
Lysosomal Dysfunction in Amyotrophic Lateral Sclerosis: A Familial Case Linked to the p.G376D TARDBP Mutation

 , , ,
, , ,  and
and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
2.1. Lysosomal Functionality Is Altered in Cells Expressing the TDP-43G376D Mutant Protein
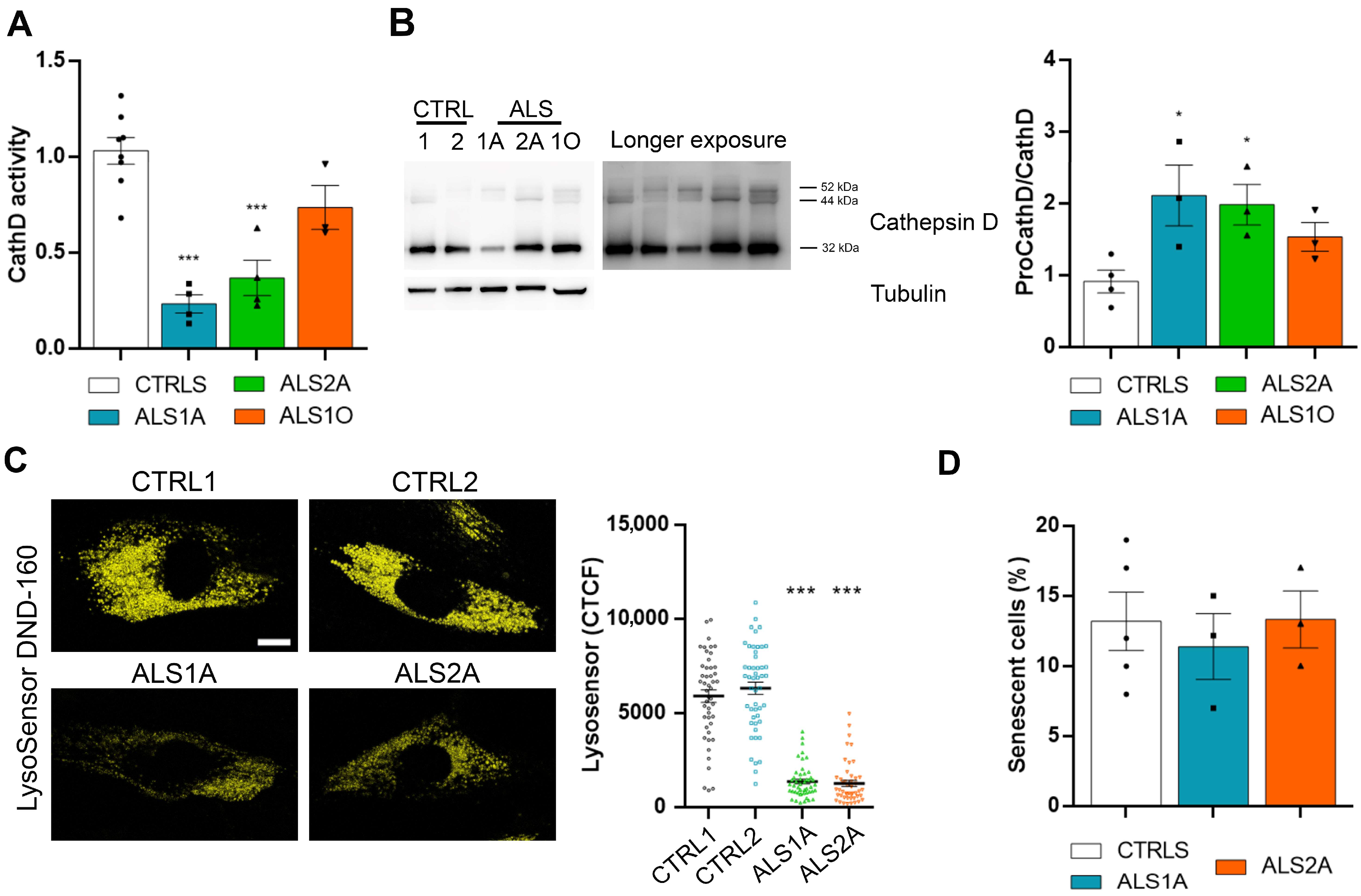
2.2. ALS Fibroblasts Display Abnormal Lysosomal Acidification
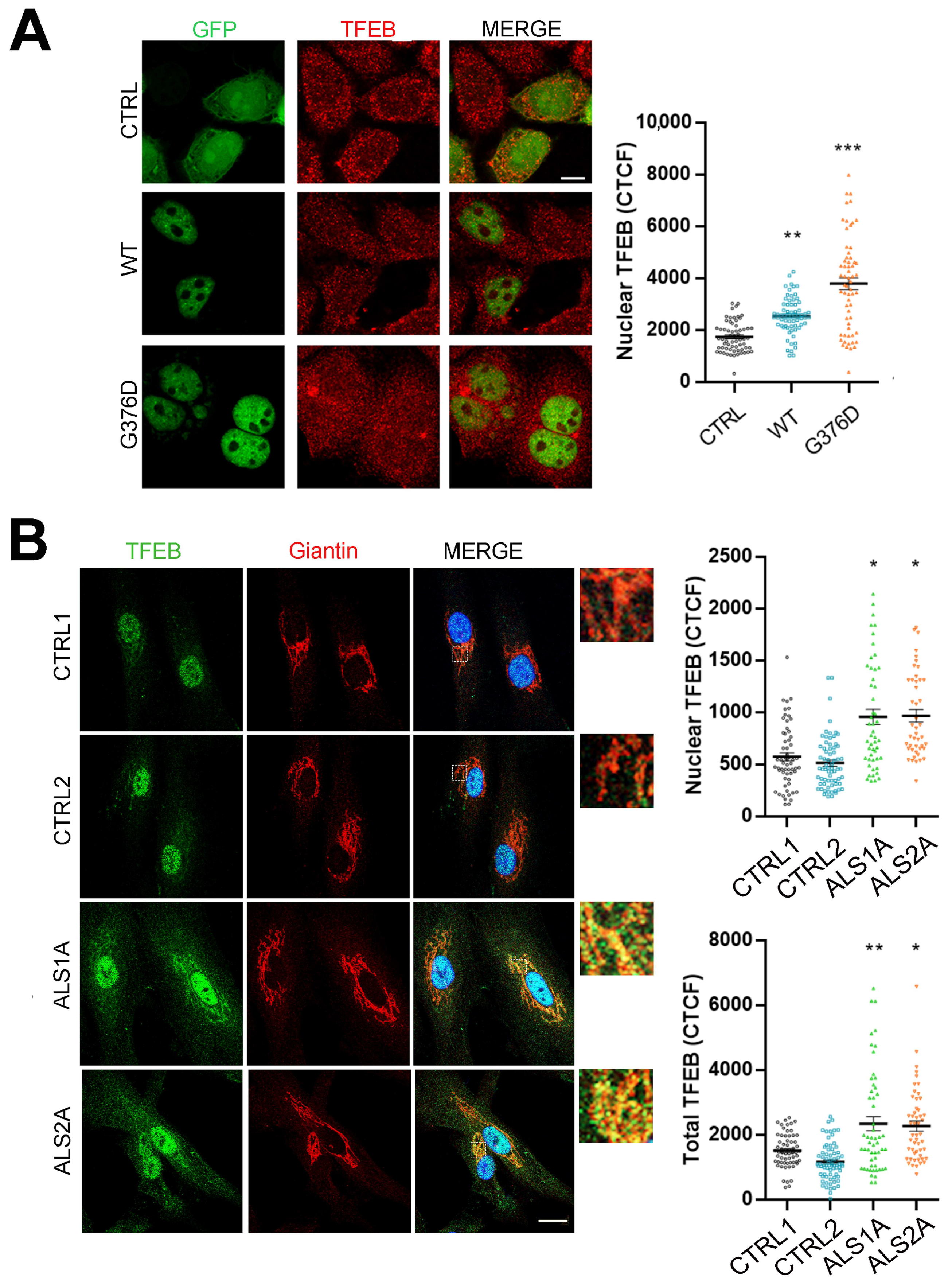
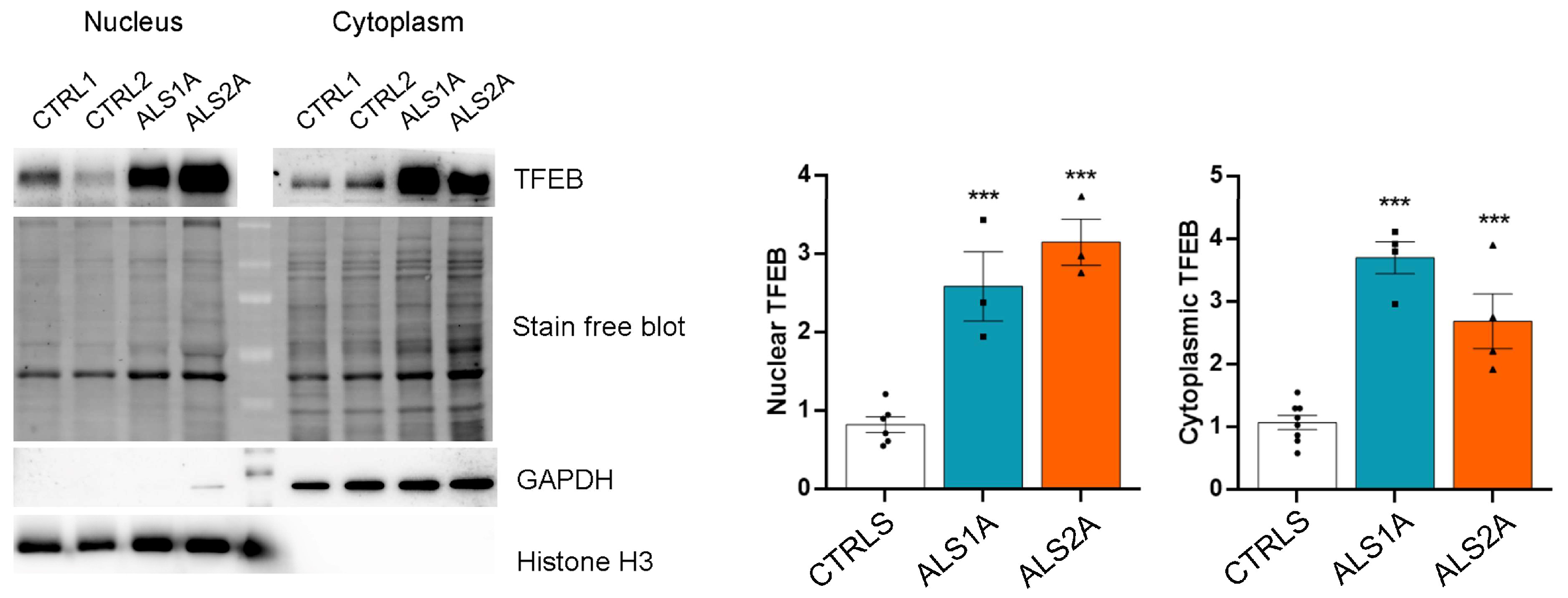
2.3. TFEB Nuclear Translocation Is Increased in Cells Expressing the TDP-43G376D Mutant Protein
2.4. Increased Nuclear Translocation of TFEB Depends on Reduced Activation of AKT
2.5. ALS Motor Neurons Carrying the G376D Mutation in TDP-43 Show Reduced Lysosomal Activity
3. Discussion
4. Materials and Methods
4.1. Antibodies and Reagents
4.2. Cell Lines
4.3. Transfection
4.4. Western Blotting
4.5. Nuclear and Cytoplasmic Fractionation
4.6. Immunofluorescence and Confocal Microscopy
4.7. DQ-BSA (Self-Quenched BODIPY Dye Conjugated of Bovine Serum Albumin) Assay and Live Cell Staining
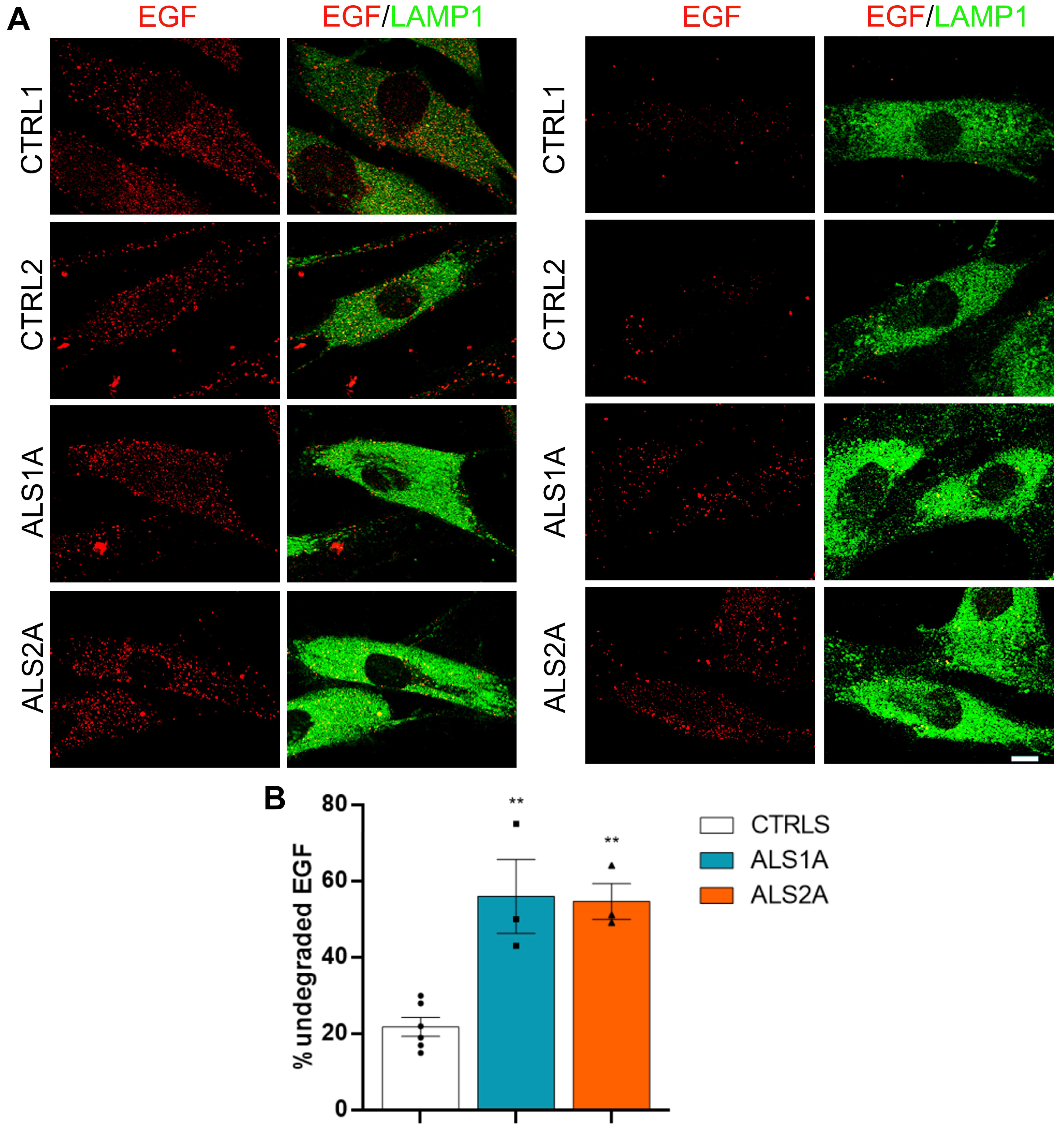
4.8. EGF Degradation Assay
4.9. Cathepsin D Activity
4.10. Real-Time PCR
4.11. Senescence-Associated Beta-Galactosidase (SA-βgal) Activity Assay
4.12. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hardiman, O.; Al-Chalabi, A.; Chio, A.; Corr, E.M.; Logroscino, G.; Robberecht, W.; Shaw, P.J.; Simmons, Z.; van den Berg, L.H. Amyotrophic lateral sclerosis. Nat. Rev. Dis. Primers 2017, 3, 17085. [Google Scholar] [CrossRef] [PubMed]
- Feldman, E.L.; Goutman, S.A.; Petri, S.; Mazzini, L.; Savelieff, M.G.; Shaw, P.J.; Sobue, G. Amyotrophic lateral sclerosis. Lancet 2022, 400, 1363–1380. [Google Scholar] [CrossRef] [PubMed]
- Prasad, A.; Bharathi, V.; Sivalingam, V.; Girdhar, A.; Patel, B.K. Molecular Mechanisms of TDP-43 Misfolding and Pathology in Amyotrophic Lateral Sclerosis. Front. Mol. Neurosci. 2019, 12, 25. [Google Scholar] [CrossRef]
- Masrori, P.; Van Damme, P. Amyotrophic lateral sclerosis: A clinical review. Eur. J. Neurol. 2020, 27, 1918–1929. [Google Scholar]
- Amin, A.; Perera, N.D.; Beart, P.M.; Turner, B.J.; Shabanpoor, F. Amyotrophic Lateral Sclerosis and Autophagy: Dysfunction and Therapeutic Targeting. Cells 2020, 9, 2413. [Google Scholar] [CrossRef]
- Suk, T.R.; Rousseaux, M.W.C. The role of TDP-43 mislocalization in amyotrophic lateral sclerosis. Mol. Neurodegener. 2020, 15, 45. [Google Scholar]
- Jo, M.; Lee, S.; Jeon, Y.M.; Kim, S.; Kwon, Y.; Kim, H.J. The role of TDP-43 propagation in neurodegenerative diseases: Integrating insights from clinical and experimental studies. Exp. Mol. Med. 2020, 52, 1652–1662. [Google Scholar] [CrossRef]
- Liao, Y.Z.; Ma, J.; Dou, J.Z. The Role of TDP-43 in Neurodegenerative Disease. Mol. Neurobiol. 2022, 59, 4223–4241. [Google Scholar] [CrossRef]
- Ayala, Y.M.; Zago, P.; D’Ambrogio, A.; Xu, Y.F.; Petrucelli, L.; Buratti, E.; Baralle, F.E. Structural determinants of the cellular localization and shuttling of TDP-43. J. Cell Sci. 2008, 121, 3778–3785. [Google Scholar] [CrossRef]
- Santamaria, N.; Alhothali, M.; Alfonso, M.H.; Breydo, L.; Uversky, V.N. Intrinsic disorder in proteins involved in amyotrophic lateral sclerosis. Cell. Mol. Life Sci. 2017, 74, 1297–1318. [Google Scholar] [CrossRef]
- Zhang, Y.J.; Xu, Y.F.; Cook, C.; Gendron, T.F.; Roettges, P.; Link, C.D.; Lin, W.L.; Tong, J.; Castanedes-Casey, M.; Ash, P.; et al. Aberrant cleavage of TDP-43 enhances aggregation and cellular toxicity. Proc. Natl. Acad. Sci. USA 2009, 106, 7607–7612. [Google Scholar] [CrossRef] [PubMed]
- Chen, A.K.; Lin, R.Y.; Hsieh, E.Z.; Tu, P.H.; Chen, R.P.; Liao, T.Y.; Chen, W.; Wang, C.H.; Huang, J.J. Induction of amyloid fibrils by the C-terminal fragments of TDP-43 in amyotrophic lateral sclerosis. J. Am. Chem. Soc. 2010, 132, 1186–1187. [Google Scholar] [CrossRef] [PubMed]
- Guo, W.; Chen, Y.; Zhou, X.; Kar, A.; Ray, P.; Chen, X.; Rao, E.J.; Yang, M.; Ye, H.; Zhu, L.; et al. An ALS-associated mutation affecting TDP-43 enhances protein aggregation, fibril formation and neurotoxicity. Nat. Struct. Mol. Biol. 2011, 18, 822–830. [Google Scholar] [CrossRef] [PubMed]
- Coyne, A.N.; Zaepfel, B.L.; Zarnescu, D.C. Failure to Deliver and Translate-New Insights into RNA Dysregulation in ALS. Front. Cell. Neurosci. 2017, 11, 243. [Google Scholar] [CrossRef]
- Birsa, N.; Bentham, M.P.; Fratta, P. Cytoplasmic functions of TDP-43 and FUS and their role in ALS. Semin. Cell Dev. Biol. 2020, 99, 193–201. [Google Scholar] [CrossRef]
- Polymenidou, M.; Lagier-Tourenne, C.; Hutt, K.R.; Huelga, S.C.; Moran, J.; Liang, T.Y.; Ling, S.C.; Sun, E.; Wancewicz, E.; Mazur, C.; et al. Long pre-mRNA depletion and RNA missplicing contribute to neuronal vulnerability from loss of TDP-43. Nat. Neurosci. 2011, 14, 459–468. [Google Scholar] [CrossRef]
- Tollervey, J.R.; Curk, T.; Rogelj, B.; Briese, M.; Cereda, M.; Kayikci, M.; Konig, J.; Hortobagyi, T.; Nishimura, A.L.; Zupunski, V.; et al. Characterizing the RNA targets and position-dependent splicing regulation by TDP-43. Nat. Neurosci. 2011, 14, 452–458. [Google Scholar] [CrossRef]
- Xiao, S.; Sanelli, T.; Dib, S.; Sheps, D.; Findlater, J.; Bilbao, J.; Keith, J.; Zinman, L.; Rogaeva, E.; Robertson, J. RNA targets of TDP-43 identified by UV-CLIP are deregulated in ALS. Mol. Cell. Neurosci. 2011, 47, 167–180. [Google Scholar] [CrossRef]
- Bose, J.K.; Huang, C.C.; Shen, C.K. Regulation of autophagy by neuropathological protein TDP-43. J. Biol. Chem. 2011, 286, 44441–44448. [Google Scholar] [CrossRef]
- Xia, Q.; Wang, H.; Hao, Z.; Fu, C.; Hu, Q.; Gao, F.; Ren, H.; Chen, D.; Han, J.; Ying, Z.; et al. TDP-43 loss of function increases TFEB activity and blocks autophagosome-lysosome fusion. EMBO J. 2016, 35, 121–142. [Google Scholar] [CrossRef]
- Root, J.; Merino, P.; Nuckols, A.; Johnson, M.; Kukar, T. Lysosome dysfunction as a cause of neurodegenerative diseases: Lessons from frontotemporal dementia and amyotrophic lateral sclerosis. Neurobiol. Dis. 2021, 154, 105360. [Google Scholar] [CrossRef] [PubMed]
- Conforti, F.L.; Sproviero, W.; Simone, I.L.; Mazzei, R.; Valentino, P.; Ungaro, C.; Magariello, A.; Patitucci, A.; La Bella, V.; Sprovieri, T.; et al. TARDBP gene mutations in south Italian patients with amyotrophic lateral sclerosis. J. Neurol. Neurosurg. Psychiatry 2011, 82, 587–588. [Google Scholar] [CrossRef] [PubMed]
- Mitsuzawa, S.; Akiyama, T.; Nishiyama, A.; Suzuki, N.; Kato, M.; Warita, H.; Izumi, R.; Osana, S.; Koyama, S.; Kato, T.; et al. TARDBP p.G376D mutation, found in rapid progressive familial ALS, induces mislocalization of TDP-43. eNeurologicalSci 2018, 11, 20–22. [Google Scholar] [CrossRef]
- Romano, R.; De Luca, M.; Del Fiore, V.S.; Pecoraro, M.; Lattante, S.; Sabatelli, M.; La Bella, V.; Bucci, C. Allele-specific silencing as therapy for familial amyotrophic lateral sclerosis caused by the p.G376D TARDBP mutation. Brain Commun. 2022, 4, fcac315. [Google Scholar] [CrossRef]
- Cheng, X.T.; Xie, Y.X.; Zhou, B.; Huang, N.; Farfel-Becker, T.; Sheng, Z.H. Revisiting LAMP1 as a marker for degradative autophagy-lysosomal organelles in the nervous system. Autophagy 2018, 14, 1472–1474. [Google Scholar] [CrossRef]
- Kobayashi, T.; Vischer, U.M.; Rosnoblet, C.; Lebrand, C.; Lindsay, M.; Parton, R.G.; Kruithof, E.K.; Gruenberg, J. The tetraspanin CD63/lamp3 cycles between endocytic and secretory compartments in human endothelial cells. Mol. Biol. Cell 2000, 11, 1829–1843. [Google Scholar] [CrossRef]
- Huotari, J.; Helenius, A. Endosome maturation. EMBO J. 2011, 30, 3481–3500. [Google Scholar] [CrossRef]
- Guerra, F.; Bucci, C. Multiple Roles of the Small GTPase Rab7. Cells 2016, 5, E34. [Google Scholar] [CrossRef]
- Collins, M.P.; Forgac, M. Regulation and function of V-ATPases in physiology and disease. Biochim. Biophys. Acta Biomembr. 2020, 1862, 183341. [Google Scholar] [CrossRef]
- De Luca, M.; Cogli, L.; Progida, C.; Nisi, V.; Pascolutti, R.; Sigismund, S.; Di Fiore, P.P.; Bucci, C. RILP regulates vacuolar ATPase through interaction with the V1G1 subunit. J. Cell Sci. 2014, 127, 2697–2708. [Google Scholar] [CrossRef]
- Mijanovic, O.; Petushkova, A.I.; Brankovic, A.; Turk, B.; Solovieva, A.B.; Nikitkina, A.I.; Bolevich, S.; Timashev, P.S.; Parodi, A.; Zamyatnin, A.A., Jr. Cathepsin D-Managing the Delicate Balance. Pharmaceutics 2021, 13, 837. [Google Scholar] [CrossRef] [PubMed]
- Lo, C.H.; Zeng, J. Defective lysosomal acidification: A new prognostic marker and therapeutic target for neurodegenerative diseases. Transl. Neurodegener. 2023, 12, 29. [Google Scholar] [CrossRef] [PubMed]
- Rovira, M.; Sereda, R.; Pladevall-Morera, D.; Ramponi, V.; Marin, I.; Maus, M.; Madrigal-Matute, J.; Diaz, A.; Garcia, F.; Munoz, J.; et al. The lysosomal proteome of senescent cells contributes to the senescence secretome. Aging Cell 2022, 21, e13707. [Google Scholar] [CrossRef] [PubMed]
- Roczniak-Ferguson, A.; Petit, C.S.; Froehlich, F.; Qian, S.; Ky, J.; Angarola, B.; Walther, T.C.; Ferguson, S.M. The transcription factor TFEB links mTORC1 signaling to transcriptional control of lysosome homeostasis. Sci. Signal. 2012, 5, ra42. [Google Scholar] [CrossRef]
- Martina, J.A.; Puertollano, R. Rag GTPases mediate amino acid-dependent recruitment of TFEB and MITF to lysosomes. J. Cell Biol. 2013, 200, 475–491. [Google Scholar] [CrossRef]
- Palmieri, M.; Pal, R.; Nelvagal, H.R.; Lotfi, P.; Stinnett, G.R.; Seymour, M.L.; Chaudhury, A.; Bajaj, L.; Bondar, V.V.; Bremner, L.; et al. mTORC1-independent TFEB activation via Akt inhibition promotes cellular clearance in neurodegenerative storage diseases. Nat. Commun. 2017, 8, 14338. [Google Scholar] [CrossRef]
- D’Anzi, A.; Altieri, F.; Perciballi, E.; Ferrari, D.; Torres, B.; Bernardini, L.; Lattante, S.; Sabatelli, M.; Vescovi, A.L.; Rosati, J. Generation of an induced pluripotent stem cell line (CSS012-A (7672)) carrying the p.G376D heterozygous mutation in the TARDBP protein. Stem Cell Res. 2021, 53, 102356. [Google Scholar] [CrossRef]
- D’Anzi, A.; Perciballi, E.; Ruotolo, G.; Ferrari, D.; Notaro, A.; Lombardi, I.; Gelati, M.; Frezza, K.; Bernardini, L.; Torrente, I.; et al. Production of CSSi013-A (9360) iPSC line from an asymptomatic subject carrying an heterozygous mutation in TDP-43 protein. Stem Cell Res. 2022, 63, 102835. [Google Scholar] [CrossRef]
- Van Acker, Z.P.; Bretou, M.; Annaert, W. Endo-lysosomal dysregulations and late-onset Alzheimer’s disease: Impact of genetic risk factors. Mol. Neurodegener. 2019, 14, 20. [Google Scholar] [CrossRef]
- Sreedharan, J.; Blair, I.P.; Tripathi, V.B.; Hu, X.; Vance, C.; Rogelj, B.; Ackerley, S.; Durnall, J.C.; Williams, K.L.; Buratti, E.; et al. TDP-43 mutations in familial and sporadic amyotrophic lateral sclerosis. Science 2008, 319, 1668–1672. [Google Scholar] [CrossRef]
- Wang, P.; Deng, J.; Dong, J.; Liu, J.; Bigio, E.H.; Mesulam, M.; Wang, T.; Sun, L.; Wang, L.; Lee, A.Y.; et al. TDP-43 induces mitochondrial damage and activates the mitochondrial unfolded protein response. PLoS Genet. 2019, 15, e1007947. [Google Scholar] [CrossRef]
- Sugai, A.; Kato, T.; Koyama, A.; Koike, Y.; Kasahara, S.; Konno, T.; Ishihara, T.; Onodera, O. Robustness and Vulnerability of the Autoregulatory System That Maintains Nuclear TDP-43 Levels: A Trade-off Hypothesis for ALS Pathology Based on in Silico Data. Front. Neurosci. 2018, 12, 28. [Google Scholar] [CrossRef]
- Highley, J.R.; Kirby, J.; Jansweijer, J.A.; Webb, P.S.; Hewamadduma, C.A.; Heath, P.R.; Higginbottom, A.; Raman, R.; Ferraiuolo, L.; Cooper-Knock, J.; et al. Loss of nuclear TDP-43 in amyotrophic lateral sclerosis (ALS) causes altered expression of splicing machinery and widespread dysregulation of RNA splicing in motor neurones. Neuropathol. Appl. Neurobiol. 2014, 40, 670–685. [Google Scholar] [CrossRef] [PubMed]
- Dang, T.N.; Lim, N.K.; Grubman, A.; Li, Q.X.; Volitakis, I.; White, A.R.; Crouch, P.J. Increased metal content in the TDP-43(A315T) transgenic mouse model of frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Front. Aging Neurosci. 2014, 6, 15. [Google Scholar] [CrossRef]
- Franco-Juarez, B.; Coronel-Cruz, C.; Hernandez-Ochoa, B.; Gomez-Manzo, S.; Cardenas-Rodriguez, N.; Arreguin-Espinosa, R.; Bandala, C.; Canseco-Avila, L.M.; Ortega-Cuellar, D. TFEB; Beyond Its Role as an Autophagy and Lysosomes Regulator. Cells 2022, 11, 3153. [Google Scholar] [CrossRef]
- Hurwitz, S.N.; Conlon, M.M.; Rider, M.A.; Brownstein, N.C.; Meckes, D.G., Jr. Nanoparticle analysis sheds budding insights into genetic drivers of extracellular vesicle biogenesis. J. Extracell. Vesicles 2016, 5, 31295. [Google Scholar] [CrossRef]
- McCluskey, G.; Morrison, K.E.; Donaghy, C.; Rene, F.; Duddy, W.; Duguez, S. Extracellular Vesicles in Amyotrophic Lateral Sclerosis. Life 2022, 13, 121. [Google Scholar] [CrossRef]
- Yadati, T.; Houben, T.; Bitorina, A.; Shiri-Sverdlov, R. The Ins and Outs of Cathepsins: Physiological Function and Role in Disease Management. Cells 2020, 9, 1679. [Google Scholar] [CrossRef]
- Vidoni, C.; Follo, C.; Savino, M.; Melone, M.A.; Isidoro, C. The Role of Cathepsin D in the Pathogenesis of Human Neurodegenerative Disorders. Med. Res. Rev. 2016, 36, 845–870. [Google Scholar] [CrossRef]
- Martini-Stoica, H.; Xu, Y.; Ballabio, A.; Zheng, H. The Autophagy-Lysosomal Pathway in Neurodegeneration: A TFEB Perspective. Trends Neurosci. 2016, 39, 221–234. [Google Scholar] [CrossRef]
- Halon-Golabek, M.; Borkowska, A.; Kaczor, J.J.; Ziolkowski, W.; Flis, D.J.; Knap, N.; Kasperuk, K.; Antosiewicz, J. hmSOD1 gene mutation-induced disturbance in iron metabolism is mediated by impairment of Akt signalling pathway. J. Cachexia Sarcopenia Muscle 2018, 9, 557–569. [Google Scholar] [CrossRef] [PubMed]
- Moors, T.E.; Morella, M.L.; Bertran-Cobo, C.; Geut, H.; Udayar, V.; Timmermans-Huisman, E.; Ingrassia, A.M.T.; Breve, J.J.P.; Bol, J.; Bonifati, V.; et al. Altered TFEB subcellular localization in nigral neurons of subjects with incidental, sporadic and GBA-related Lewy body diseases. Acta Neuropathol. 2024, 147, 67. [Google Scholar] [CrossRef] [PubMed]
- Garone, M.G.; de Turris, V.; Soloperto, A.; Brighi, C.; De Santis, R.; Pagani, F.; Di Angelantonio, S.; Rosa, A. Conversion of Human Induced Pluripotent Stem Cells (iPSCs) into Functional Spinal and Cranial Motor Neurons Using PiggyBac Vectors. J. Vis. Exp. 2019, 147, e59321. [Google Scholar] [CrossRef]
- Nishimura, A.L.; Shum, C.; Scotter, E.L.; Abdelgany, A.; Sardone, V.; Wright, J.; Lee, Y.B.; Chen, H.J.; Bilican, B.; Carrasco, M.; et al. Allele-specific knockdown of ALS-associated mutant TDP-43 in neural stem cells derived from induced pluripotent stem cells. PLoS ONE 2014, 9, e91269. [Google Scholar] [CrossRef]
- Romano, R.; Del Fiore, V.S.; Saveri, P.; Palama, I.E.; Pisciotta, C.; Pareyson, D.; Bucci, C.; Guerra, F. Autophagy and Lysosomal Functionality in CMT2B Fibroblasts Carrying the RAB7(K126R) Mutation. Cells 2022, 11, 496. [Google Scholar] [CrossRef]
- De Luca, M.; Romano, R.; Bucci, C. Role of the V1G1 subunit of V-ATPase in breast cancer cell migration. Sci. Rep. 2021, 11, 4615. [Google Scholar] [CrossRef]
- Romano, R.; Rivellini, C.; De Luca, M.; Tonlorenzi, R.; Beli, R.; Manganelli, F.; Nolano, M.; Santoro, L.; Eskelinen, E.L.; Previtali, S.C.; et al. Alteration of the late endocytic pathway in Charcot-Marie-Tooth type 2B disease. Cell. Mol. Life Sci. 2021, 78, 351–372. [Google Scholar] [CrossRef]
- Romano, R.; Calcagnile, M.; Margiotta, A.; Franci, L.; Chiariello, M.; Alifano, P.; Bucci, C. RAB7A Regulates Vimentin Phosphorylation through AKT and PAK. Cancers 2021, 13, 2220. [Google Scholar] [CrossRef]
- Tala, A.; Guerra, F.; Calcagnile, M.; Romano, R.; Resta, S.C.; Paiano, A.; Chiariello, M.; Pizzolante, G.; Bucci, C.; Alifano, P. HrpA anchors meningococci to the dynein motor and affects the balance between apoptosis and pyroptosis. J. Biomed. Sci. 2022, 29, 45. [Google Scholar] [CrossRef]
- Kim, H.N.; Seo, B.R.; Kim, H.; Koh, J.Y. Cilostazol restores autophagy flux in bafilomycin A1-treated, cultured cortical astrocytes through lysosomal reacidification: Roles of PKA, zinc and metallothionein 3. Sci. Rep. 2020, 10, 9175. [Google Scholar] [CrossRef]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Romano, R.; Del Fiore, V.S.; Ruotolo, G.; Mazzoni, M.; Rosati, J.; Conforti, F.L.; Bucci, C. Lysosomal Dysfunction in Amyotrophic Lateral Sclerosis: A Familial Case Linked to the p.G376D TARDBP Mutation. Int. J. Mol. Sci. 2025, 26, 2867. https://doi.org/10.3390/ijms26072867
Romano R, Del Fiore VS, Ruotolo G, Mazzoni M, Rosati J, Conforti FL, Bucci C. Lysosomal Dysfunction in Amyotrophic Lateral Sclerosis: A Familial Case Linked to the p.G376D TARDBP Mutation. International Journal of Molecular Sciences. 2025; 26(7):2867. https://doi.org/10.3390/ijms26072867
Chicago/Turabian StyleRomano, Roberta, Victoria Stefania Del Fiore, Giorgia Ruotolo, Martina Mazzoni, Jessica Rosati, Francesca Luisa Conforti, and Cecilia Bucci. 2025. "Lysosomal Dysfunction in Amyotrophic Lateral Sclerosis: A Familial Case Linked to the p.G376D TARDBP Mutation" International Journal of Molecular Sciences 26, no. 7: 2867. https://doi.org/10.3390/ijms26072867
APA StyleRomano, R., Del Fiore, V. S., Ruotolo, G., Mazzoni, M., Rosati, J., Conforti, F. L., & Bucci, C. (2025). Lysosomal Dysfunction in Amyotrophic Lateral Sclerosis: A Familial Case Linked to the p.G376D TARDBP Mutation. International Journal of Molecular Sciences, 26(7), 2867. https://doi.org/10.3390/ijms26072867