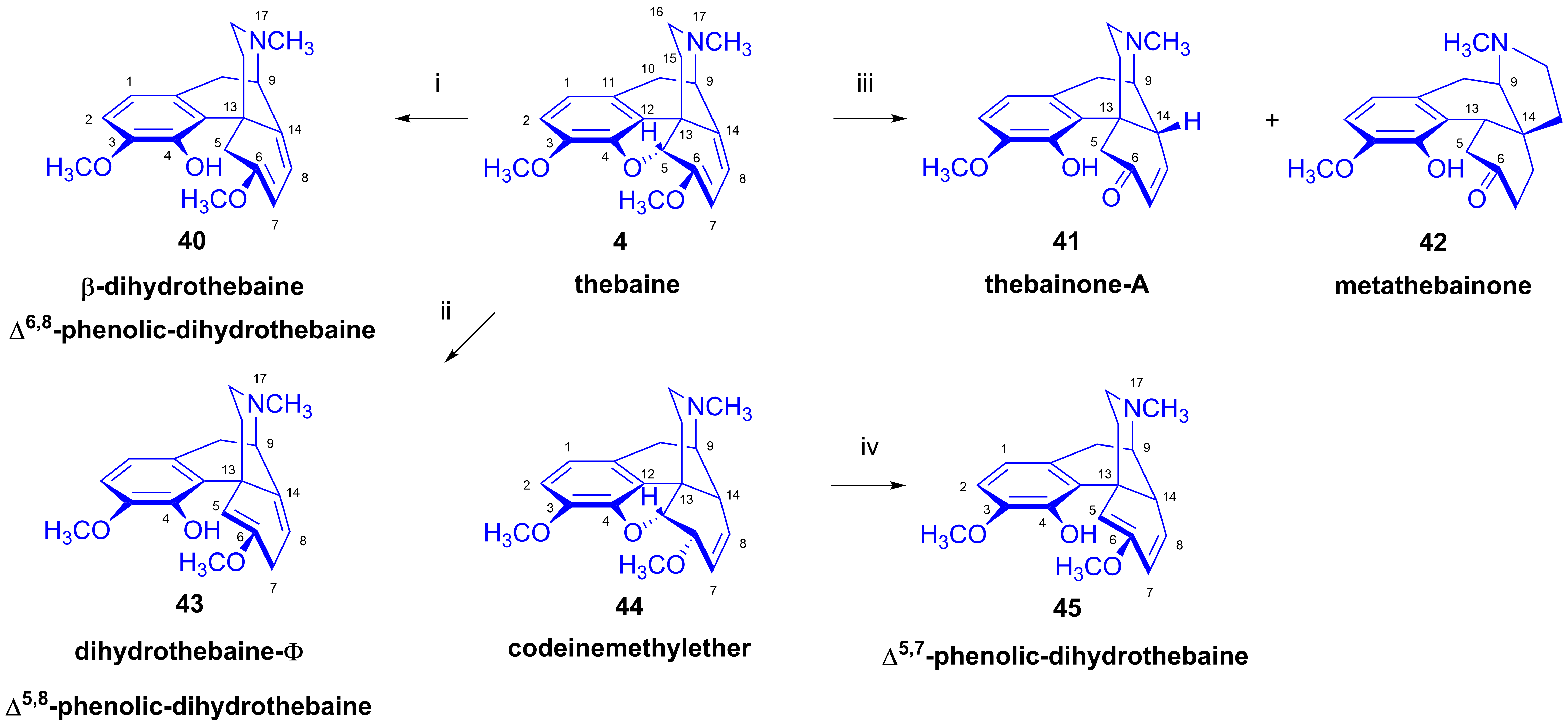
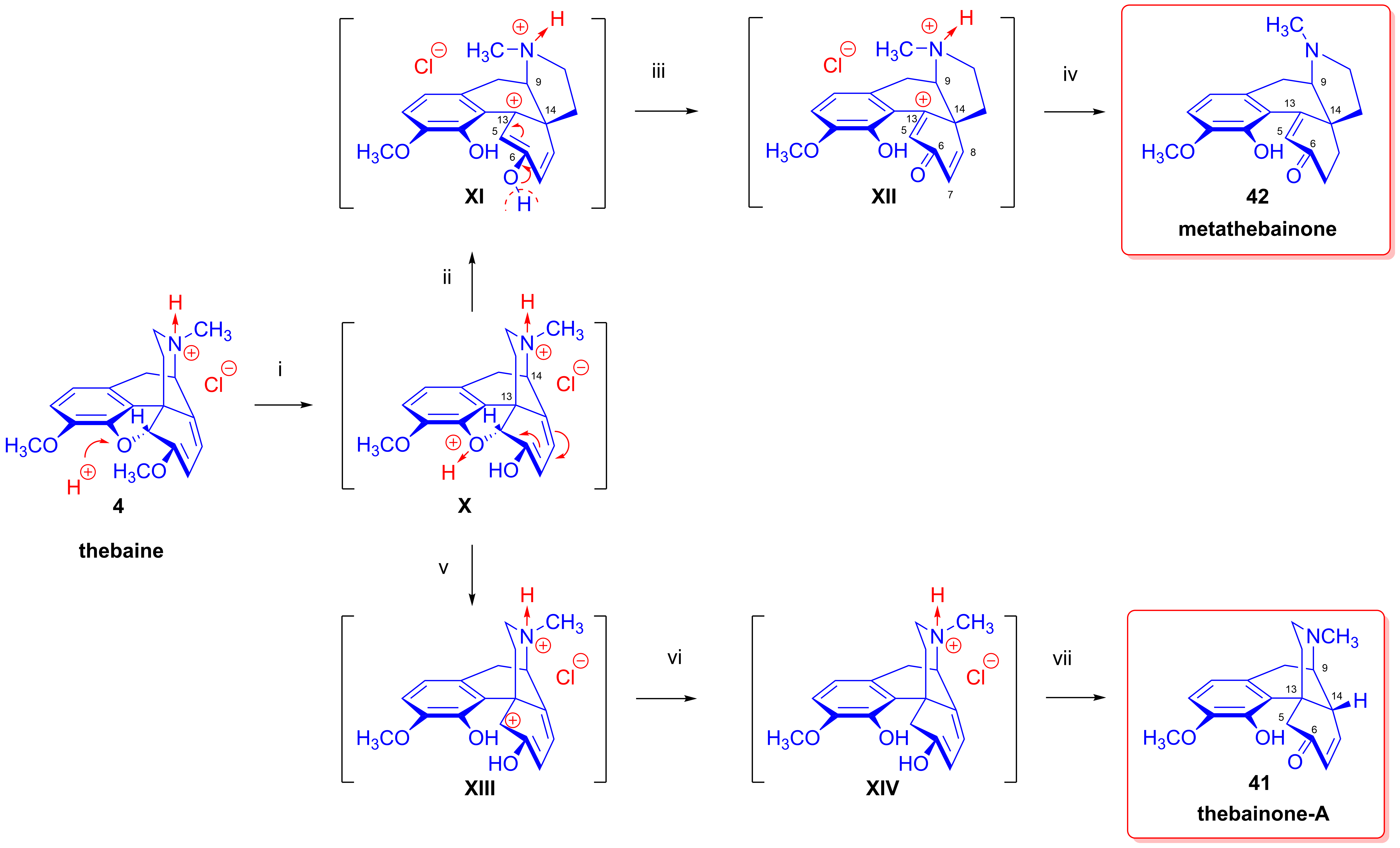
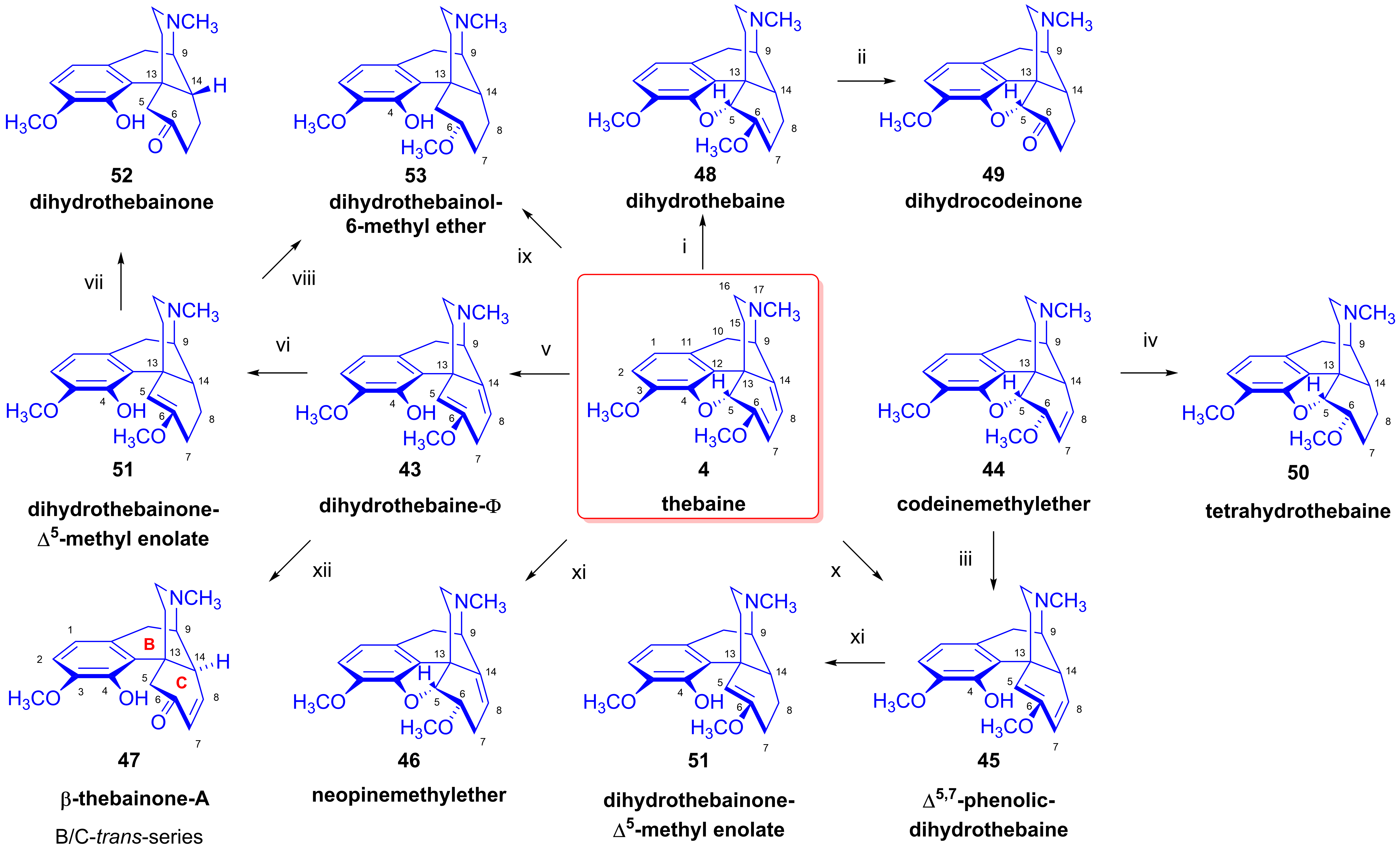
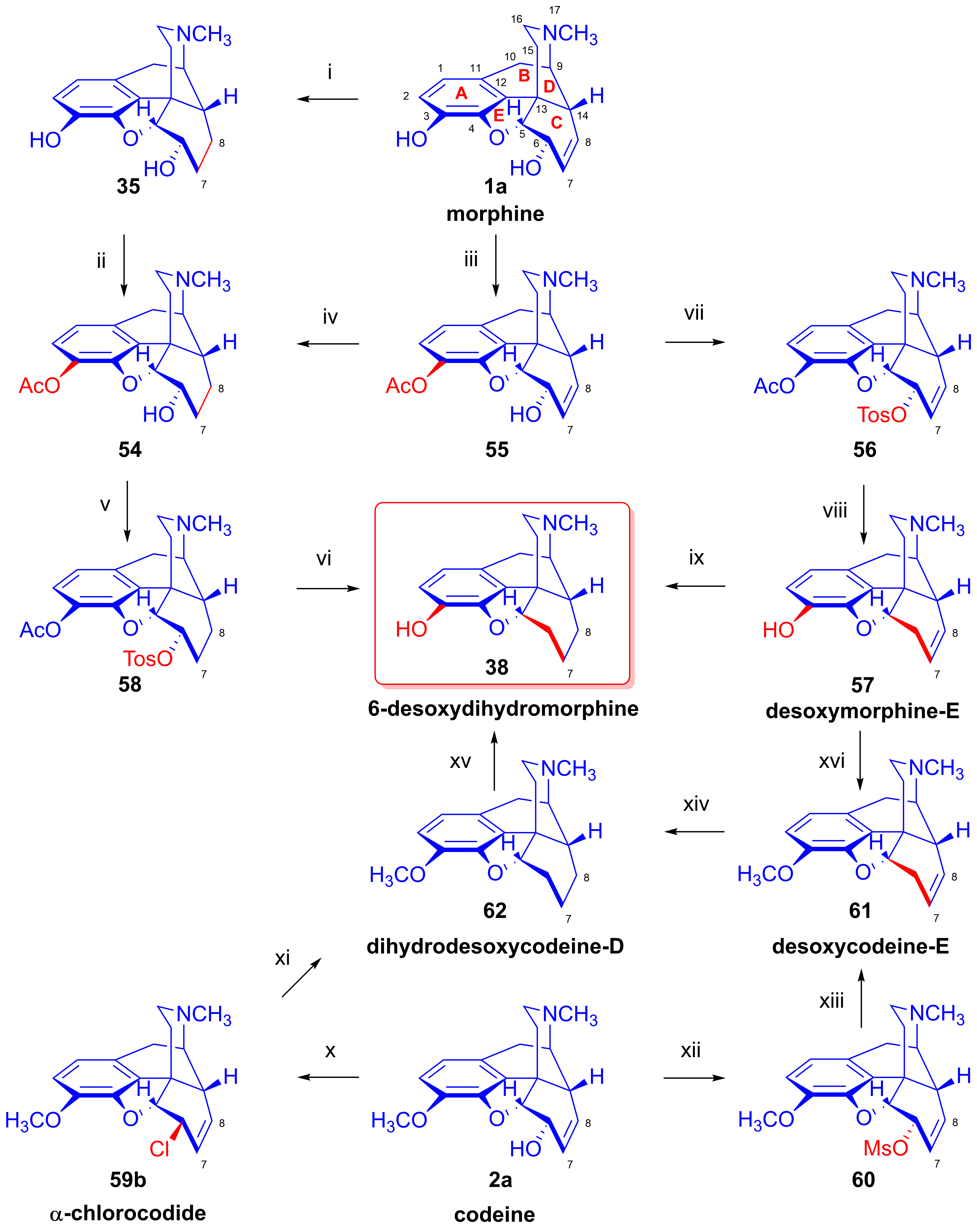
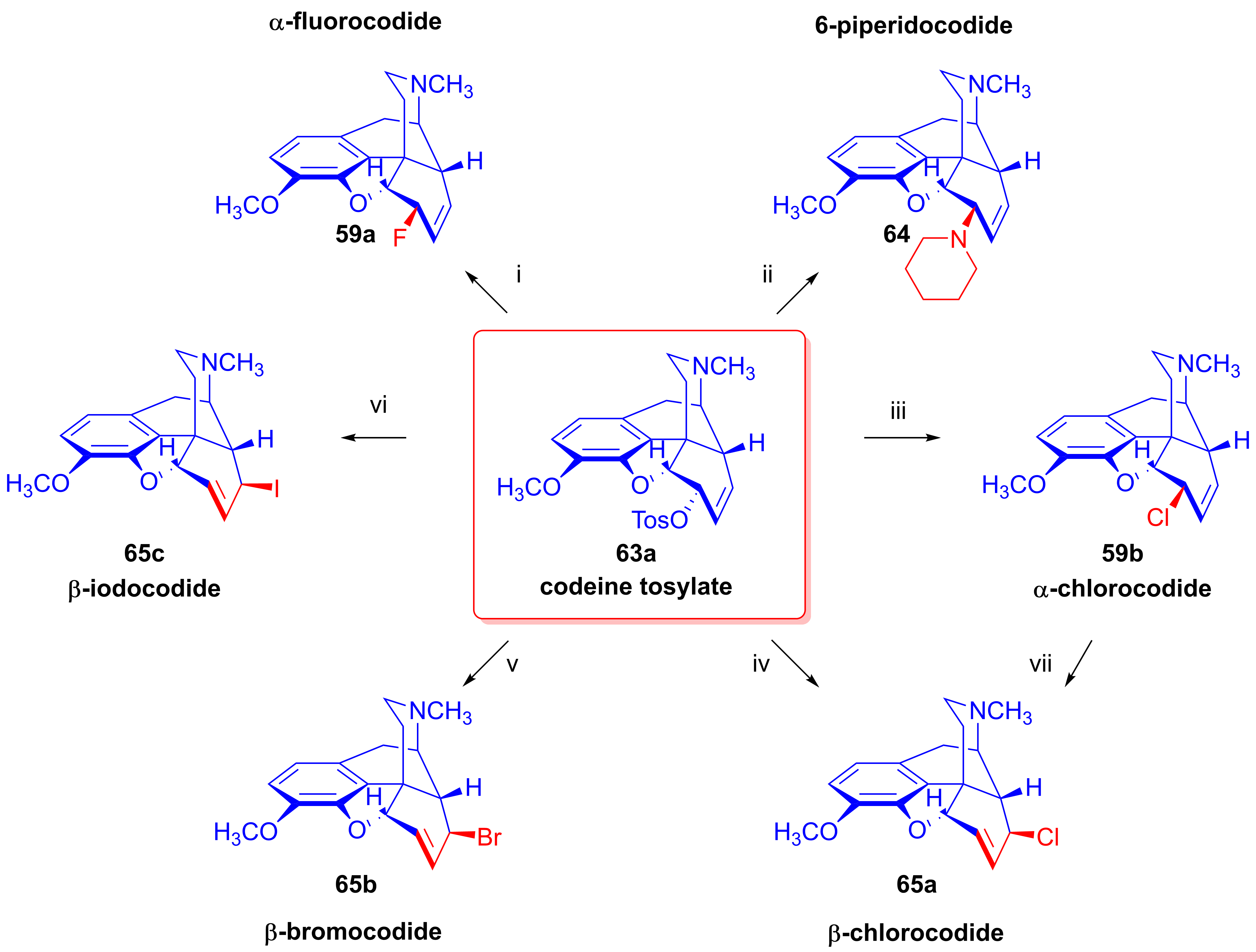
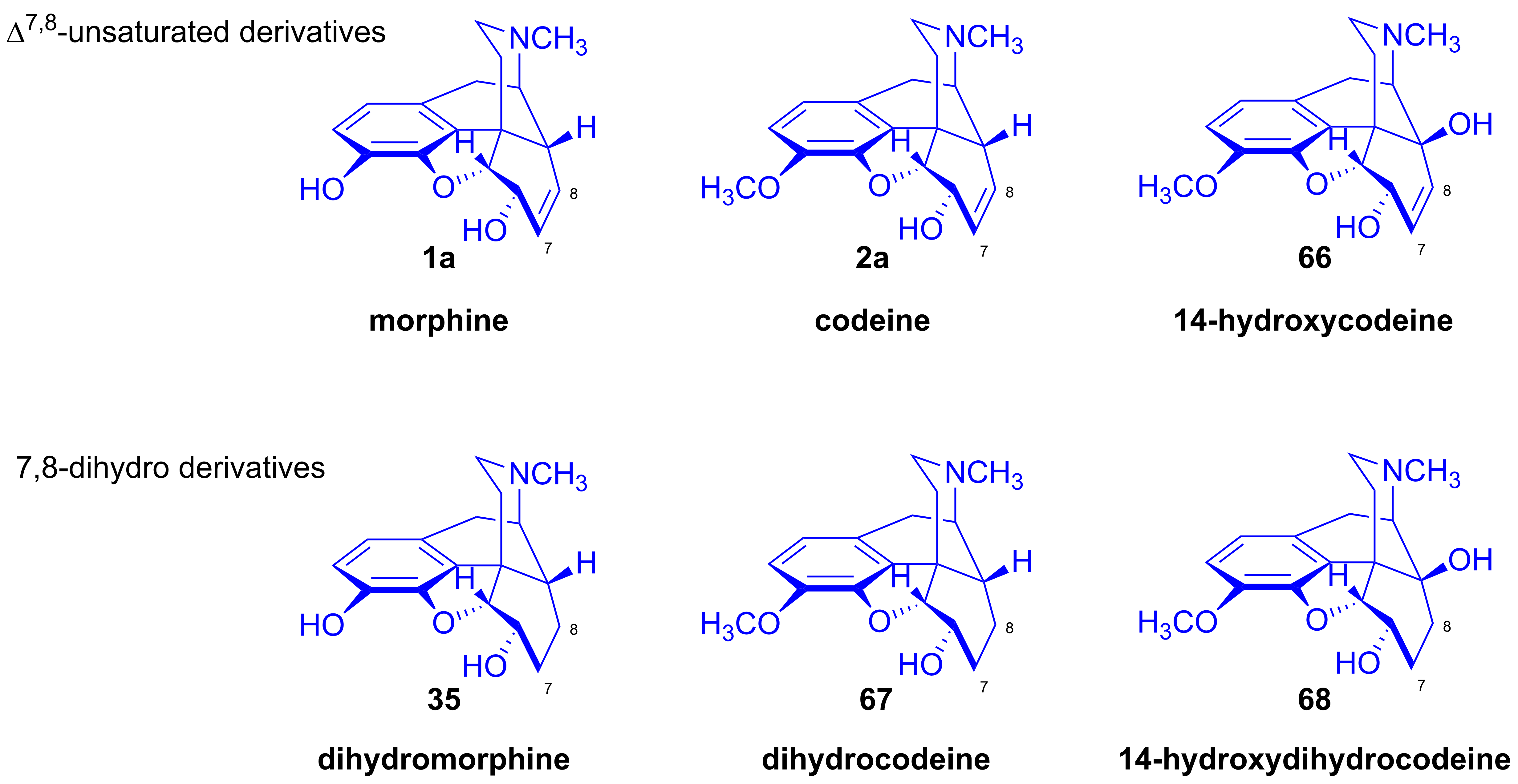
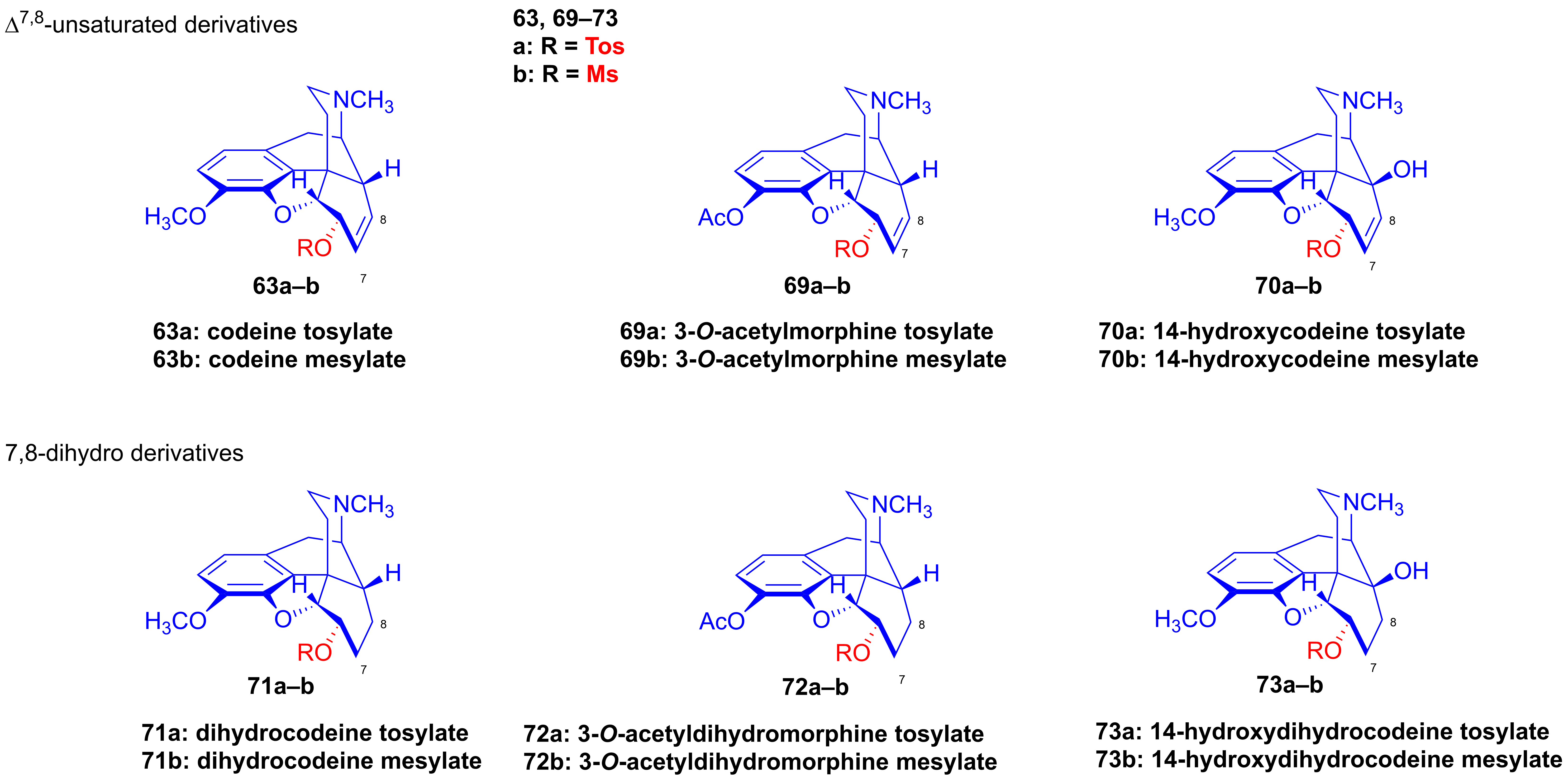

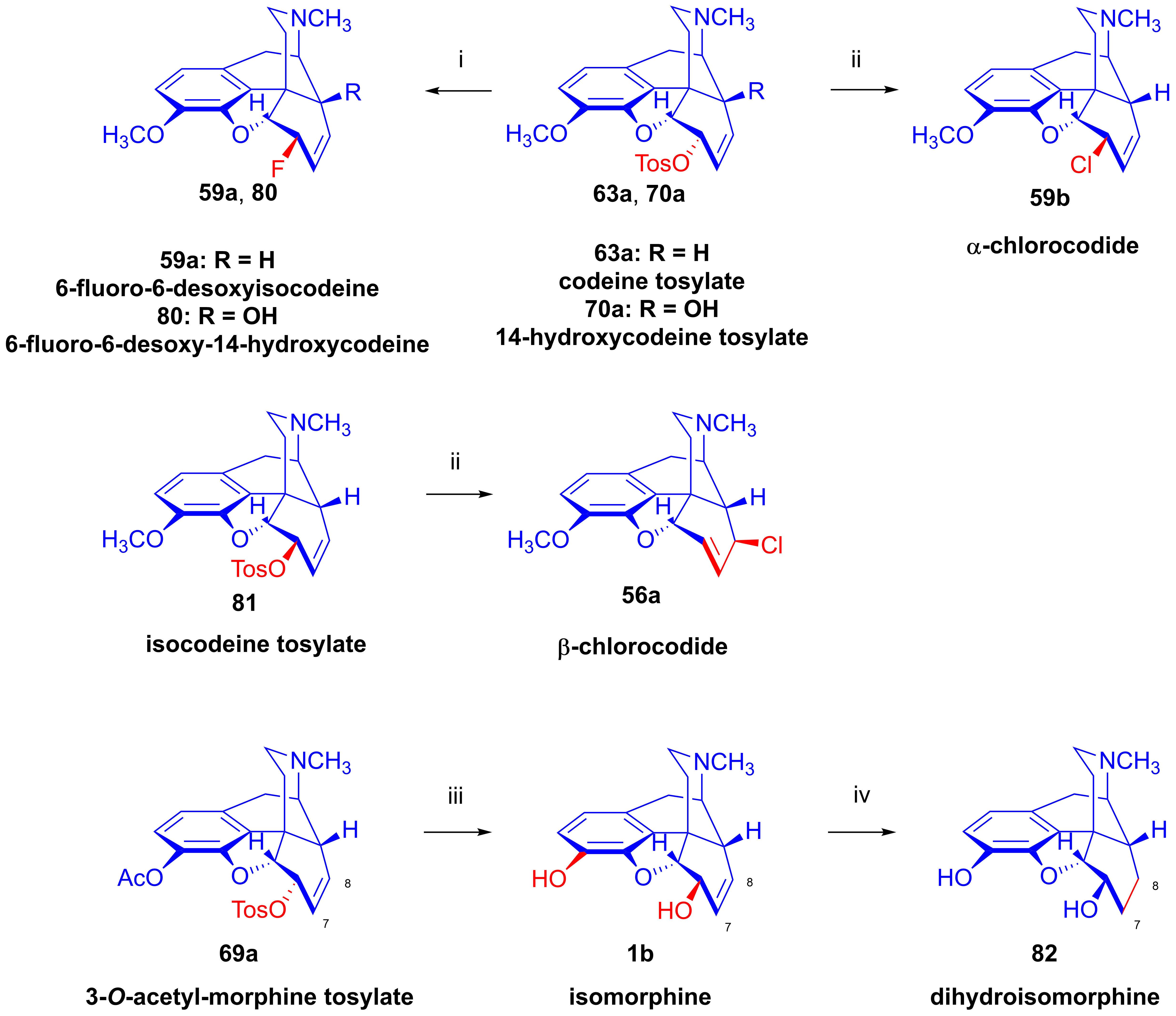
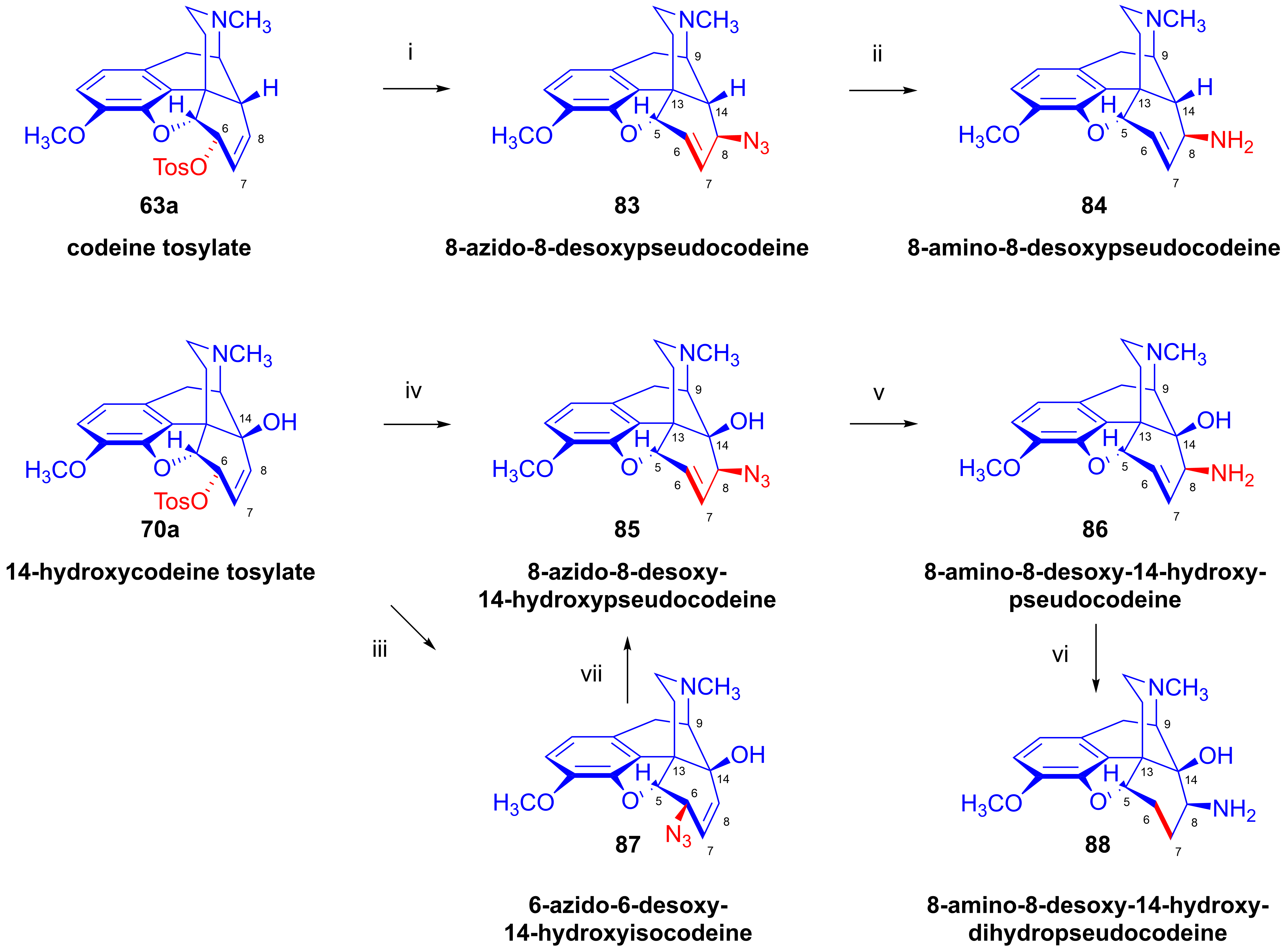

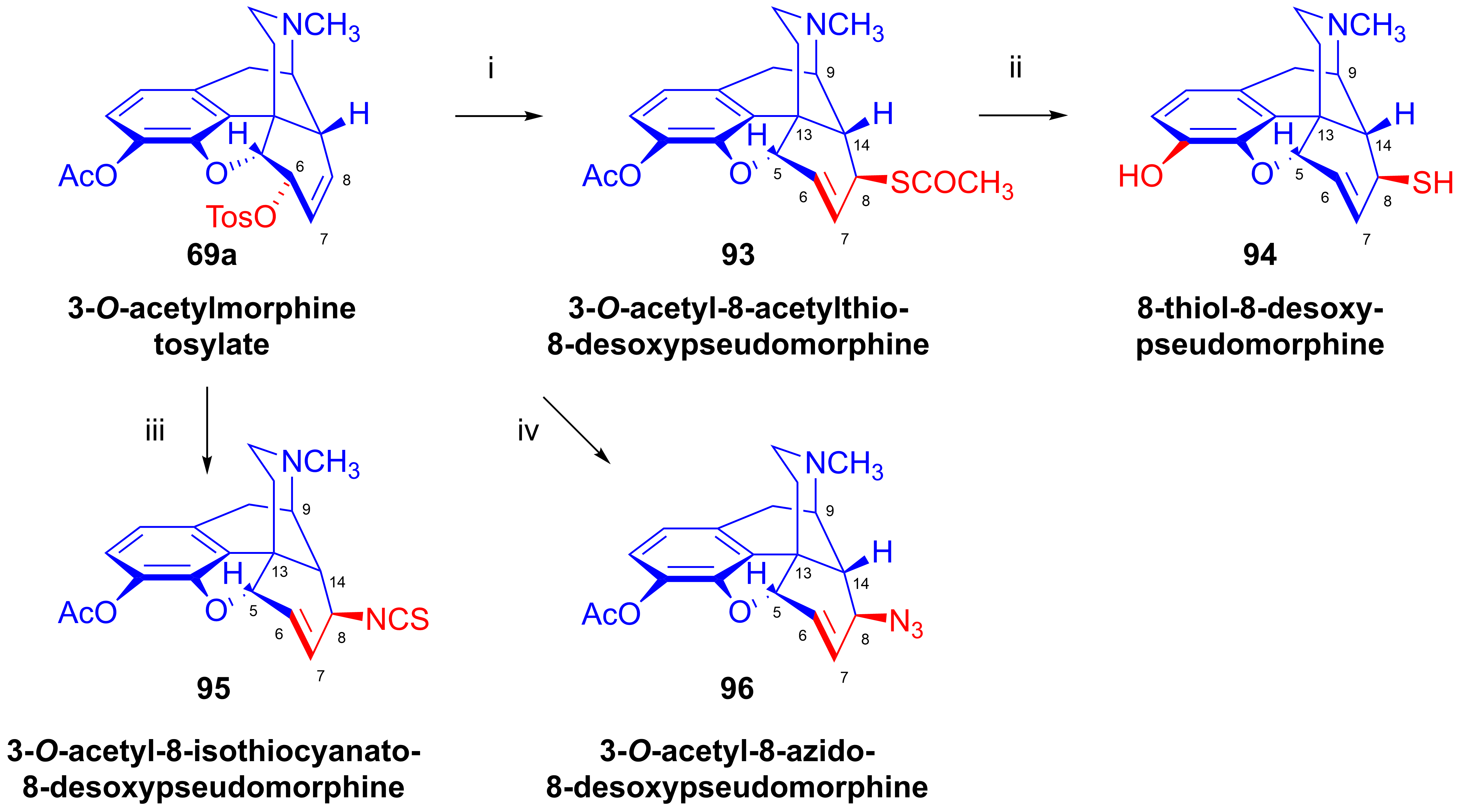

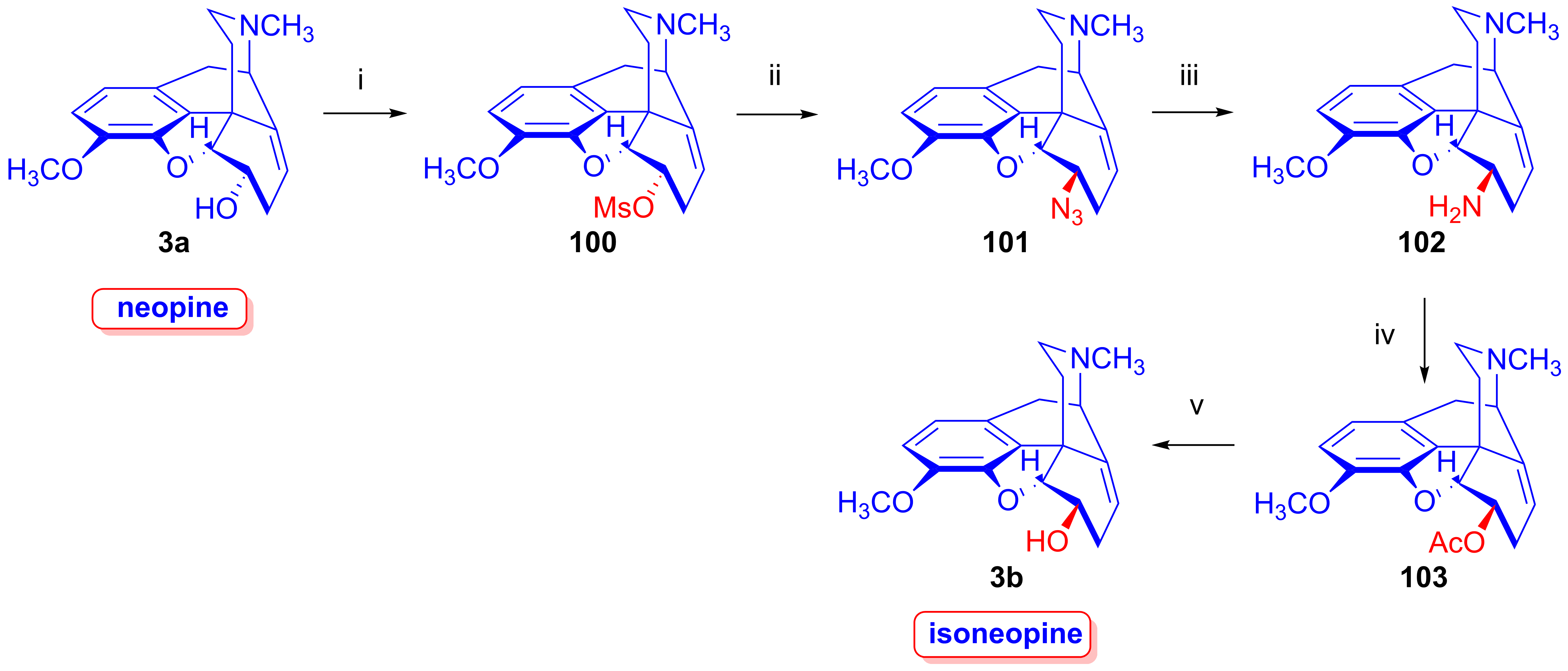

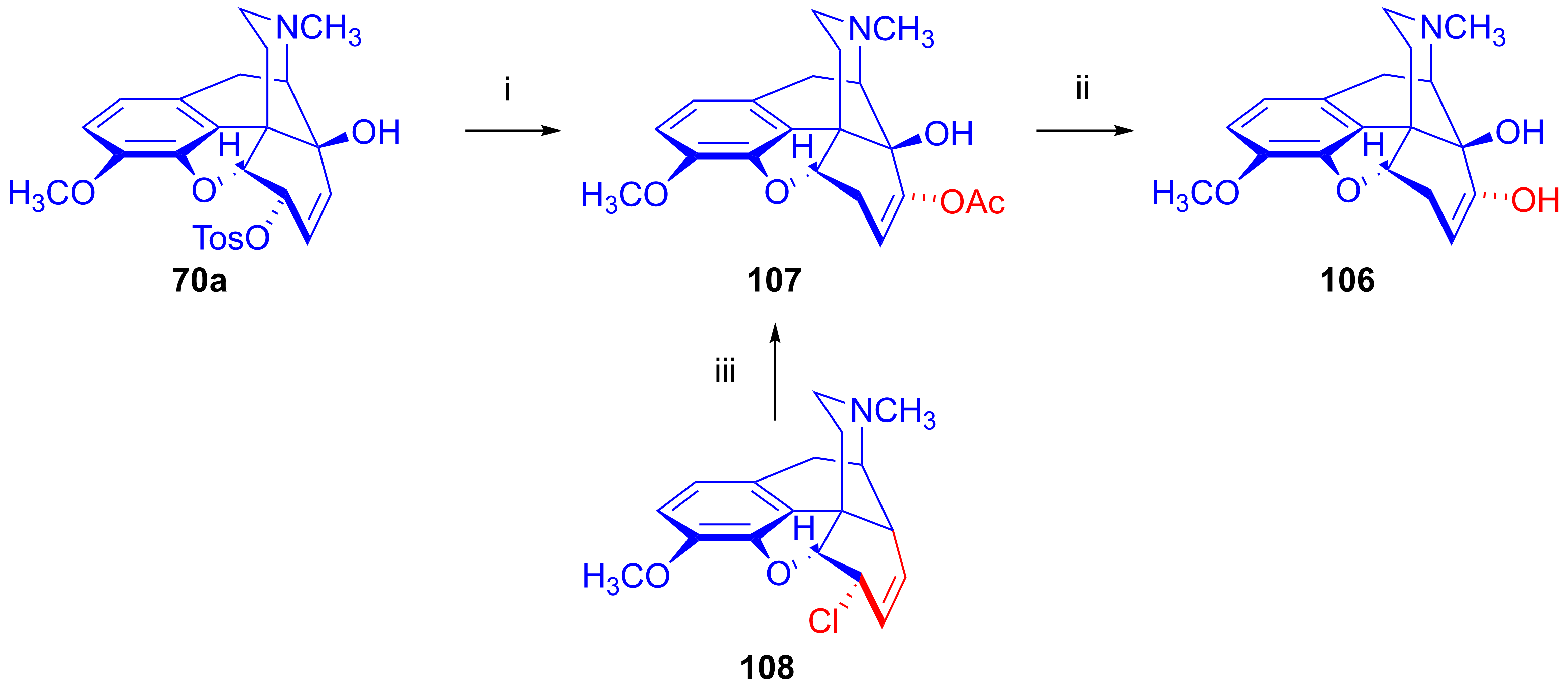
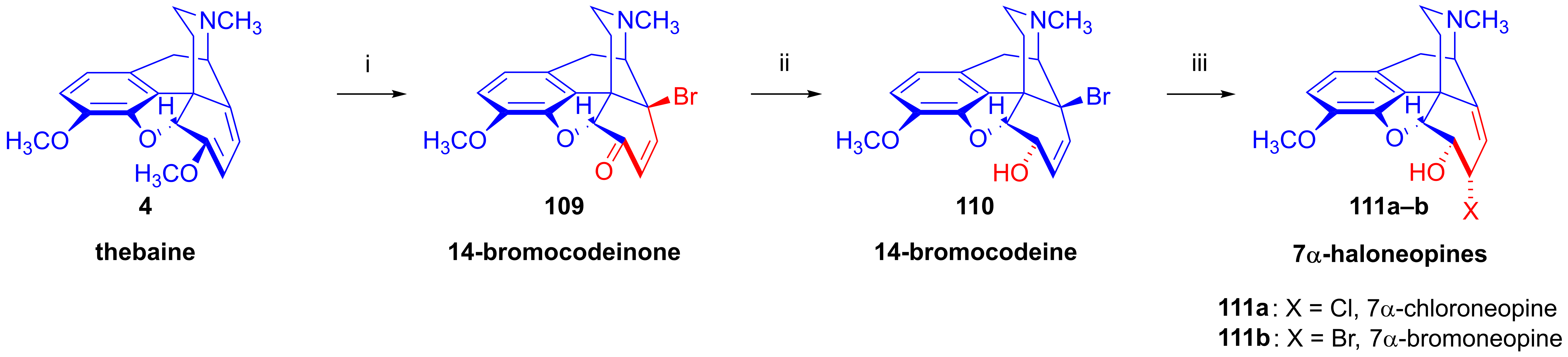



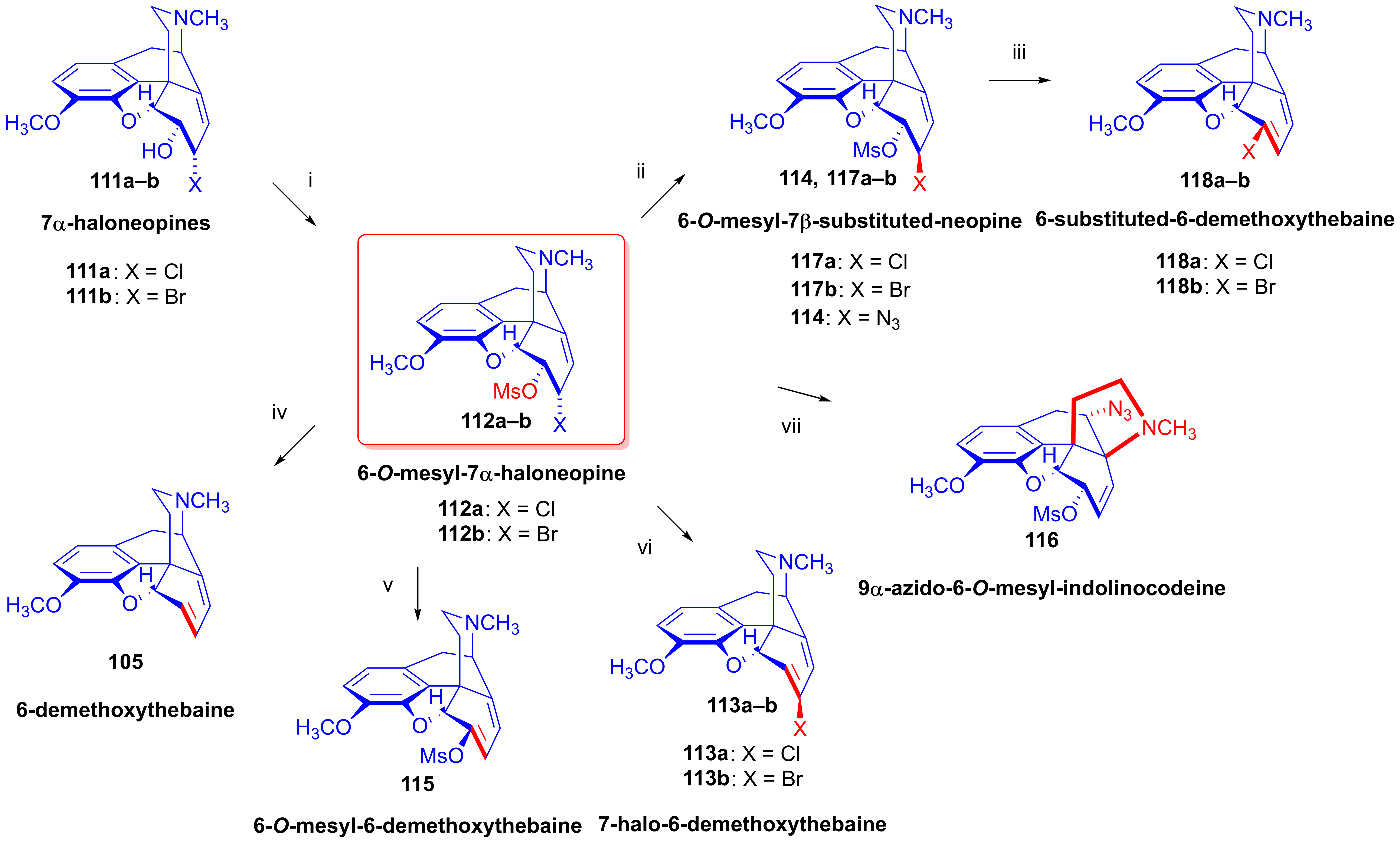
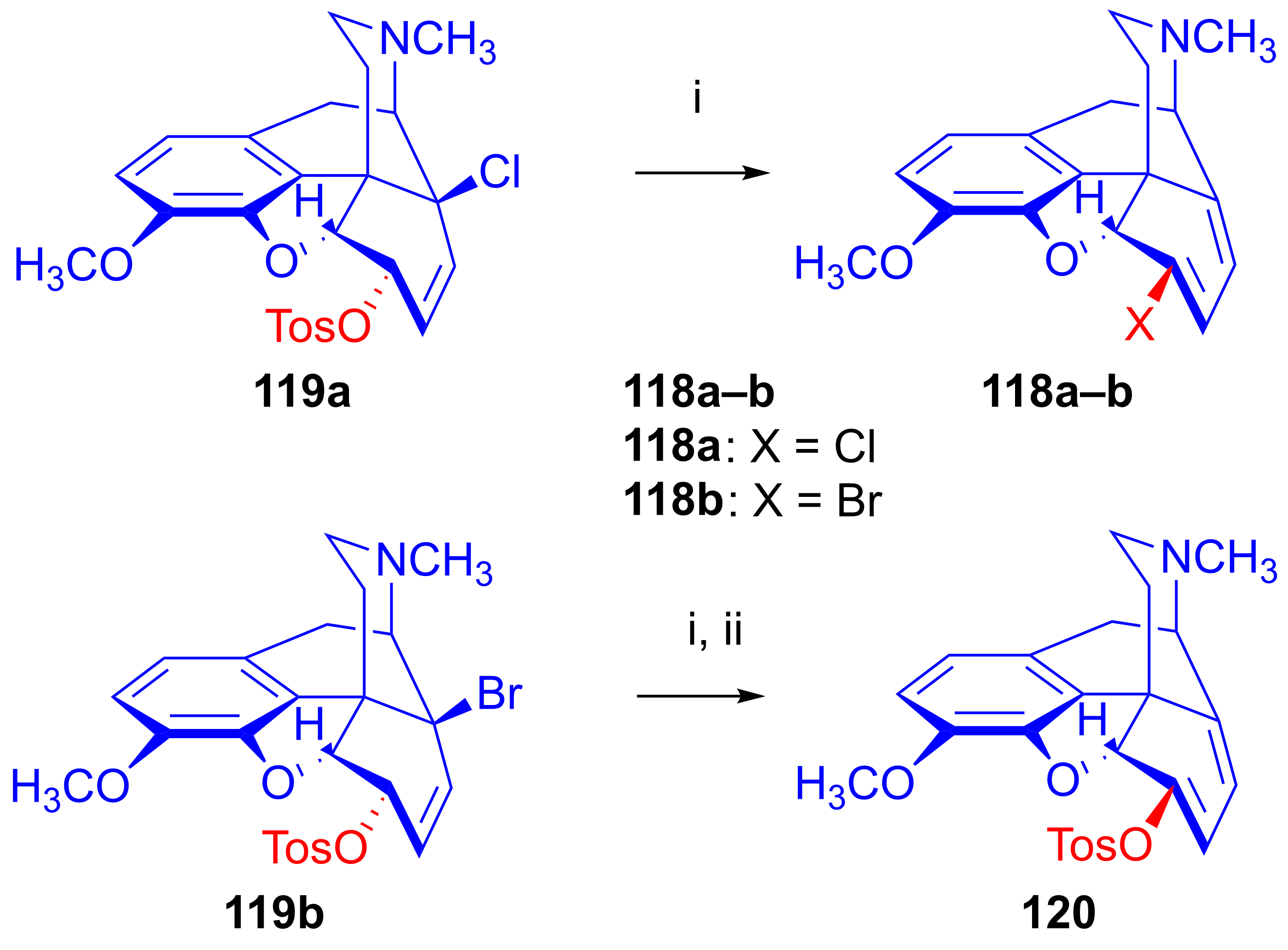

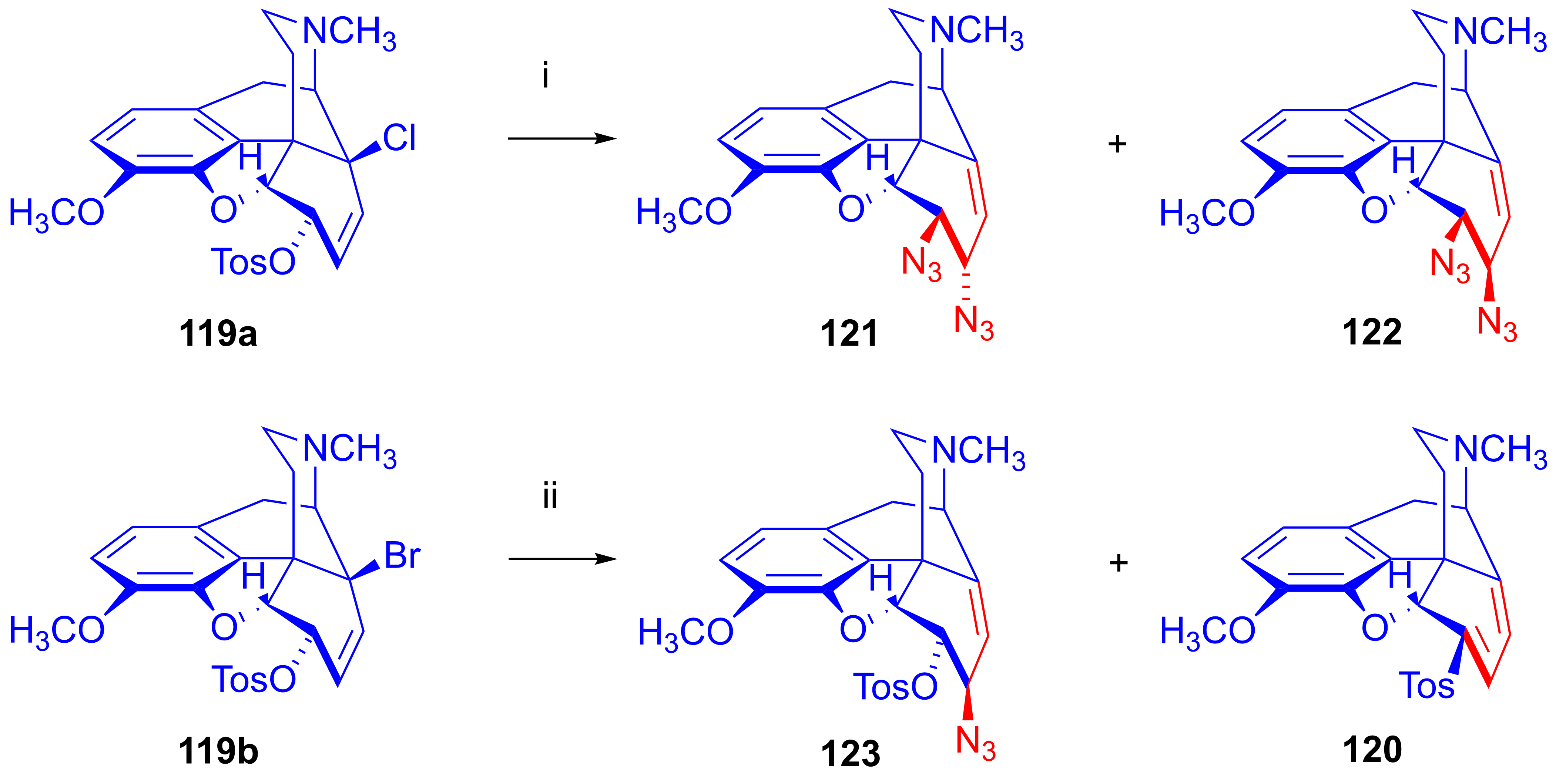
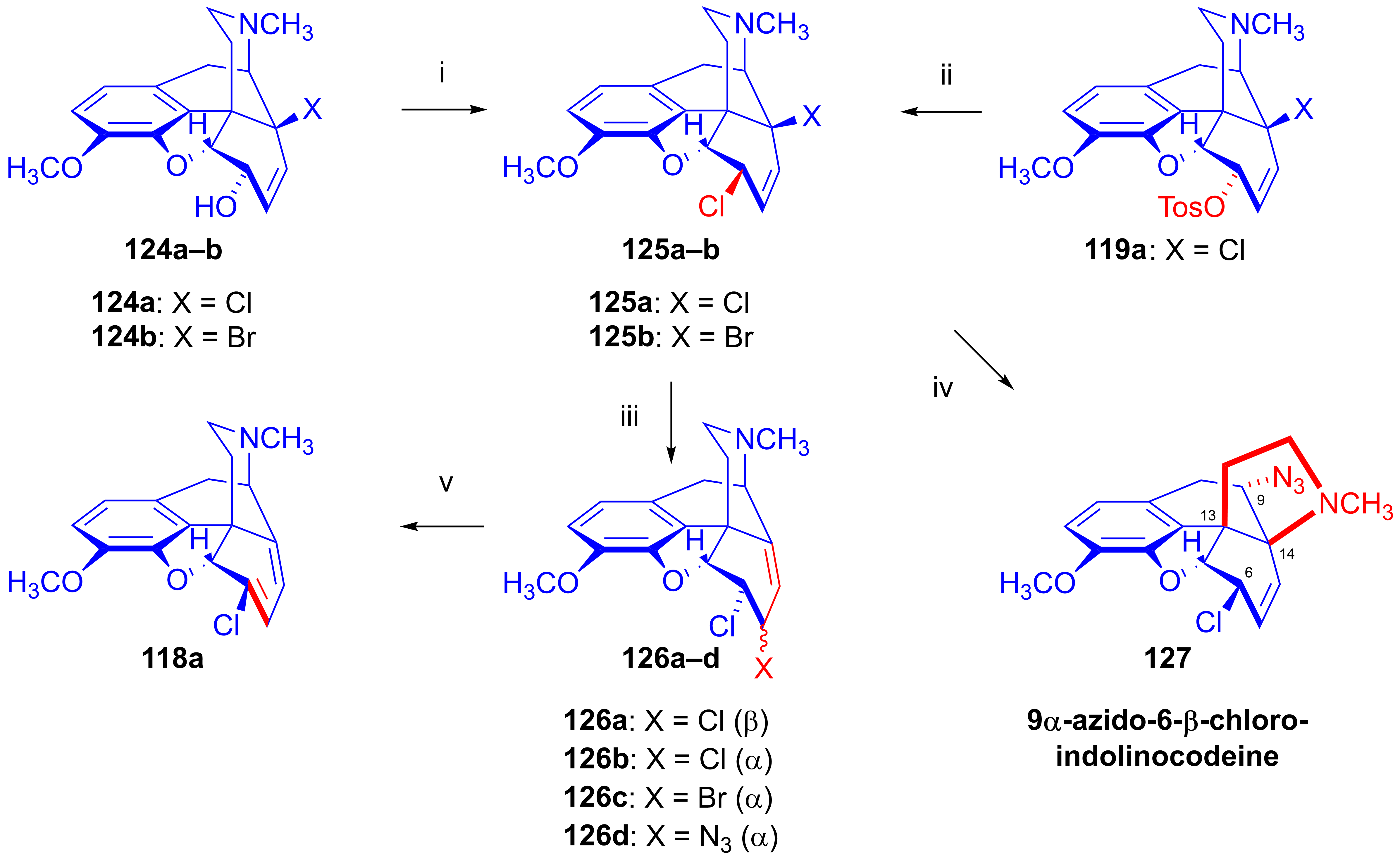
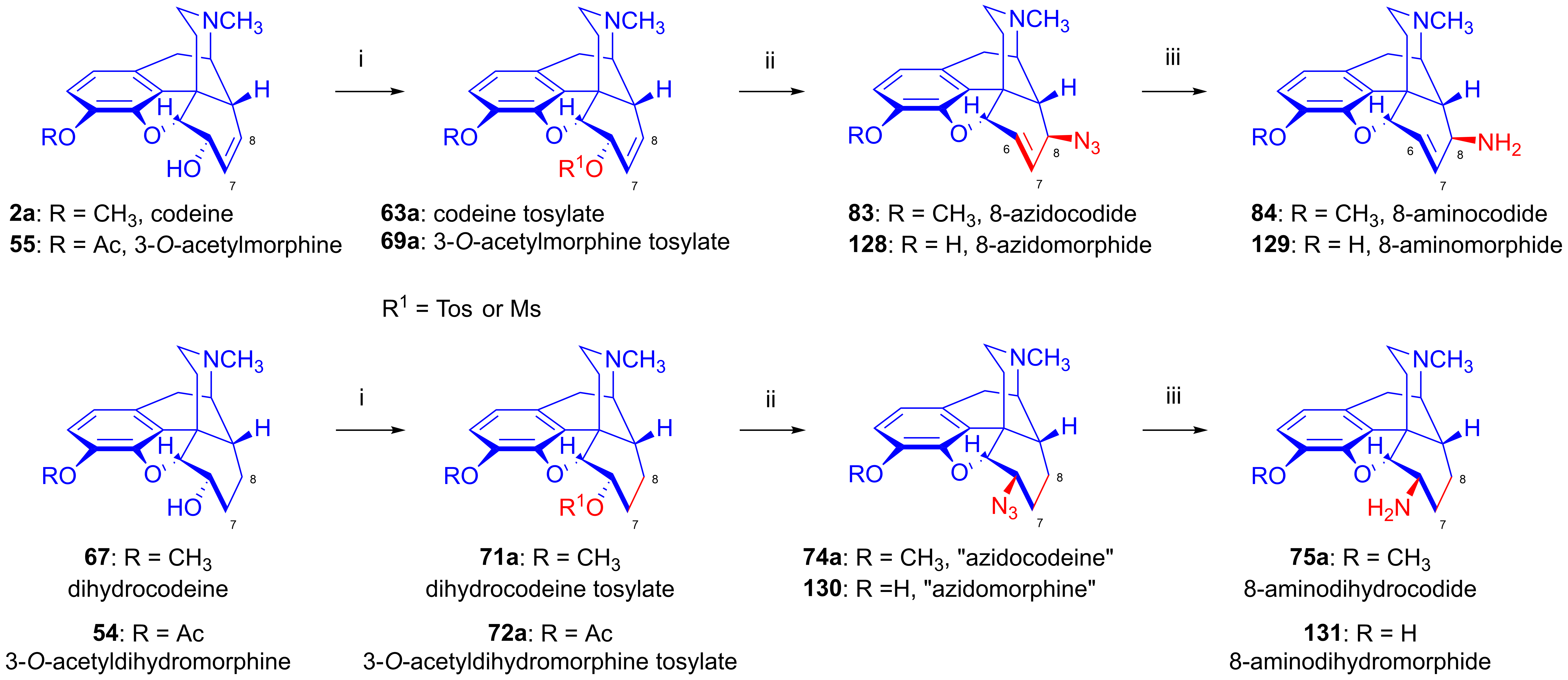
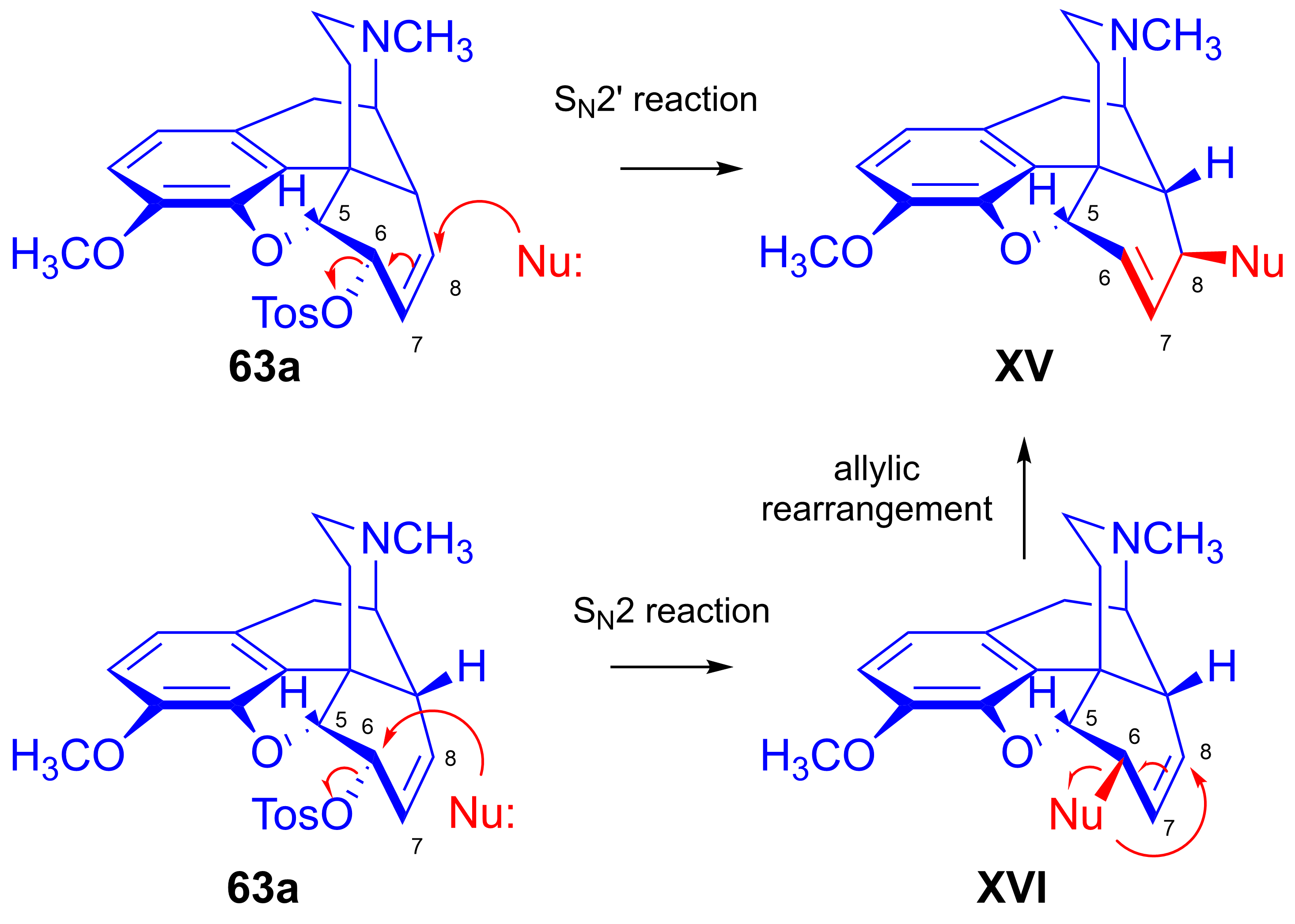
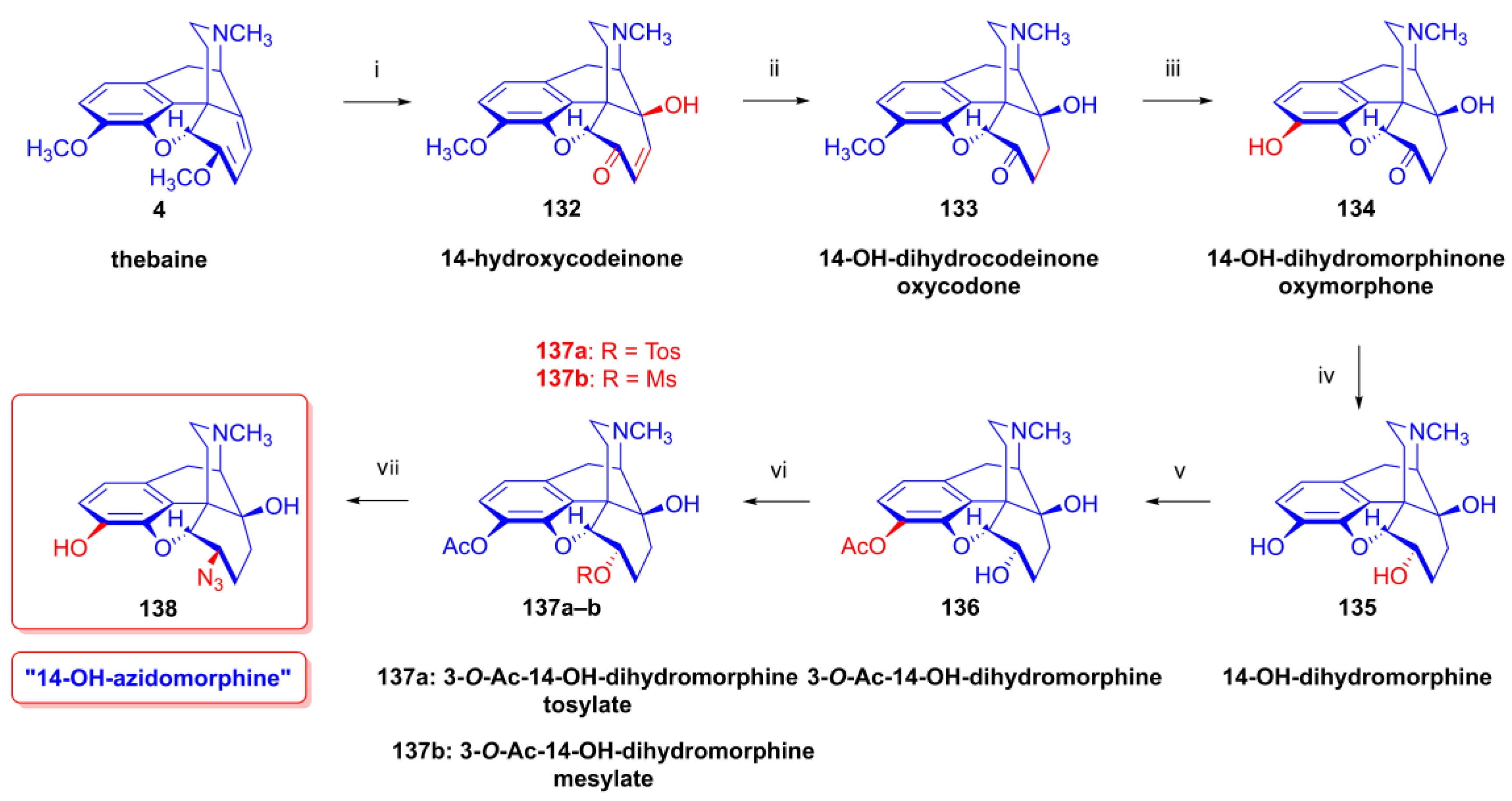
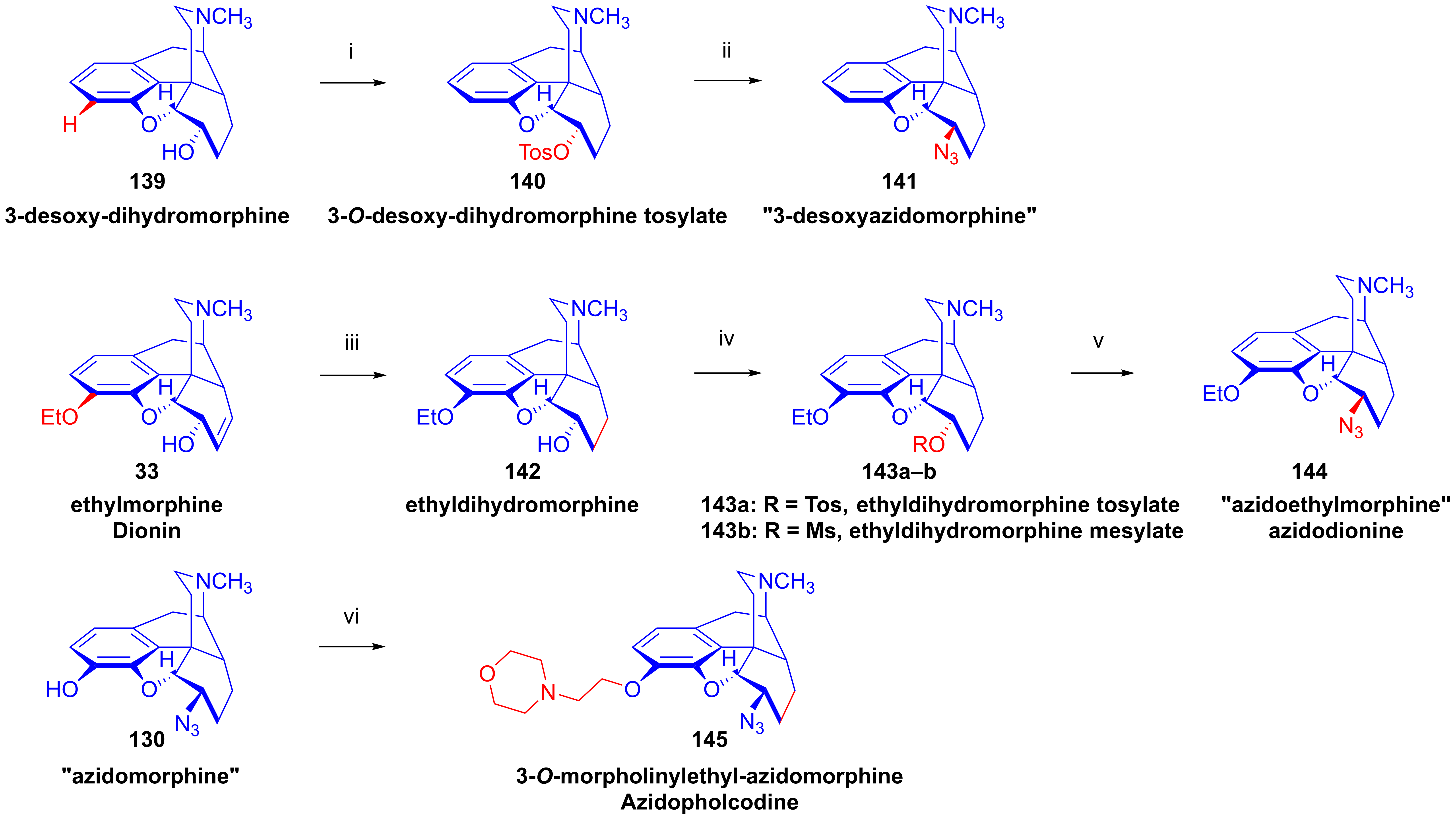

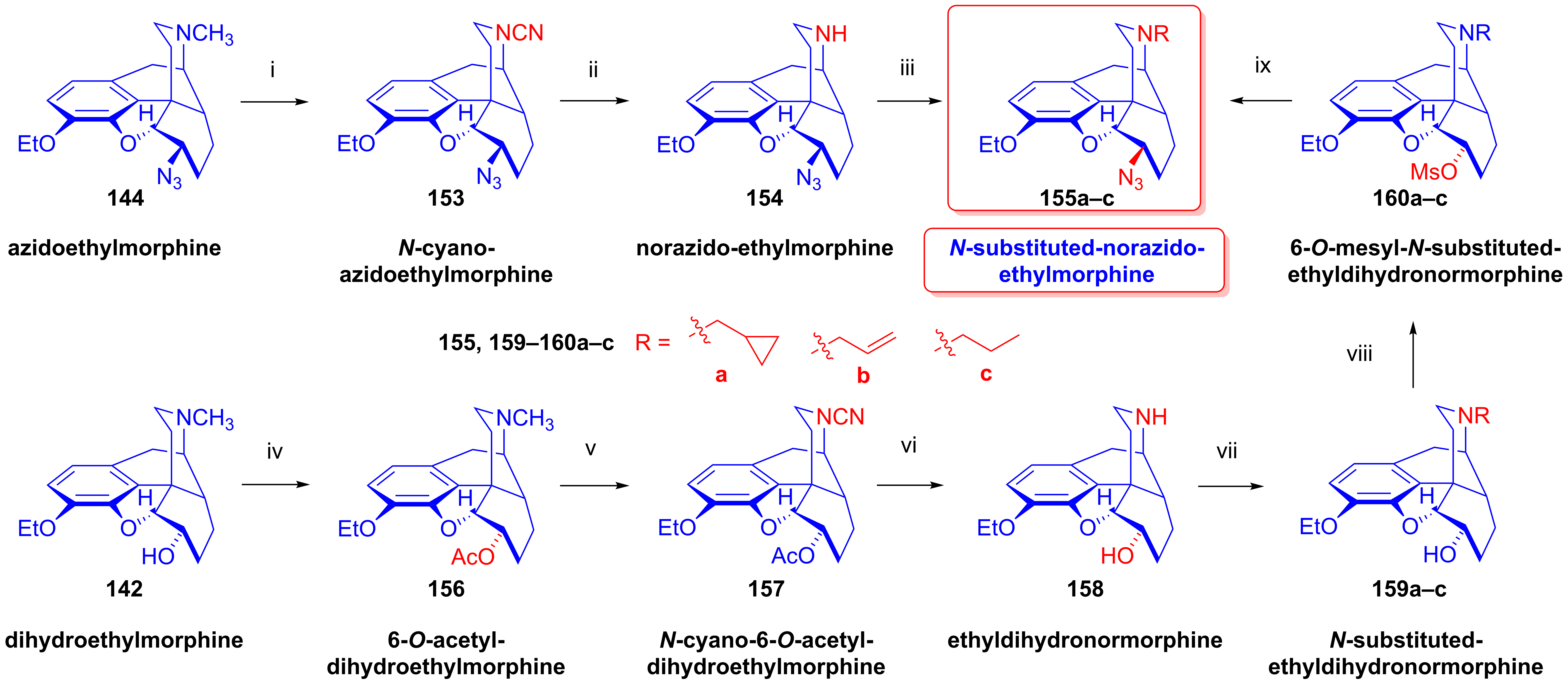

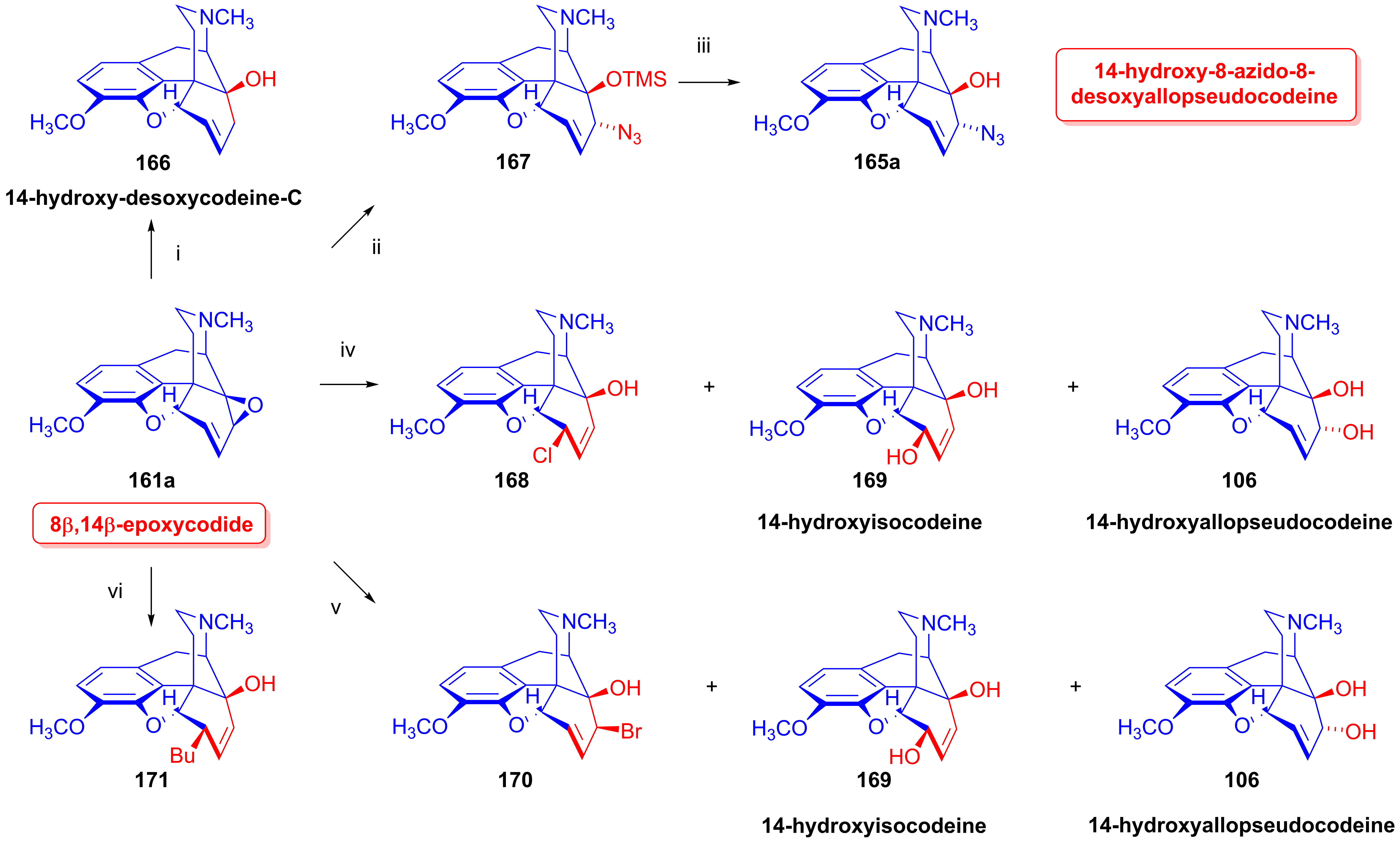

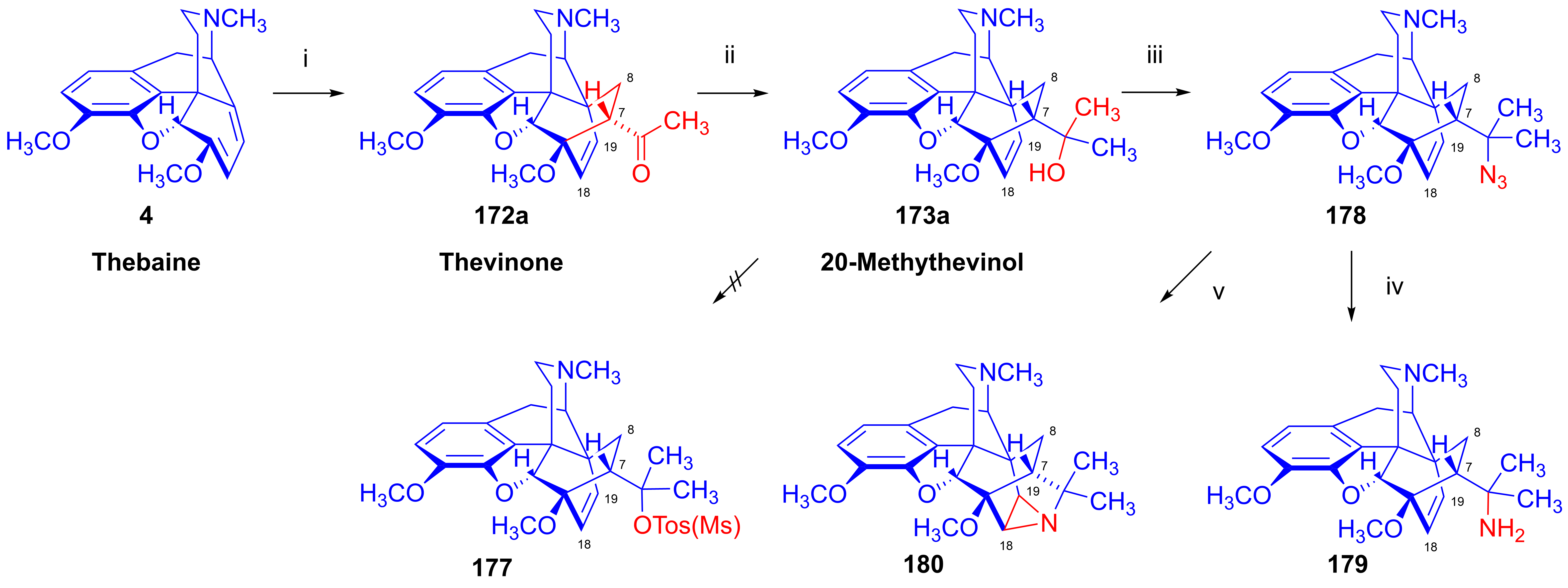
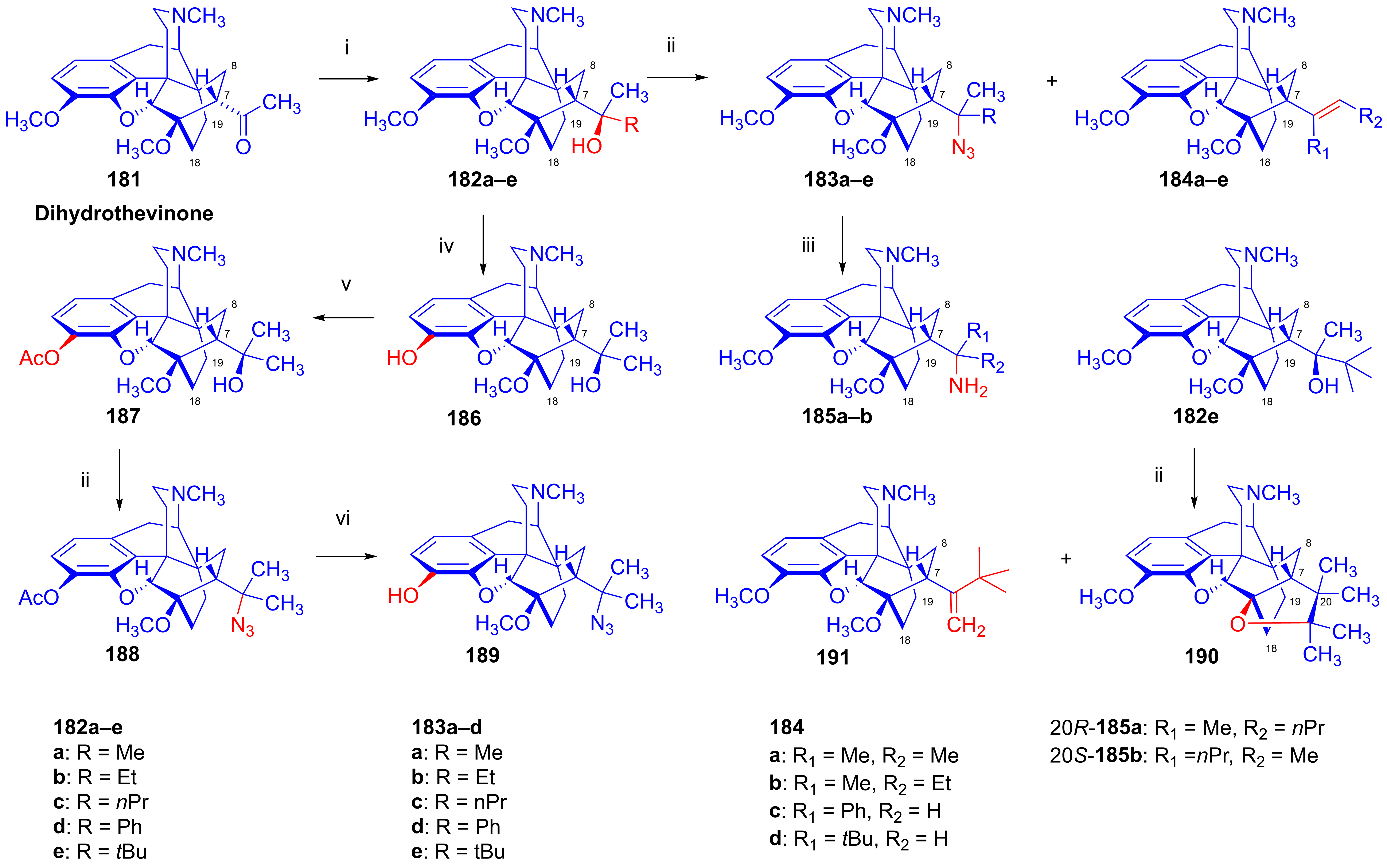




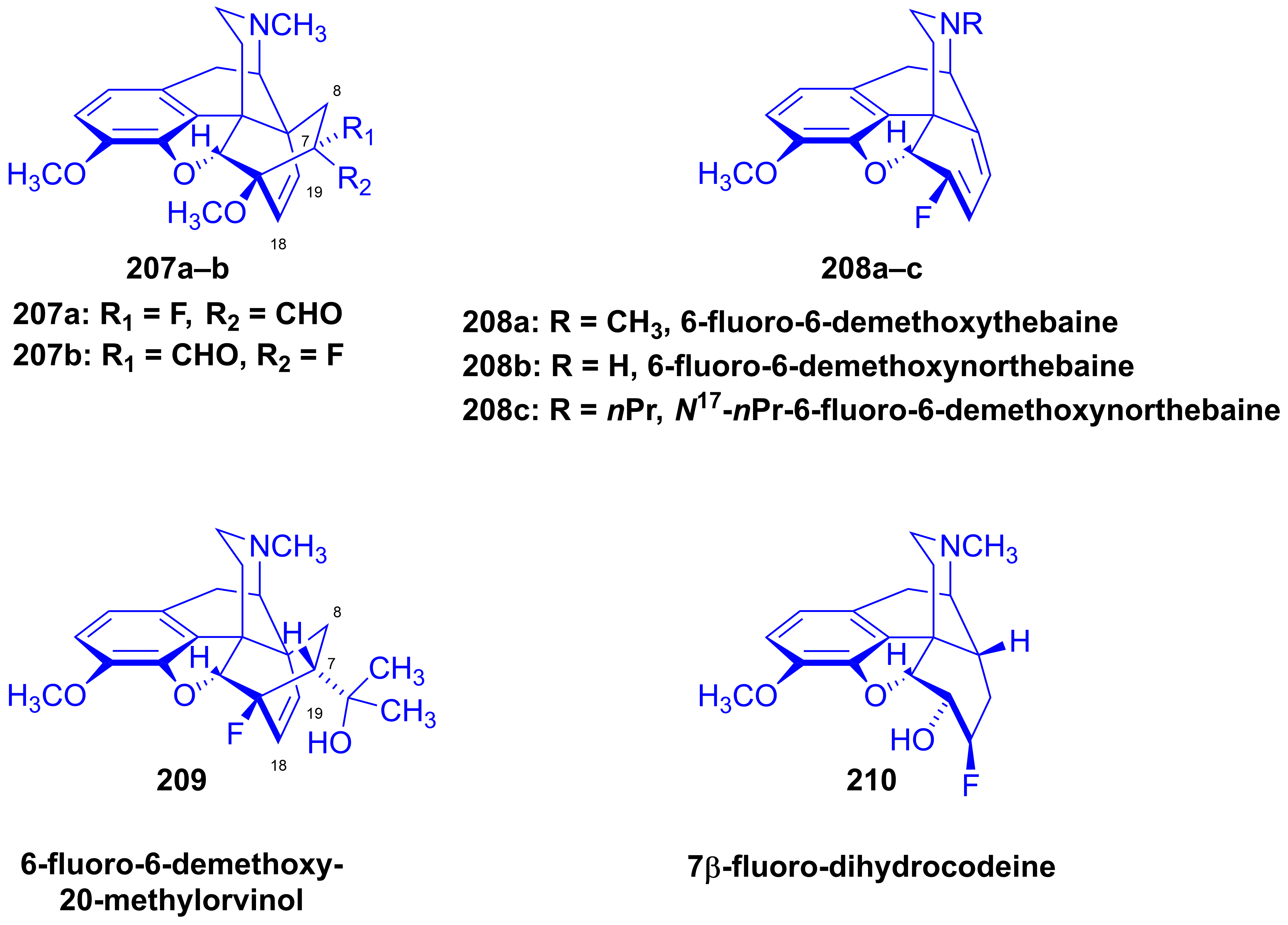
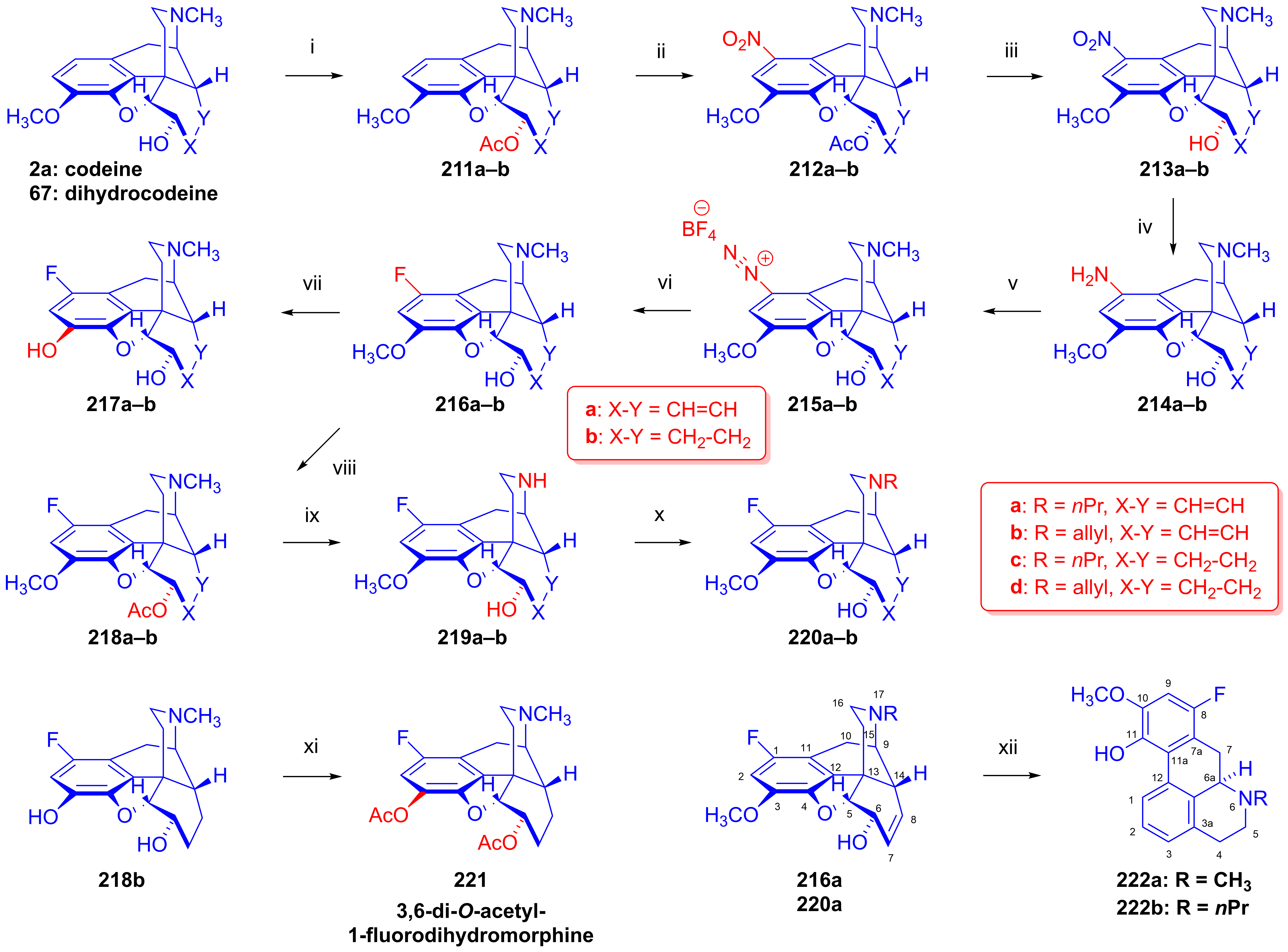
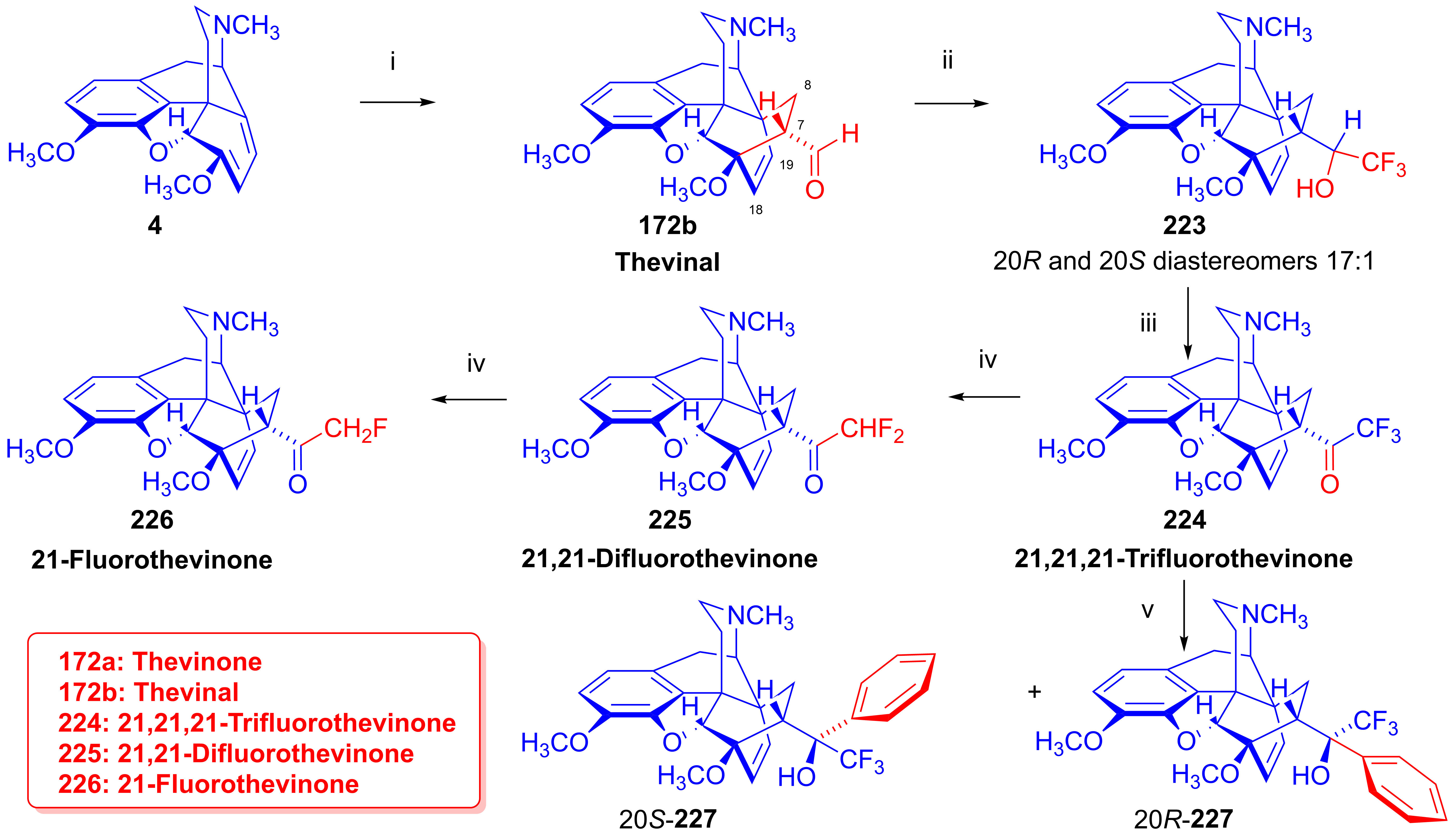
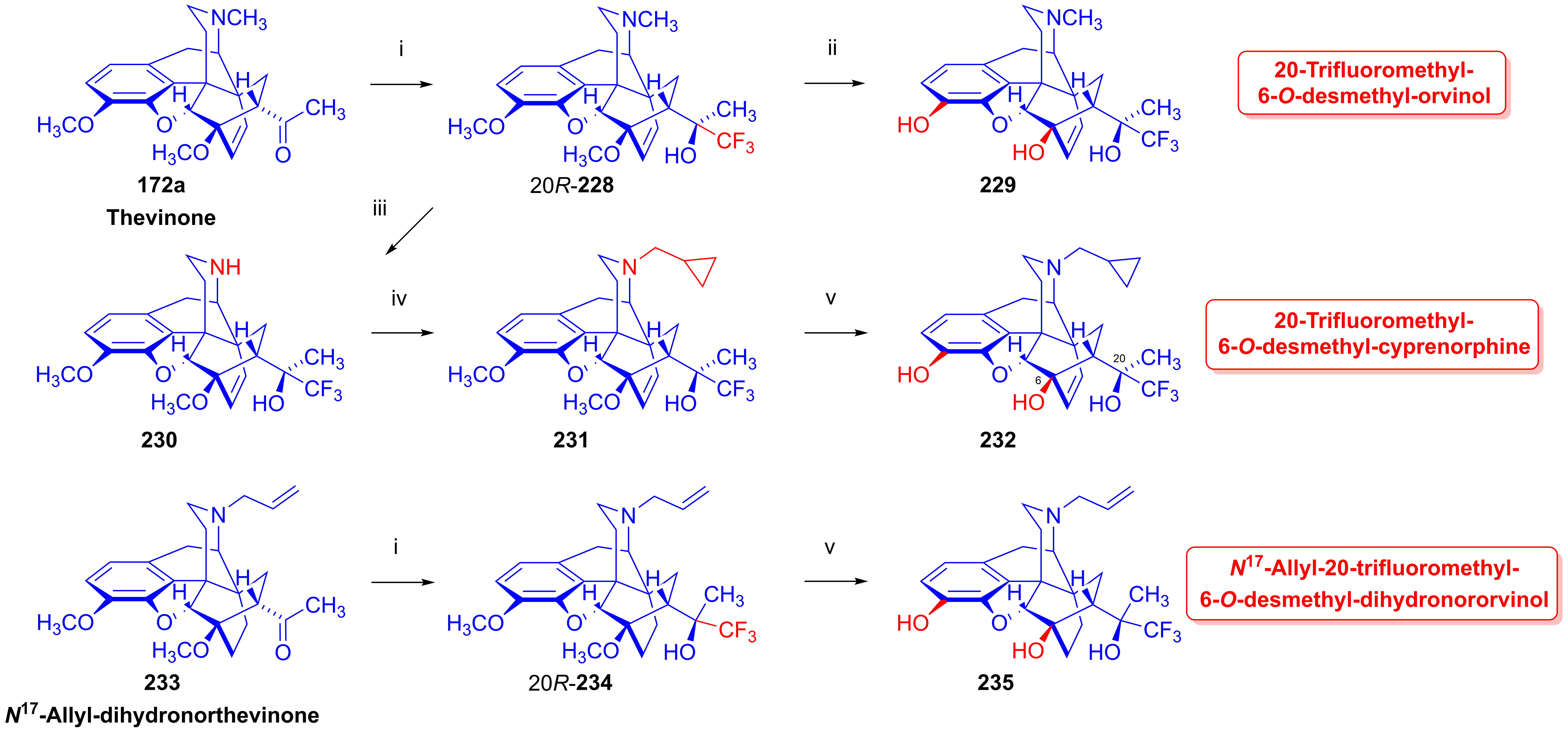

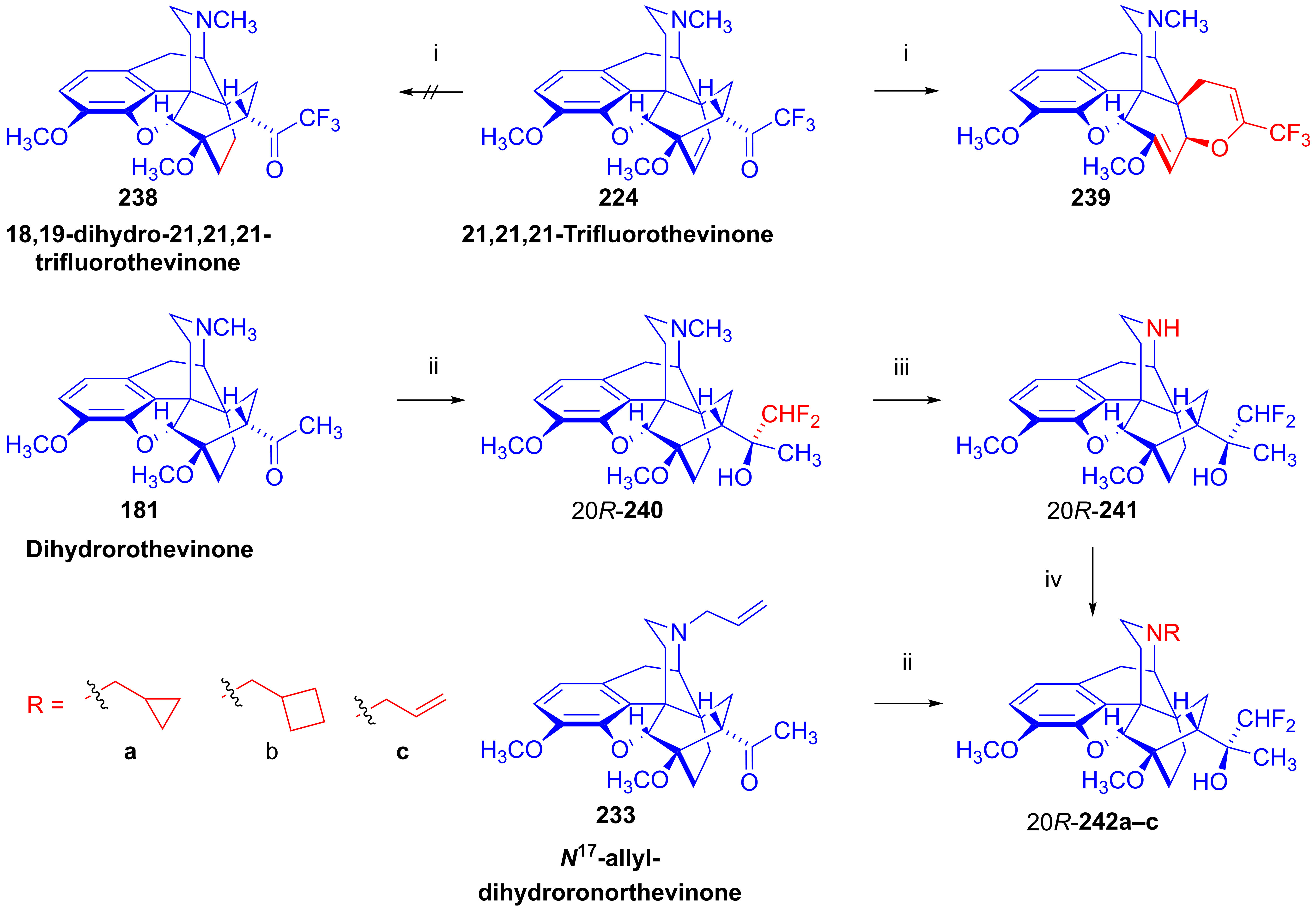
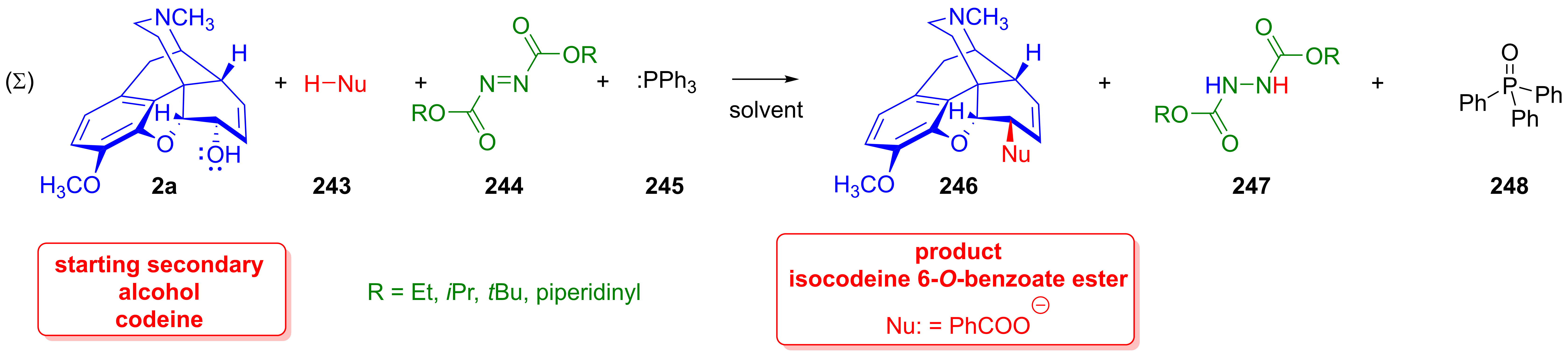

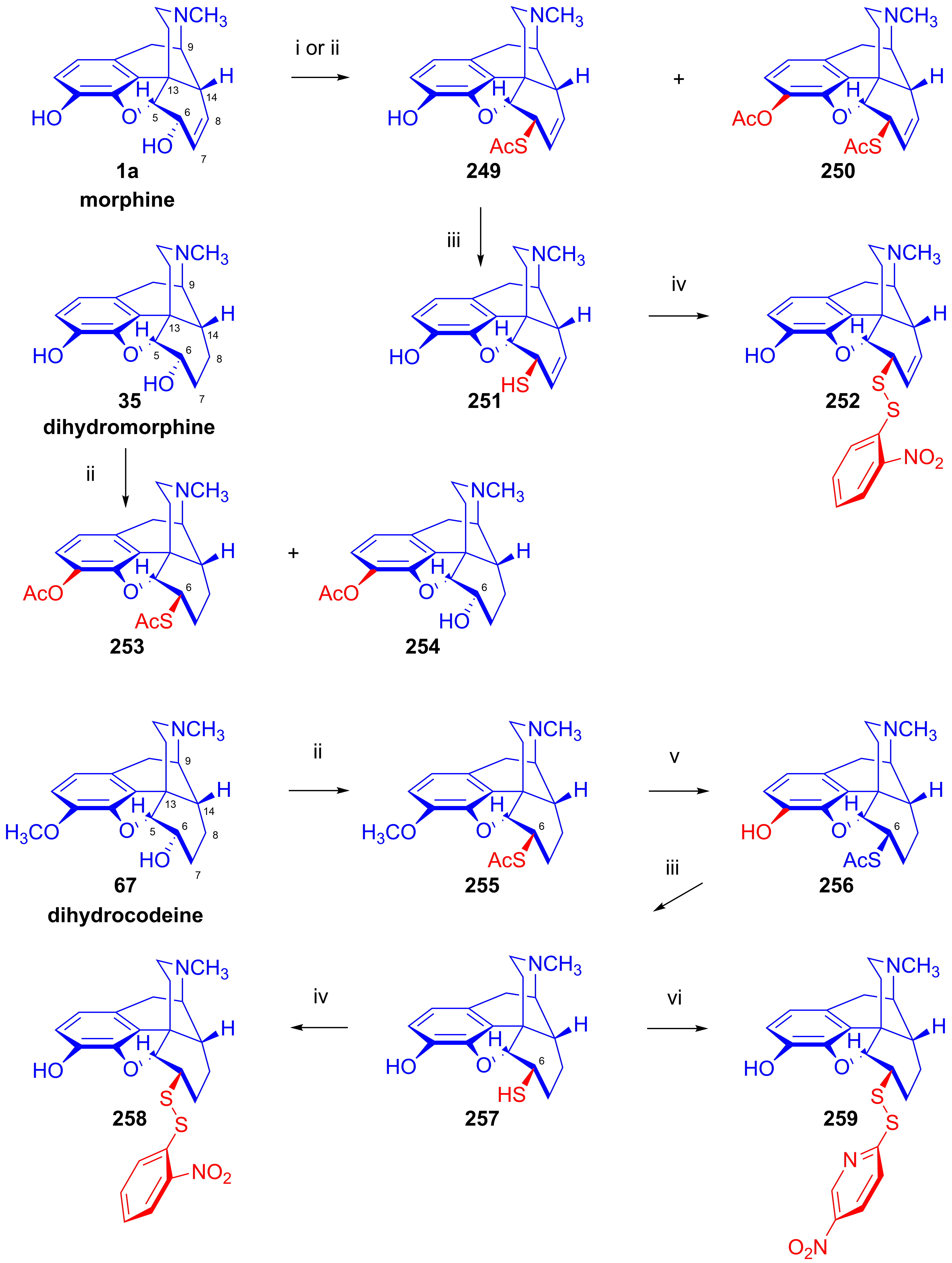
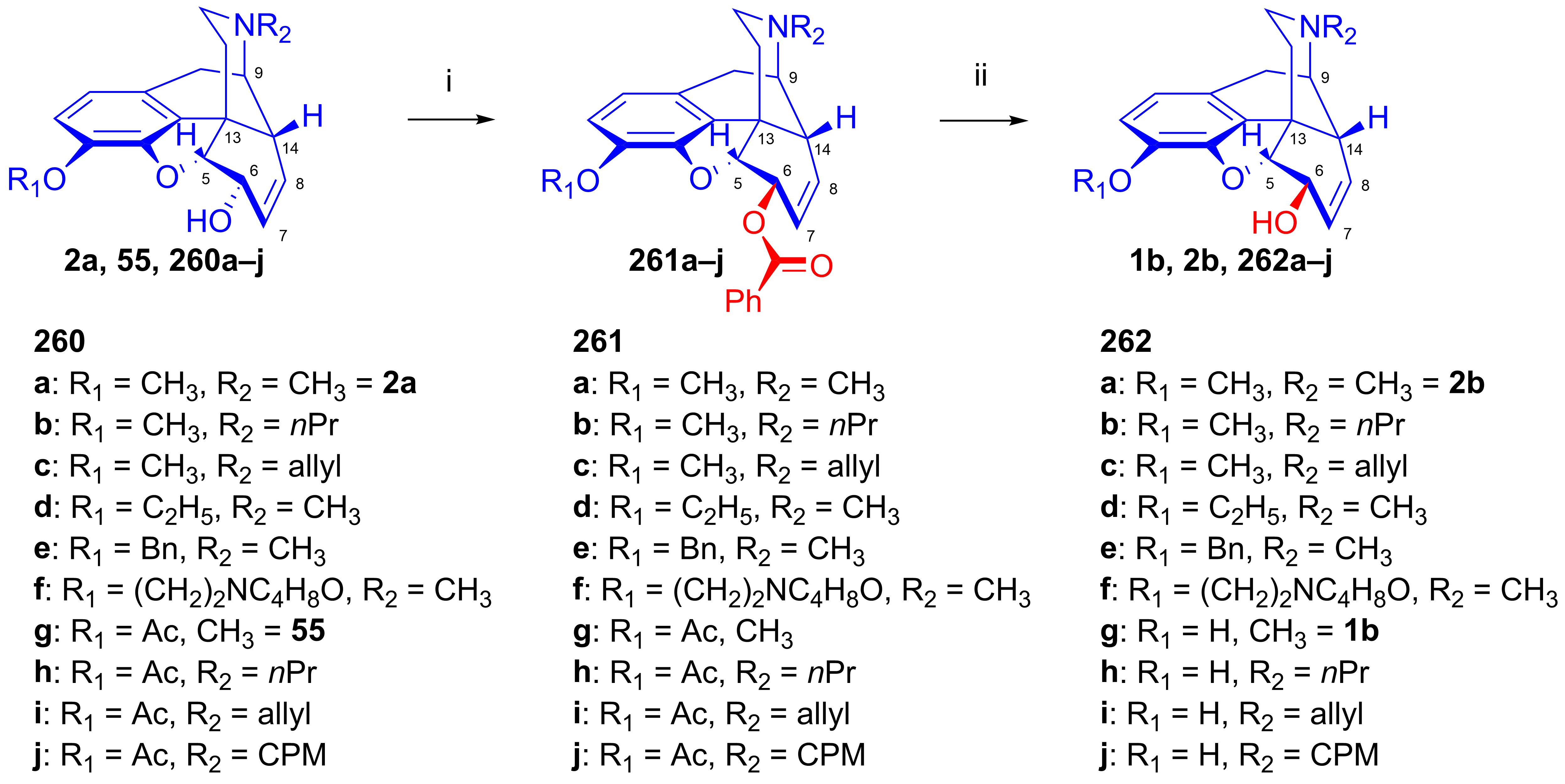
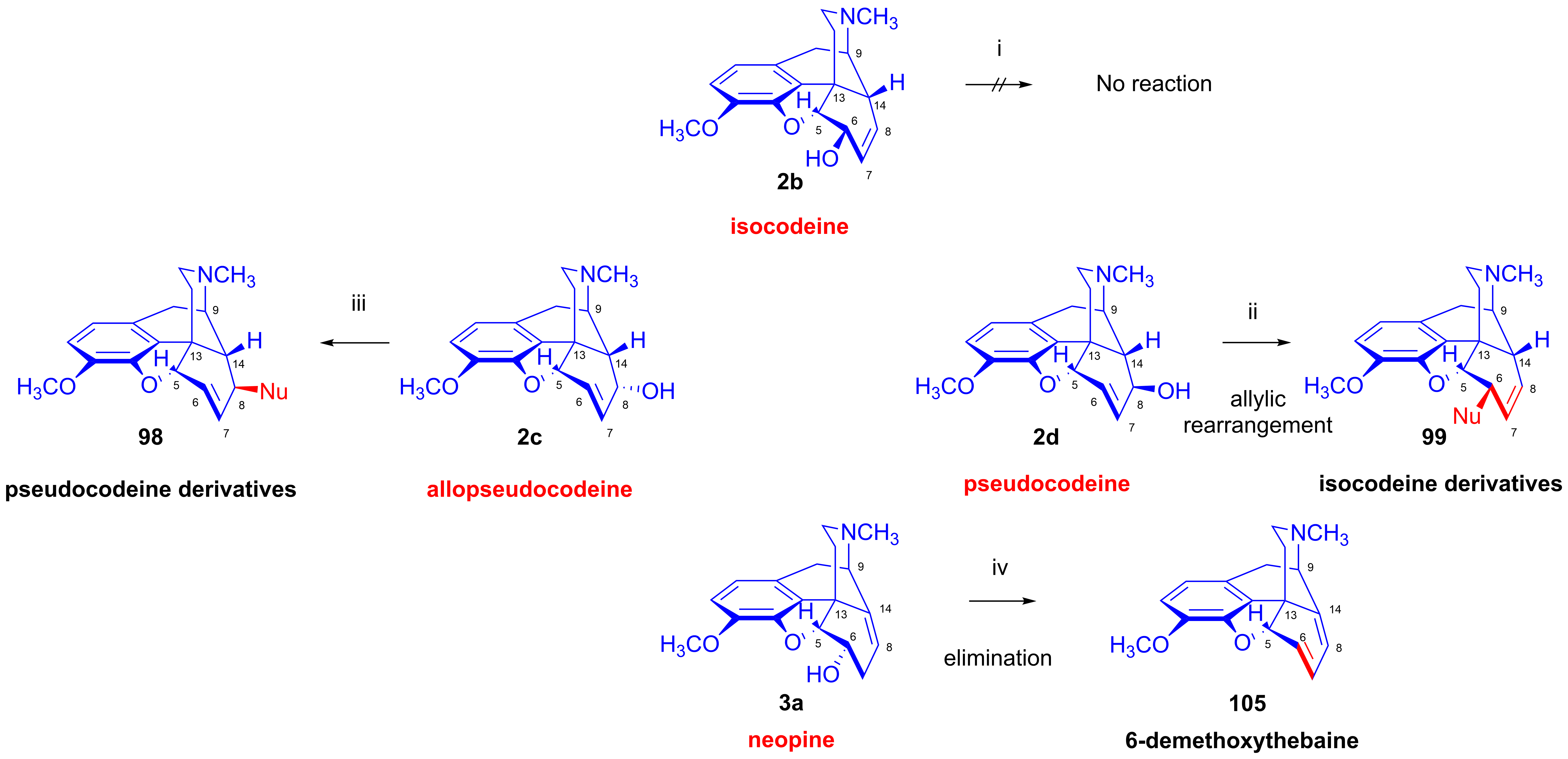

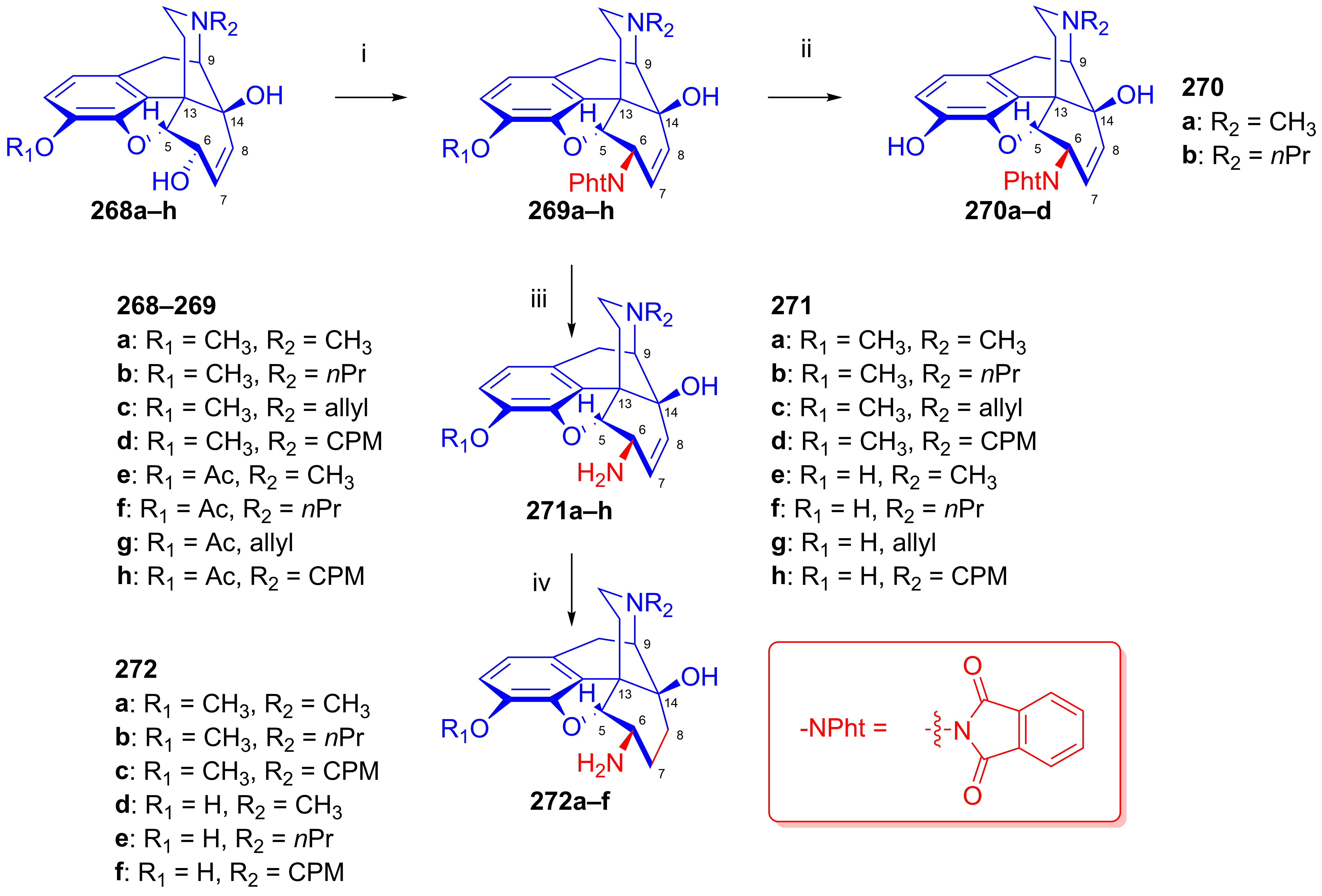
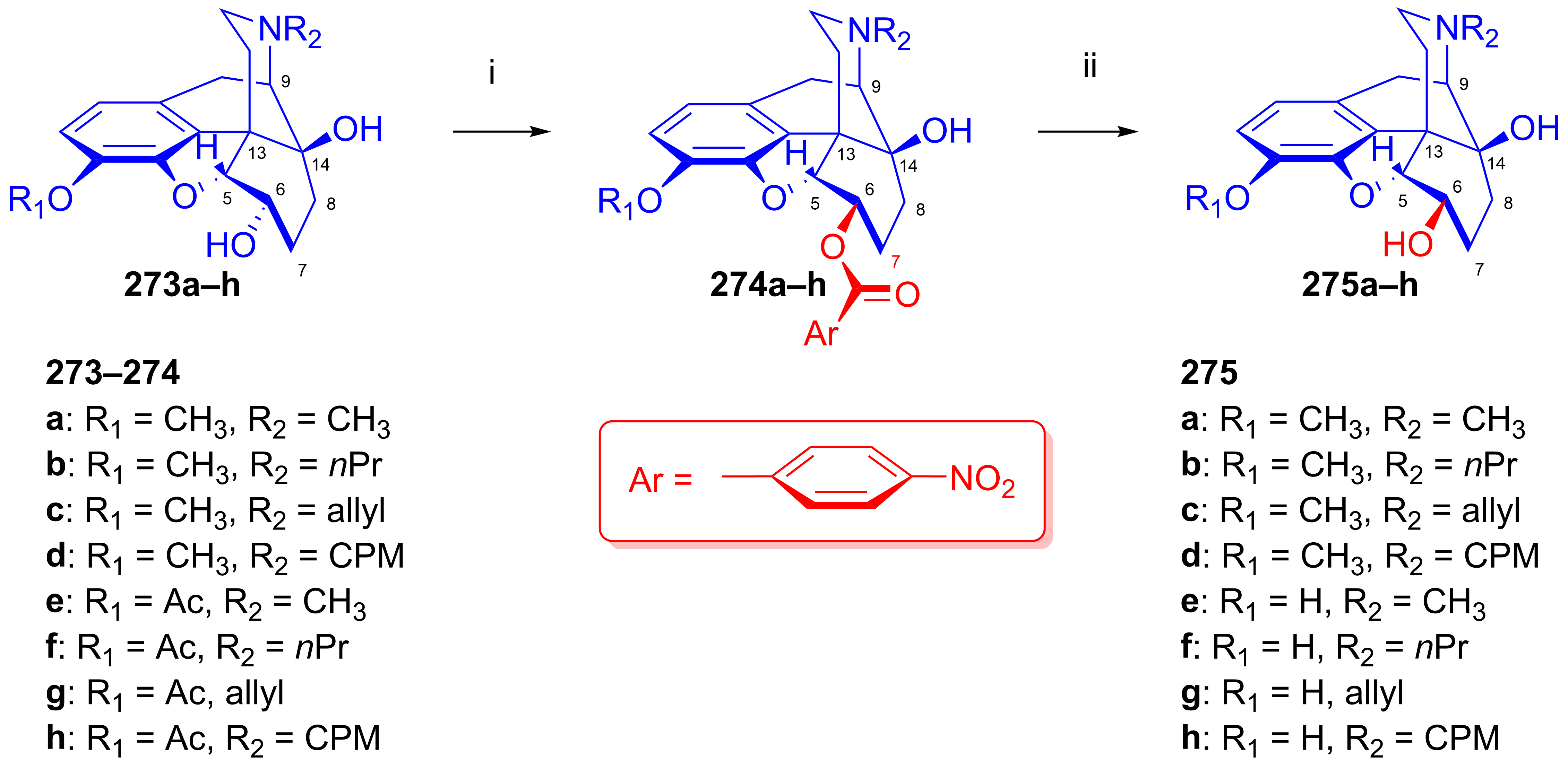

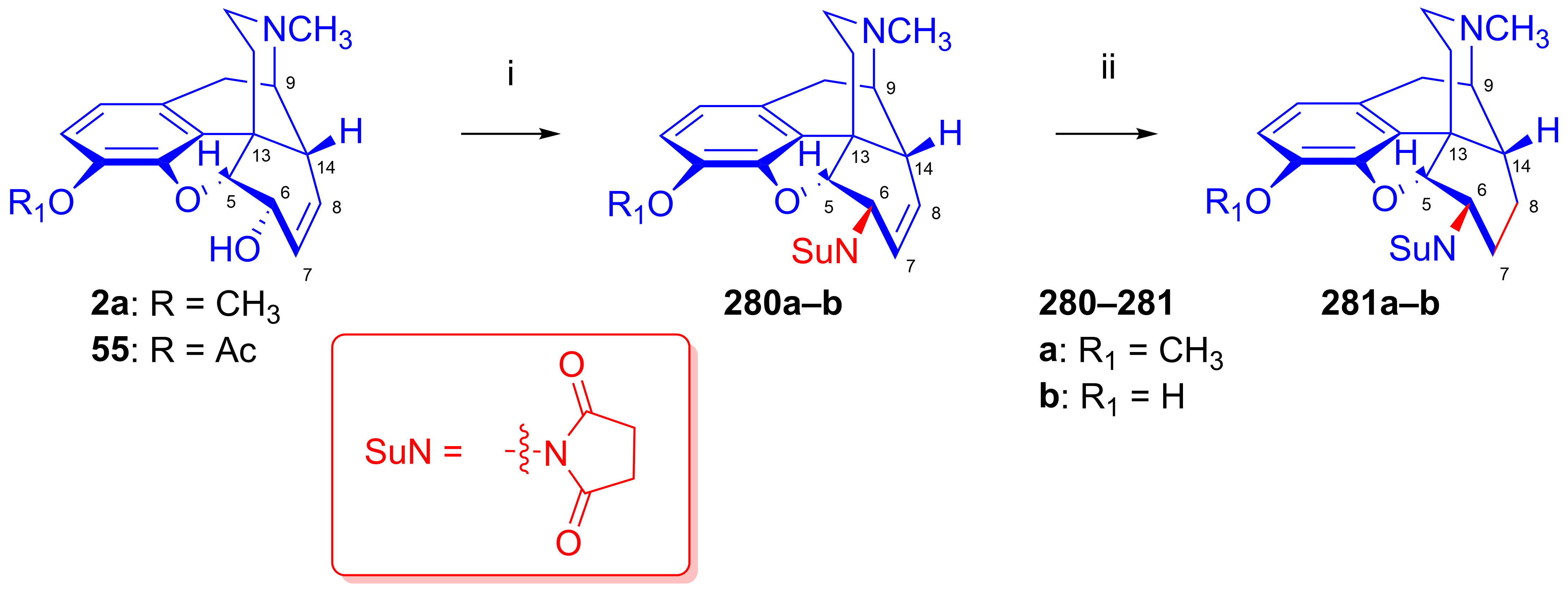
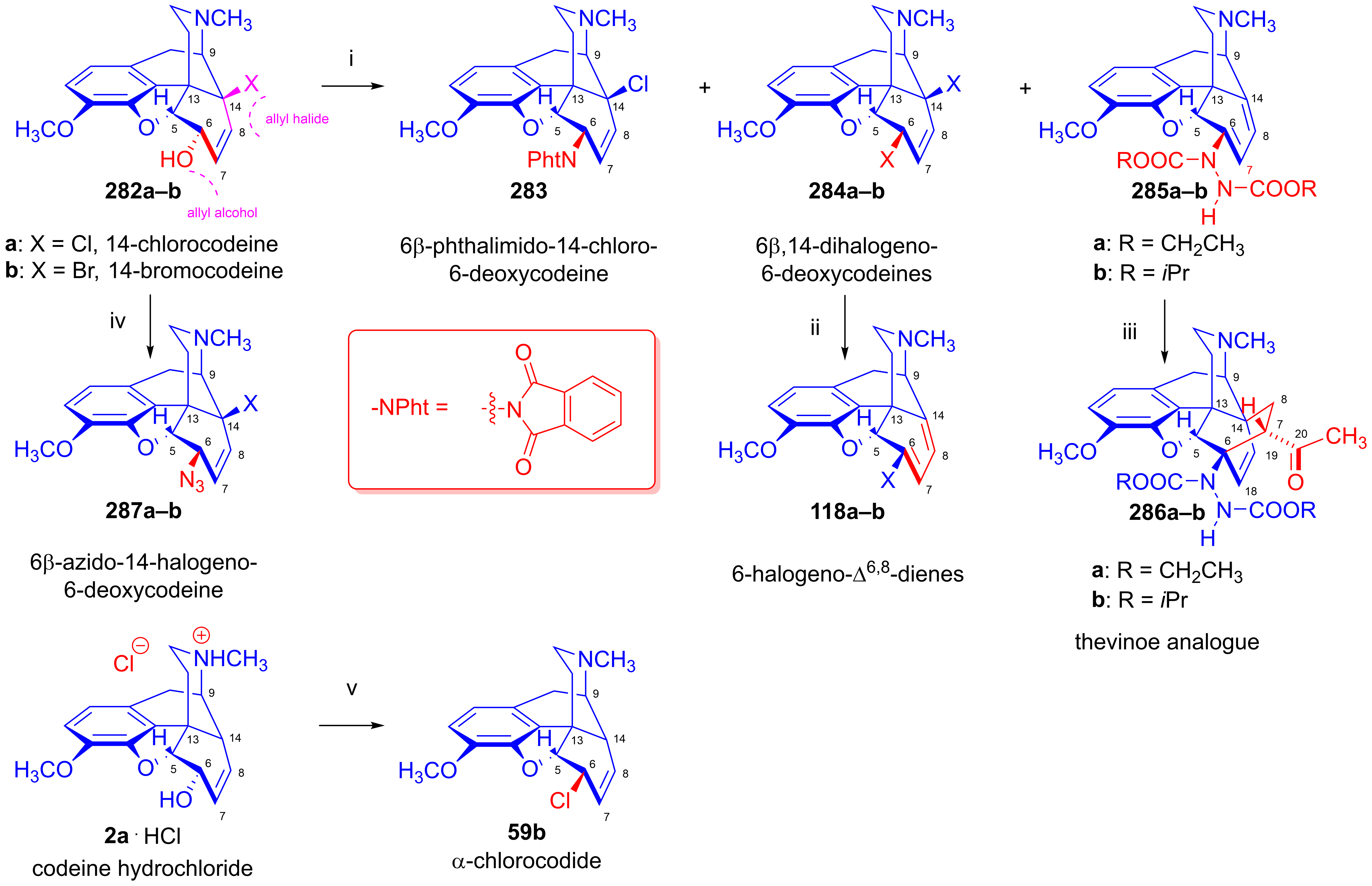
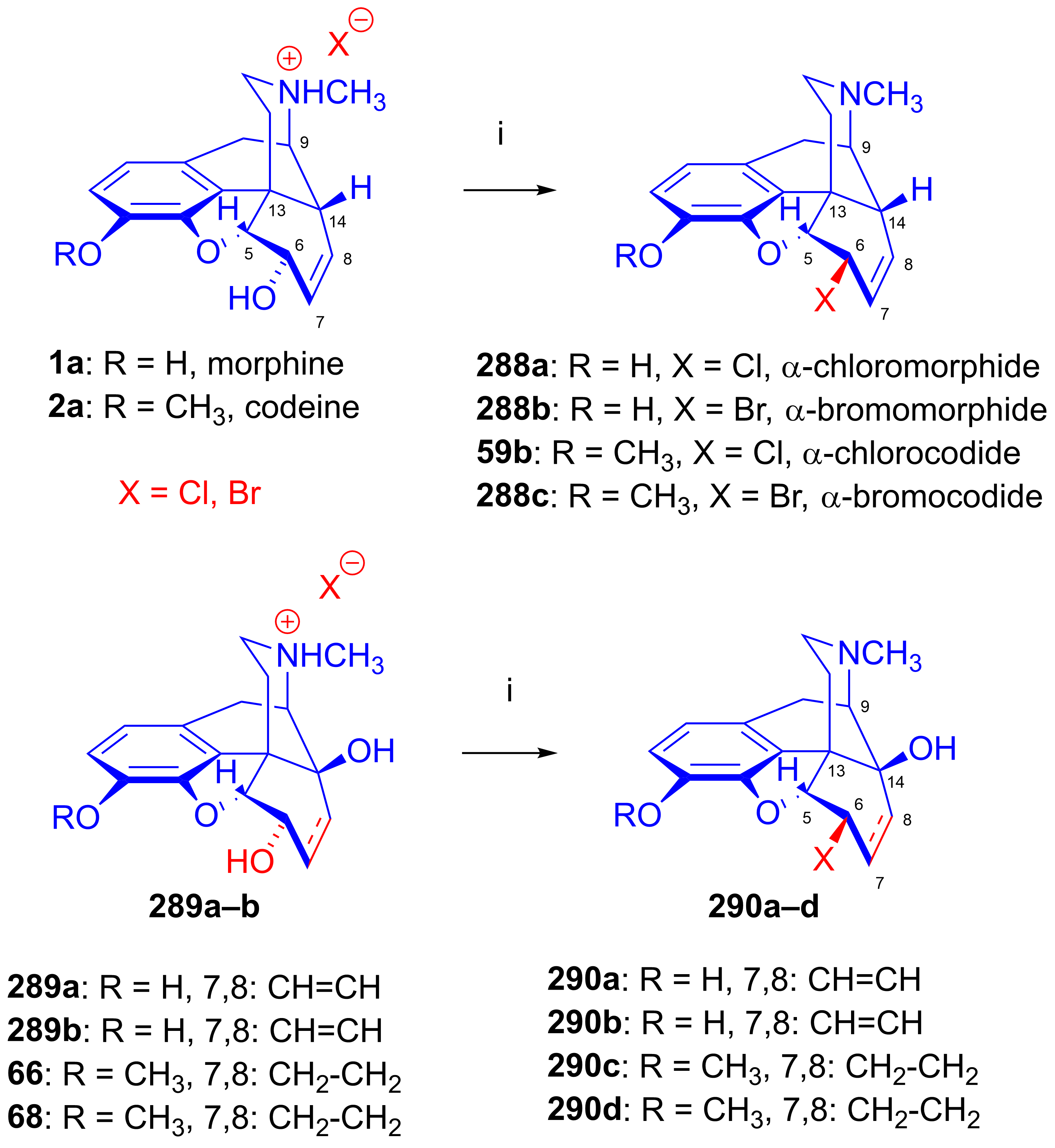
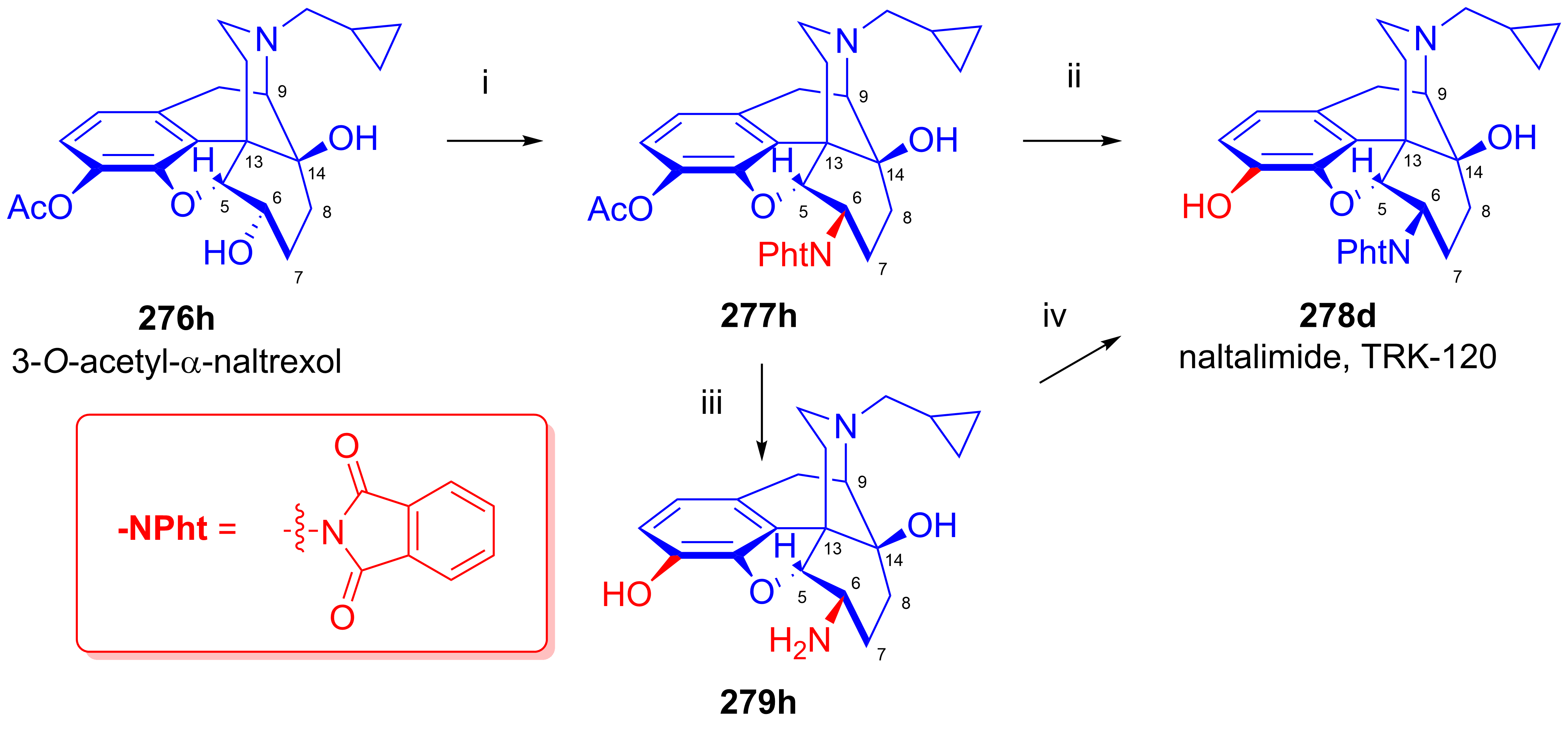
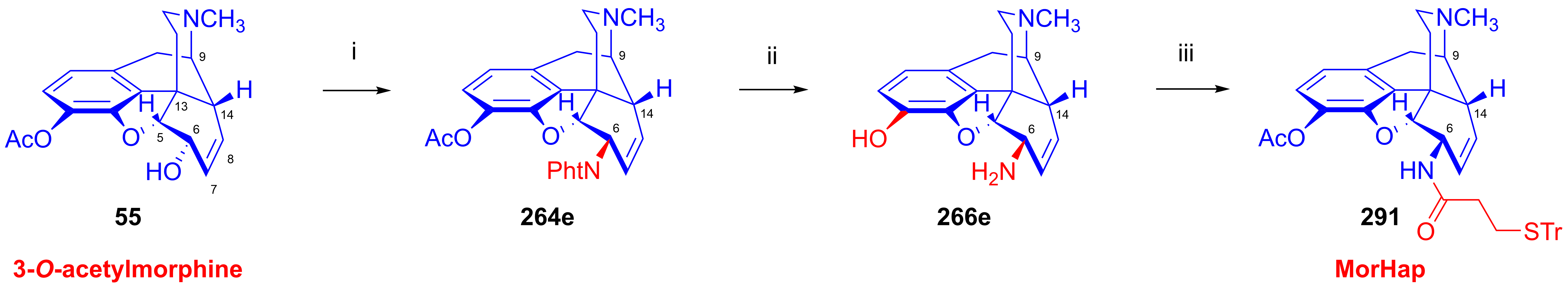
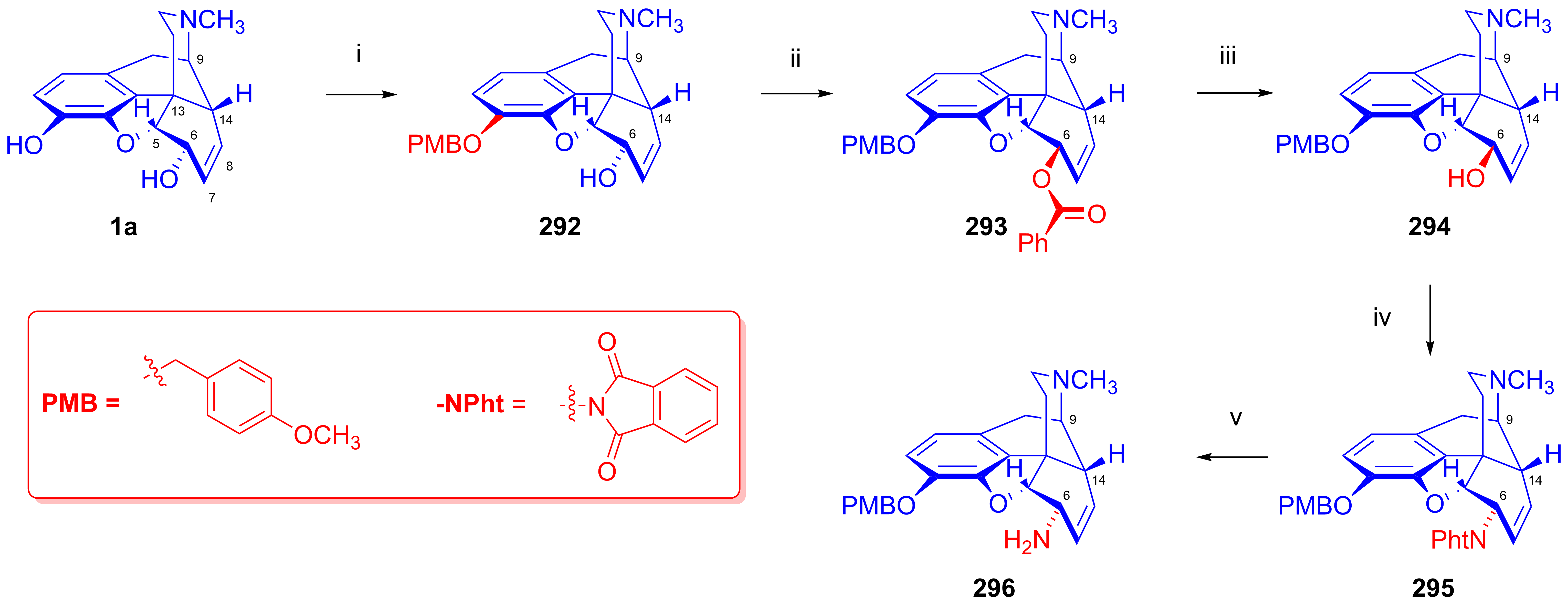
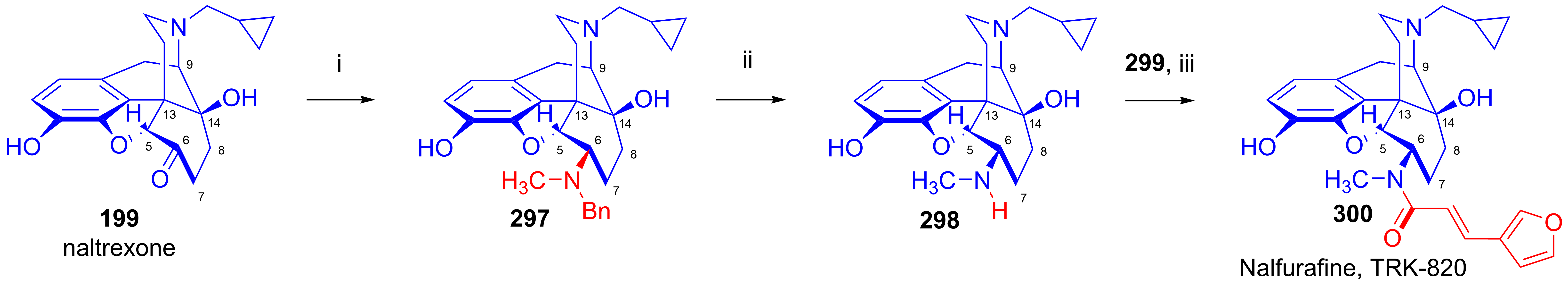


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Comp. | Name | Position of the C=C Double Bond | Position | |||||
|---|---|---|---|---|---|---|---|---|
| 5 | 6 | 8 | 9 | 13 | 14 | |||
| 1a | morphine | Δ7,8 | 5R | 6S | - | 9R | 13S | 14R |
| 1b | isomorphine * | Δ7,8 | 5R | 6R | - | 9R | 13S | 14R |
| 1c | allopseudomorphine ** | Δ6,7 | 5S | - | 8R | 9R | 13S | 14R |
| 1d | γ-isomorphine | Δ6,7 | 5S | - | 8S | 9R | 13S | 14R |
| 2a | codeine | Δ7,8 | 5R | 6S | - | 9R | 13S | 14R |
| 2b | isocodeine | Δ7,8 | 5R | 6R | - | 9R | 13S | 14R |
| 2c | allopseudocodeine | Δ6,7 | 5S | - | 8R | 9R | 13S | 14R |
| 2d | pseudocodeine | Δ6,7 | 5S | - | 8S | 9R | 13S | 14R |
| 3a | neopine | Δ8,14 | 5R | 6S | - | 9R | 13S | - |
| 3b | isoneopine | Δ8,14 | 5R | 6R | - | 9R | 13S | - |
| 3c | neomorphine | Δ8,14 | 5R | 6S | - | 9R | 13S | - |
| 3d | isoneomorphine | Δ8,14 | 5R | 6R | - | 9R | 13S | - |
| 4 | thebaine | Δ6,7 and Δ8,14 | 5R | - | - | 9R | 13S | - |
| 5 | oripavine | Δ6,7 and Δ8,14 | 5R | - | - | 9R | 13S | - |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Marton, J.; Cumming, P.; Rice, K.C.; Linders, J.T.M. Morphinan Alkaloids and Their Transformations: A Historical Perspective of a Century of Opioid Research in Hungary. Int. J. Mol. Sci. 2025, 26, 2736. https://doi.org/10.3390/ijms26062736
Marton J, Cumming P, Rice KC, Linders JTM. Morphinan Alkaloids and Their Transformations: A Historical Perspective of a Century of Opioid Research in Hungary. International Journal of Molecular Sciences. 2025; 26(6):2736. https://doi.org/10.3390/ijms26062736
Chicago/Turabian StyleMarton, János, Paul Cumming, Kenner C. Rice, and Joannes T. M. Linders. 2025. "Morphinan Alkaloids and Their Transformations: A Historical Perspective of a Century of Opioid Research in Hungary" International Journal of Molecular Sciences 26, no. 6: 2736. https://doi.org/10.3390/ijms26062736
APA StyleMarton, J., Cumming, P., Rice, K. C., & Linders, J. T. M. (2025). Morphinan Alkaloids and Their Transformations: A Historical Perspective of a Century of Opioid Research in Hungary. International Journal of Molecular Sciences, 26(6), 2736. https://doi.org/10.3390/ijms26062736