

Genetic Alterations in Atypical Cerebral Palsy Identified Through Chromosomal Microarray and Exome Sequencing

Abstract
:1. Introduction
2. Results
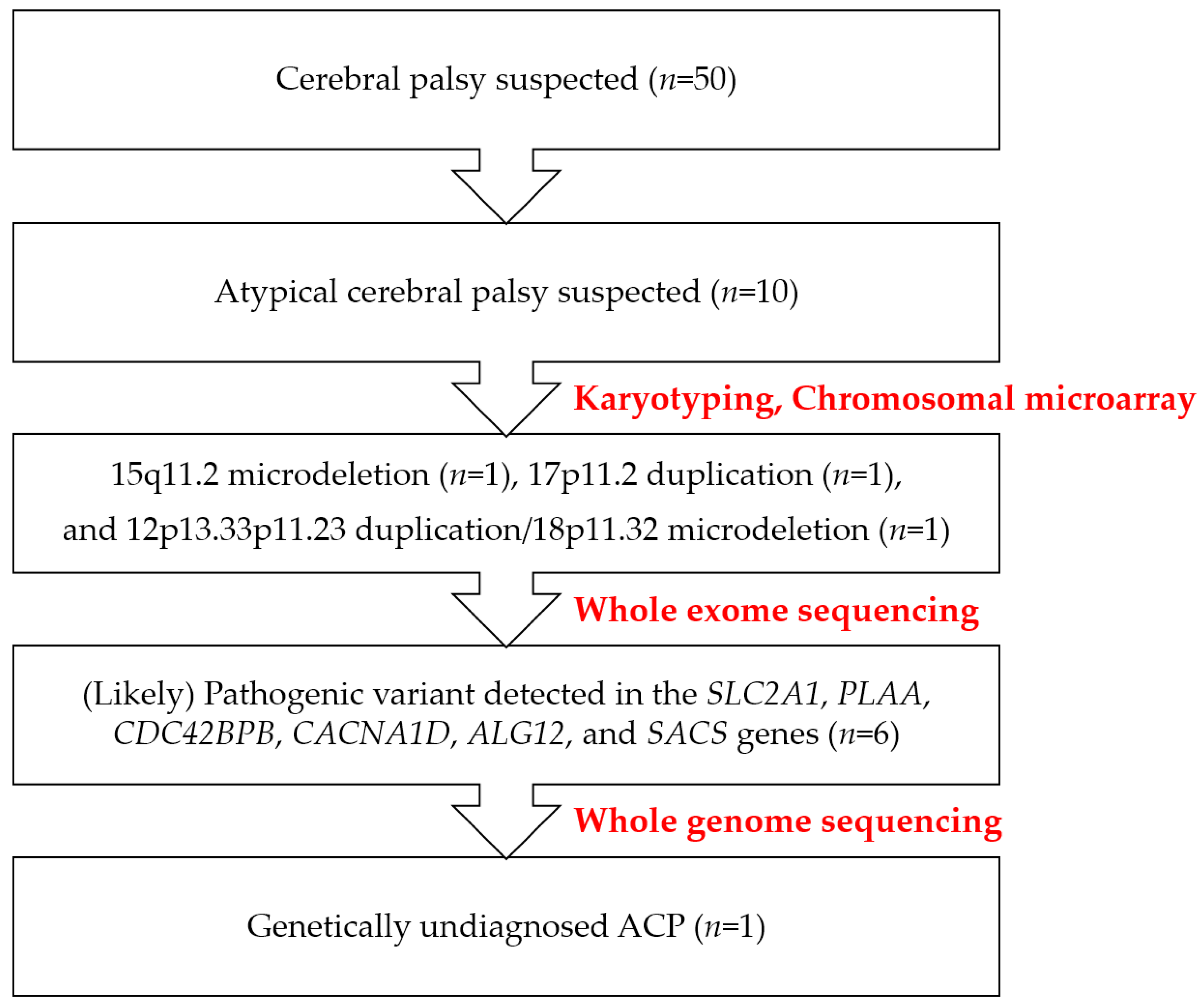
2.1. Presentation of a Genetically Diagnosed Case Series in Atypical Cerebral Palsy
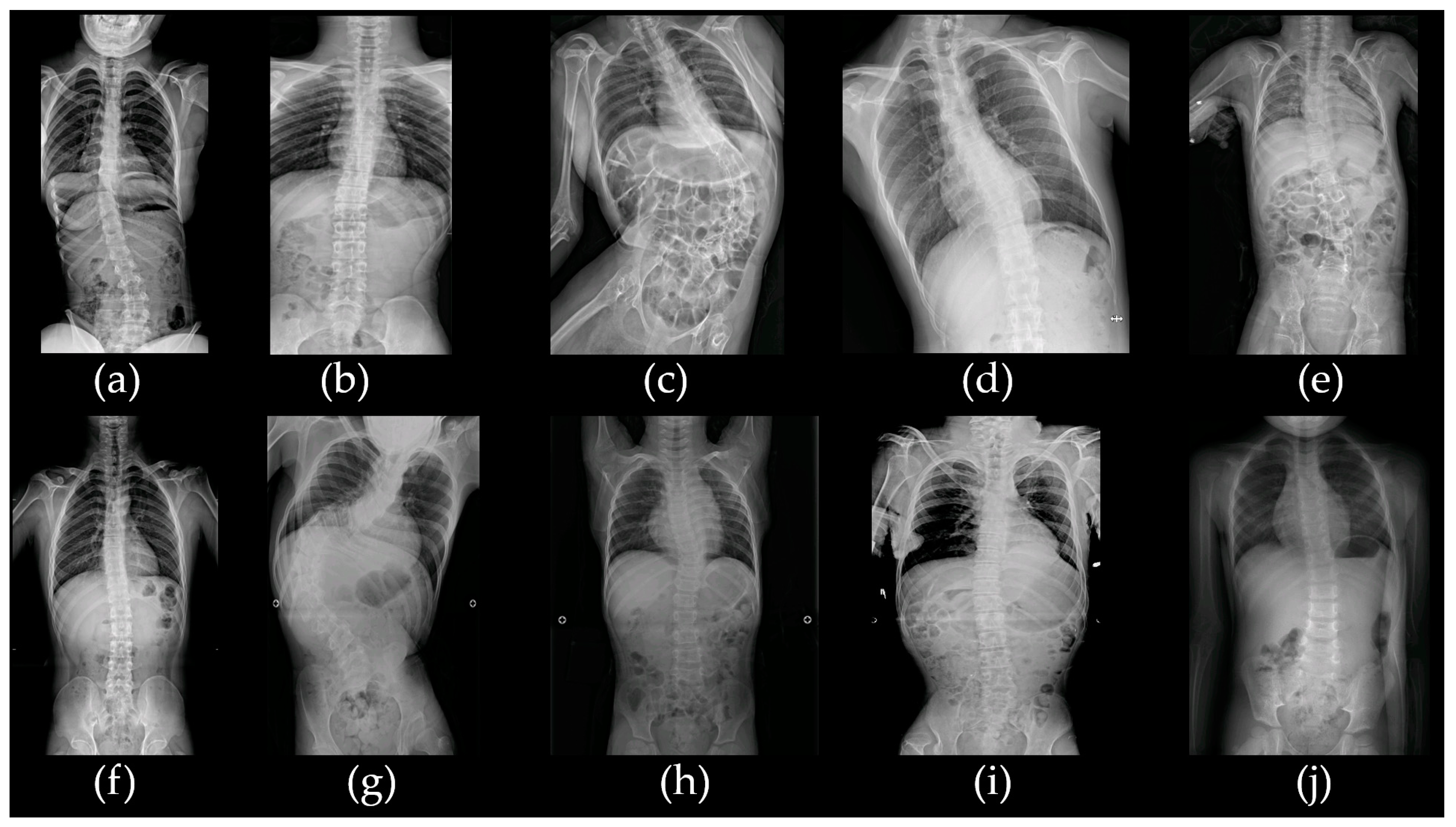
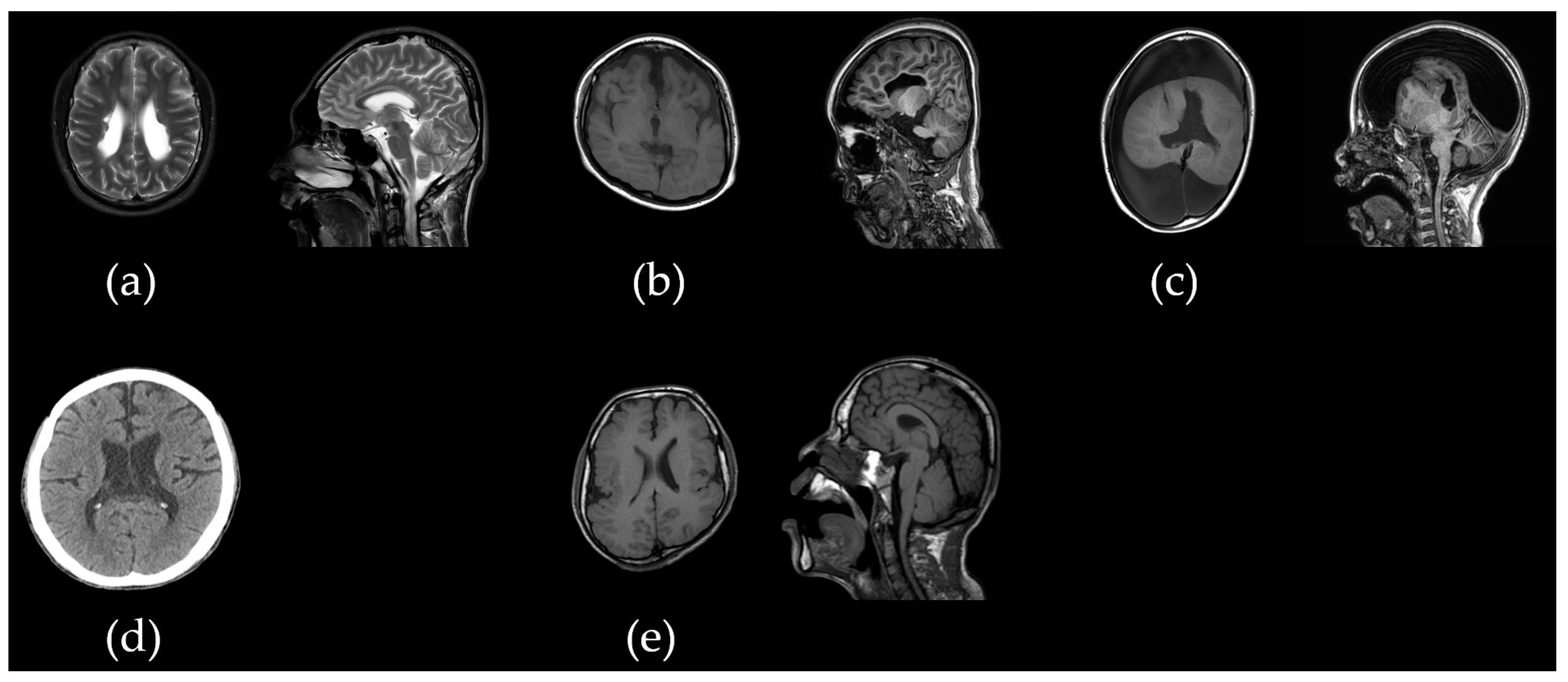
2.1.1. Patient acp01k with SLC2A1 c.277C>T/p.Arg93Trp Variant
2.1.2. Patient acp02s with 15p11.2 Microdeletion
2.1.3. Patient acp03k with Bi-Allelic PLAA c.1039+1G>A and c.1834C>T/p.Pro612Ser Variants
2.1.4. Patient acp05y with CDC42BPB c.4049G>A/p.Arg1350Gln Variant
2.1.5. Patient acp06l with 17p11.2 Duplication
2.1.6. Patient acp07l with CACNA1D c.1846T>C/p.Cys616Arg Variant
2.1.7. Patient acp08p with Bi-Allelic ALG12 c.437G>A/p.Arg146Gln and c.788A>G/p.Tyr263Cys Variants
2.1.8. Patient acp09k with 12p13.33p11.23 Duplication and 18p11.32 Microdeletion
2.1.9. Patient acp10k with Bi-Allelic SACS c.11101T>C/p.Trp3701Arg and c.12973C>T/p.Arg4325Ter Variants
3. Discussion
4. Materials and Methods
4.1. Patients
4.2. Single Nucleotide Polymorphism (SNP) Microarray and Data Interpretation
4.3. Library Preparation, Exome Sequencing, Genome Sequencing, and Bioinformatic Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Rosenbaum, P.; Paneth, N.; Leviton, A.; Goldstein, M.; Bax, M.; Damiano, D.; Dan, B.; Jacobsson, B. A report: The definition and classification of cerebral palsy April 2006. Dev. Med. Child Neurol. Suppl. 2007, 109, 8–14. [Google Scholar] [PubMed]
- Colver, A.; Fairhurst, C.; Pharoah, P.O. Cerebral palsy. Lancet 2014, 383, 1240–1249. [Google Scholar] [CrossRef] [PubMed]
- MacLennan, A.H.; Thompson, S.C.; Gecz, J. Cerebral palsy: Causes, pathways, and the role of genetic variants. Am. J. Obstet. Gynecol. 2015, 213, 779–788. [Google Scholar] [CrossRef] [PubMed]
- McIntyre, S.; Morgan, C.; Walker, K.; Novak, I. Cerebral palsy—Don’t delay. Dev. Disabil. Res. Rev. 2011, 17, 114–129. [Google Scholar]
- McIntyre, S.; Goldsmith, S.; Webb, A.; Ehlinger, V.; Hollung, S.J.; McConnell, K.; Arnaud, C.; Smithers-Sheedy, H.; Oskoui, M.; Khandaker, G. Global prevalence of cerebral palsy: A systematic analysis. Dev. Med. Child Neurol. 2022, 64, 1494–1506. [Google Scholar]
- Maudsley, G.; Hutton, J.L.; Pharoah, P.O. Cause of death in cerebral palsy: A descriptive study. Arch. Dis. Child. 1999, 81, 390–394. [Google Scholar]
- Moreno-De-Luca, A.; Ledbetter, D.H.; Martin, C.L. Genetic insights into the causes and classification of the cerebral palsies. Lancet Neurol. 2012, 11, 283–292. [Google Scholar] [CrossRef]
- McMichael, G.; Bainbridge, M.N.; Haan, E.; Corbett, M.; Gardner, A.; Thompson, S.; van Bon, B.W.; van Eyk, C.L.; Broadbent, J.; Reynolds, C.; et al. Whole-exome sequencing points to considerable genetic heterogeneity of cerebral palsy. Mol. Psychiatry 2015, 20, 176–182. [Google Scholar] [CrossRef]
- Leach, E.L.; Shevell, M.; Bowden, K.; Stockler-Ipsiroglu, S.; van Karnebeek, C.D. Treatable inborn errors of metabolism presenting as cerebral palsy mimics: Systematic literature review. Orphanet J. Rare Dis. 2014, 9, 197. [Google Scholar] [CrossRef]
- Fahey, M.C.; Maclennan, A.H.; Kretzschmar, D.; Gecz, J.; Kruer, M.C. The genetic basis of cerebral palsy. Dev. Med. Child Neurol. 2017, 59, 462–469. [Google Scholar] [CrossRef]
- Boycott, K.M.; Rath, A.; Chong, J.X.; Hartley, T.; Alkuraya, F.S.; Baynam, G.; Brookes, A.J.; Brudno, M.; Carracedo, A.; den Dunnen, J.T.; et al. International Cooperation to Enable the Diagnosis of All Rare Genetic Diseases. Am. J. Hum. Genet. 2017, 100, 695–705. [Google Scholar] [CrossRef] [PubMed]
- Tarailo-Graovac, M.; Shyr, C.; Ross, C.J.; Horvath, G.A.; Salvarinova, R.; Ye, X.C.; Zhang, L.H.; Bhavsar, A.P.; Lee, J.J.; Drögemöller, B.I.; et al. Exome Sequencing and the Management of Neurometabolic Disorders. N. Engl. J. Med. 2016, 374, 2246–2255. [Google Scholar] [CrossRef] [PubMed]
- Novak, I.; Morgan, C.; Adde, L.; Blackman, J.; Boyd, R.N.; Brunstrom-Hernandez, J.; Cioni, G.; Damiano, D.; Darrah, J.; Eliasson, A.C.; et al. Early, Accurate Diagnosis and Early Intervention in Cerebral Palsy: Advances in Diagnosis and Treatment. JAMA Pediatr. 2017, 171, 897–907. [Google Scholar] [CrossRef] [PubMed]
- Korzeniewski, S.J.; Slaughter, J.; Lenski, M.; Haak, P.; Paneth, N. The complex aetiology of cerebral palsy. Nat. Rev. Neurol. 2018, 14, 528–543. [Google Scholar] [CrossRef]
- Hollung, S.J.; Bakken, I.J.; Vik, T.; Lydersen, S.; Wiik, R.; Aaberg, K.M.; Andersen, G.L. Comorbidities in cerebral palsy: A patient registry study. Dev. Med. Child Neurol. 2020, 62, 97–103. [Google Scholar] [CrossRef]
- McGuire, D.O.; Tian, L.H.; Yeargin-Allsopp, M.; Dowling, N.F.; Christensen, D.L. Prevalence of cerebral palsy, intellectual disability, hearing loss, and blindness, National Health Interview Survey, 2009–2016. Disabil. Health J. 2019, 12, 443–451. [Google Scholar] [CrossRef]
- Berger, S.M.; Bartsch, D. The role of L-type voltage-gated calcium channels Cav1.2 and Cav1.3 in normal and pathological brain function. Cell Tissue Res. 2014, 357, 463–476. [Google Scholar] [CrossRef]
- Krägeloh-Mann, I.; Horber, V. The role of magnetic resonance imaging in elucidating the pathogenesis of cerebral palsy: A systematic review. Dev. Med. Child Neurol. 2007, 49, 144–151. [Google Scholar]
- Srivastava, S.; Love-Nichols, J.A.; Dies, K.A.; Ledbetter, D.H.; Martin, C.L.; Chung, W.K.; Firth, H.V.; Frazier, T.; Hansen, R.L.; Prock, L.; et al. Meta-analysis and multidisciplinary consensus statement: Exome sequencing is a first-tier clinical diagnostic test for individuals with neurodevelopmental disorders. Genet. Med. 2019, 21, 2413–2421. [Google Scholar] [CrossRef]
- Lewis, S.A.; Shetty, S.; Wilson, B.A.; Huang, A.J.; Jin, S.C.; Smithers-Sheedy, H.; Fahey, M.C.; Kruer, M.C. Insights from Genetic Studies of Cerebral Palsy. Front. Neurol. 2020, 11, 625428. [Google Scholar] [CrossRef]
- Gonzalez-Mantilla, P.J.; Hu, Y.; Myers, S.M.; Finucane, B.M.; Ledbetter, D.H.; Martin, C.L.; Moreno-De-Luca, A. Diagnostic Yield of Exome Sequencing in Cerebral Palsy and Implications for Genetic Testing Guidelines: A Systematic Review and Meta-analysis. JAMA Pediatr. 2023, 177, 472–478. [Google Scholar] [CrossRef] [PubMed]
- Cox, D.M.; Butler, M.G. The 15q11.2 BP1-BP2 microdeletion syndrome: A review. Int. J. Mol. Sci. 2015, 16, 4068–4082. [Google Scholar] [CrossRef] [PubMed]
- Han, J.Y.; Park, J. Phenotypic Diversity of 15q11.2 BP1–BP2 Deletion in Three Korean Families with Development Delay and/or Intellectual Disability: A Case Series and Literature Review. Diagnostics 2021, 11, 722. [Google Scholar] [CrossRef] [PubMed]
- Grama, A.; Sîrbe, C.; Miclea, D.; Cǎinap, S.S.; Huniadi, D.; Bulata, B.; Pop, T.L. Case Report: Potocki-Lupski Syndrome in Five Siblings. Front. Pediatr. 2021, 9, 698629. [Google Scholar] [CrossRef]
- Ciaccio, C.; Pantaleoni, C.; Milani, D.; Alfei, E.; Sciacca, F.L.; Canafoglia, L.; Erbetta, A.; D’Arrigo, S. Neurological phenotype of Potocki-Lupski syndrome. Am. J. Med. Genet. A 2020, 182, 2317–2324. [Google Scholar] [CrossRef]
- Shuib, S.; Saaid, N.N.; Zakaria, Z.; Ismail, J.; Abdul Latiff, Z. Duplication 17p11.2 (Potocki-Lupski Syndrome) in a child with developmental delay. Malays. J. Pathol. 2017, 39, 77–81. [Google Scholar]
- Oliveira, J.S.; Joaquim, T.M.; Silva, R.; Souza, D.H.; Martelli, L.R.; Moretti-Ferreira, D. Non-mosaic partial duplication 12p in a patient with dysmorphic characteristics and developmental delay. Genet. Mol. Biol. 2020, 43, e20180285. [Google Scholar] [CrossRef]
- Halperin, R.; Arnon, L.; Nasirov, S.; Friedensohn, L.; Gershinsky, M.; Telerman, A.; Friedman, E.; Bernstein-Molho, R.; Tirosh, A. Germline CDKN1B variant type and site are associated with phenotype in MEN4. Endocr.-Relat. Cancer 2023, 30, e220174. [Google Scholar] [CrossRef]
- Poirsier, C.; Landais, E.; Bednarek, N.; Nobecourt, J.M.; Khoury, M.; Schmidt, P.; Morville, P.; Gruson, N.; Clomes, S.; Michel, N.; et al. Report on 3 patients with 12p duplication including GRIN2B. Eur. J. Med. Genet. 2014, 57, 185–194. [Google Scholar] [CrossRef]
- Wang, F.; Ning, S.; Yu, B.; Wang, Y. USP14: Structure, Function, and Target Inhibition. Front. Pharmacol. 2021, 12, 801328. [Google Scholar] [CrossRef]
- Kantaputra, P.N.; Limwongse, C.; Tochareontanaphol, C.; Mutirangura, A.; Mevatee, U.; Praphanphoj, V. Contiguous gene syndrome of holoprosencephaly and hypotrichosis simplex: Association with an 18p11.3 deletion. Am. J. Med. Genet. A 2006, 140, 2598–2602. [Google Scholar] [CrossRef] [PubMed]
- Verrotti, A.; Palka, C.; Prezioso, G.; Alfonsi, M.; Calabrese, G.; Palka, G.; Chiarelli, F. Deletion 18p11.32p11.31 in a Child with Global Developmental Delay and Atypical, Drug-Resistant Absence Seizures. Cytogenet. Genome Res. 2015, 146, 115–119. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, S.; Cohen, J.S.; Vernon, H.; Barañano, K.; McClellan, R.; Jamal, L.; Naidu, S.; Fatemi, A. Clinical whole exome sequencing in child neurology practice. Ann. Neurol. 2014, 76, 473–483. [Google Scholar] [PubMed]
- Finucane, B.M.; Myers, S.M.; Martin, C.L.; Ledbetter, D.H. Long overdue: Including adults with brain disorders in precision health initiatives. Curr. Opin. Genet. Dev. 2020, 65, 47–52. [Google Scholar] [CrossRef]
- Sanchez Fernandez, I.; Loddenkemper, T.; Gainza-Lein, M.; Sheidley, B.R.; Poduri, A. Diagnostic yield of genetic tests in epilepsy: A meta-analysis and cost-effectiveness study. Neurology 2019, 92, e418–e428. [Google Scholar]
- Moreno-De-Luca, A.; Millan, F.; Pesacreta, D.R.; Elloumi, H.Z.; Oetjens, M.T.; Teigen, C.; Wain, K.E.; Scuffins, J.; Myers, S.M.; Torene, R.I.; et al. Molecular Diagnostic Yield of Exome Sequencing in Patients With Cerebral Palsy. JAMA 2021, 325, 467–475. [Google Scholar] [CrossRef]
- Cooper, M.S.; Fahey, M.C.; Mackay, M.T. Making waves: The changing tide of cerebral palsy. J. Paediatr. Child Health 2022, 58, 1929–1934. [Google Scholar]
- Koch, H.; Weber, Y.G. The glucose transporter type 1 (Glut1) syndromes. Epilepsy Behav. 2019, 91, 90–93. [Google Scholar] [CrossRef]
- Leen, W.G.; Klepper, J.; Verbeek, M.M.; Leferink, M.; Hofste, T.; van Engelen, B.G.; Wevers, R.A.; Arthur, T.; Bahi-Buisson, N.; Ballhausen, D.; et al. Glucose transporter-1 deficiency syndrome: The expanding clinical and genetic spectrum of a treatable disorder. Brain 2010, 133, 655–670. [Google Scholar] [CrossRef]
- Gras, D.; Roze, E.; Caillet, S.; Méneret, A.; Doummar, D.; Billette de Villemeur, T.; Vidailhet, M.; Mochel, F. GLUT1 deficiency syndrome: An update. Rev. Neurol. 2014, 170, 91–99. [Google Scholar] [CrossRef]
- Papadopoulos, C.; Kirchner, P.; Bug, M.; Grum, D.; Koerver, L.; Schulze, N.; Poehler, R.; Dressler, A.; Fengler, S.; Arhzaouy, K.; et al. VCP/p97 cooperates with YOD1, UBXD1 and PLAA to drive clearance of ruptured lysosomes by autophagy. EMBO J. 2017, 36, 135–150. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; Sha, J.; Wood, T.G.; Galindo, C.L.; Garner, H.R.; Burkart, M.F.; Suarez, G.; Sierra, J.C.; Agar, S.L.; Peterson, J.W.; et al. Alteration in the activation state of new inflammation-associated targets by phospholipase A2-activating protein (PLAA). Cell. Signal. 2008, 20, 844–861. [Google Scholar] [CrossRef] [PubMed]
- Hall, E.A.; Nahorski, M.S.; Murray, L.M.; Shaheen, R.; Perkins, E.; Dissanayake, K.N.; Kristaryanto, Y.; Jones, R.A.; Vogt, J.; Rivagorda, M.; et al. PLAA Mutations Cause a Lethal Infantile Epileptic Encephalopathy by Disrupting Ubiquitin-Mediated Endolysosomal Degradation of Synaptic Proteins. Am. J. Hum. Genet. 2017, 100, 706–724. [Google Scholar] [CrossRef]
- Falik Zaccai, T.C.; Savitzki, D.; Zivony-Elboum, Y.; Vilboux, T.; Fitts, E.C.; Shoval, Y.; Kalfon, L.; Samra, N.; Keren, Z.; Gross, B.; et al. Phospholipase A2-activating protein is associated with a novel form of leukoencephalopathy. Brain 2017, 140, 370–386. [Google Scholar] [CrossRef]
- Dai, C.; Zeng, S.; Tan, Z.; Yang, X.; Du, J.; Lu, G.; Wang, J. Neurodevelopmental disorder with progressive microcephaly, spasticity, and brain anomalies in China caused by novel mutations of PLAA. Clin. Genet. 2019, 96, 380–381. [Google Scholar] [CrossRef]
- Heikkila, T.; Wheatley, E.; Crighton, D.; Schroder, E.; Boakes, A.; Kaye, S.J.; Mezna, M.; Pang, L.; Rushbrooke, M.; Turnbull, A.; et al. Co-crystal structures of inhibitors with MRCKβ, a key regulator of tumor cell invasion. PLoS ONE 2011, 6, e24825. [Google Scholar] [CrossRef]
- Chilton, I.; Okur, V.; Vitiello, G.; Selicorni, A.; Mariani, M.; Goldenberg, A.; Husson, T.; Campion, D.; Lichtenbelt, K.D.; van Gassen, K.; et al. De novo heterozygous missense and loss-of-function variants in CDC42BPB are associated with a neurodevelopmental phenotype. Am. J. Med. Genet. A 2020, 182, 962–973. [Google Scholar] [CrossRef]
- Bock, G.; Gebhart, M.; Scharinger, A.; Jangsangthong, W.; Busquet, P.; Poggiani, C.; Sartori, S.; Mangoni, M.E.; Sinnegger-Brauns, M.J.; Herzig, S.; et al. Functional properties of a newly identified C-terminal splice variant of Cav1.3 L-type Ca2+ channels. J. Biol. Chem. 2011, 286, 42736–42748. [Google Scholar] [CrossRef]
- Scholl, U.I.; Goh, G.; Stölting, G.; de Oliveira, R.C.; Choi, M.; Overton, J.D.; Fonseca, A.L.; Korah, R.; Starker, L.F.; Kunstman, J.W.; et al. Somatic and germline CACNA1D calcium channel mutations in aldosterone-producing adenomas and primary aldosteronism. Nat. Genet. 2013, 45, 1050–1054. [Google Scholar] [CrossRef]
- Ortner, N.J.; Kaserer, T.; Copeland, J.N.; Striessnig, J. De novo CACNA1D Ca2+ channelopathies: Clinical phenotypes and molecular mechanism. Pflugers Arch. 2020, 472, 755–773. [Google Scholar] [CrossRef]
- Marquardt, T.; Denecke, J. Congenital disorders of glycosylation: Review of their molecular bases, clinical presentations and specific therapies. Eur. J. Pediatr. 2003, 162, 359–379. [Google Scholar] [PubMed]
- Chantret, I.; Dupré, T.; Delenda, C.; Bucher, S.; Dancourt, J.; Barnier, A.; Charollais, A.; Heron, D.; Bader-Meunier, B.; Danos, O. Congenital disorders of glycosylation type Ig is defined by a deficiency in dolichyl-P-mannose: Man7GlcNAc2-PP-dolichyl mannosyltransferase. J. Biol. Chem. 2002, 277, 25815–25822. [Google Scholar]
- Eklund, E.A.; Newell, J.W.; Sun, L.; Seo, N.S.; Alper, G.; Willert, J.; Freeze, H.H. Molecular and clinical description of the first US patients with congenital disorder of glycosylation Ig. Mol. Genet. Metab. 2005, 84, 25–31. [Google Scholar] [CrossRef]
- Kranz, C.; Basinger, A.A.; Güçsavaş-Çalıkoğlu, M.; Sun, L.; Powell, C.M.; Henderson, F.W.; Aylsworth, A.S.; Freeze, H.H. Expanding spectrum of congenital disorder of glycosylation Ig (CDG-Ig): Sibs with a unique skeletal dysplasia, hypogammaglobulinemia, cardiomyopathy, genital malformations, and early lethality. Am. J. Med. Genet. Part A 2007, 143, 1371–1378. [Google Scholar]
- Bouhlal, Y.; Amouri, R.; El Euch-Fayeche, G.; Hentati, F. Autosomal recessive spastic ataxia of Charlevoix–Saguenay: An overview. Park. Relat. Disord. 2011, 17, 418–422. [Google Scholar]
- Aly, K.A.; Moutaoufik, M.T.; Zilocchi, M.; Phanse, S.; Babu, M. Insights into SACS pathological attributes in autosomal recessive spastic ataxia of Charlevoix-Saguenay (ARSACS). Curr. Opin. Chem. Biol. 2022, 71, 102211. [Google Scholar] [CrossRef]
- Lessard, I.; Côté, I.; St-Gelais, R.; Hébert, L.J.; Brais, B.; Mathieu, J.; Rodrigue, X.; Gagnon, C. Natural history of autosomal recessive spastic ataxia of Charlevoix-Saguenay: A 4-year longitudinal study. Cerebellum 2024, 23, 489–501. [Google Scholar]
- Scaravilli, A.; Negroni, D.; Senatore, C.; Ugga, L.; Cosottini, M.; Ricca, I.; Bender, B.; Traschütz, A.; Başak, A.N.; Vural, A. MRI-ARSACS: An Imaging Index for Autosomal Recessive Spastic Ataxia of Charlevoix-Saguenay (ARSACS) Identification Based on the Multicenter PROSPAX Study. Mov. Disord. 2024, 39, 1343–1351. [Google Scholar]
- Takezawa, Y.; Kikuchi, A.; Haginoya, K.; Niihori, T.; Numata-Uematsu, Y.; Inui, T.; Yamamura-Suzuki, S.; Miyabayashi, T.; Anzai, M.; Suzuki-Muromoto, S.; et al. Genomic analysis identifies masqueraders of full-term cerebral palsy. Ann. Clin. Transl. Neurol. 2018, 5, 538–551. [Google Scholar] [CrossRef]
- Matthews, A.M.; Blydt-Hansen, I.; Al-Jabri, B.; Andersen, J.; Tarailo-Graovac, M.; Price, M.; Selby, K.; Demos, M.; Connolly, M.; Drögemoller, B.; et al. Atypical cerebral palsy: Genomics analysis enables precision medicine. Genet. Med. 2019, 21, 1621–1628. [Google Scholar] [CrossRef]
- Zouvelou, V.; Yubero, D.; Apostolakopoulou, L.; Kokkinou, E.; Bilanakis, M.; Dalivigka, Z.; Nikas, I.; Kollia, E.; Perez-Dueñas, B.; Macaya, A.; et al. The genetic etiology in cerebral palsy mimics: The results from a Greek tertiary care center. Eur. J. Paediatr. Neurol. 2019, 23, 427–437. [Google Scholar] [CrossRef] [PubMed]
- Nejabat, M.; Inaloo, S.; Sheshdeh, A.T.; Bahramjahan, S.; Sarvestani, F.M.; Katibeh, P.; Nemati, H.; Tabei, S.M.B.; Faghihi, M.A. Genetic Testing in Various Neurodevelopmental Disorders Which Manifest as Cerebral Palsy: A Case Study From Iran. Front. Pediatr. 2021, 9, 734946. [Google Scholar] [CrossRef]
- Rosello, M.; Caro-Llopis, A.; Orellana, C.; Oltra, S.; Alemany-Albert, M.; Marco-Hernandez, A.V.; Monfort, S.; Pedrola, L.; Martinez, F.; Tomás, M. Hidden etiology of cerebral palsy: Genetic and clinical heterogeneity and efficient diagnosis by next-generation sequencing. Pediatr. Res. 2021, 90, 284–288. [Google Scholar] [CrossRef] [PubMed]
- Chopra, M.; Gable, D.L.; Love-Nichols, J.; Tsao, A.; Rockowitz, S.; Sliz, P.; Barkoudah, E.; Bastianelli, L.; Coulter, D.; Davidson, E.; et al. Mendelian etiologies identified with whole exome sequencing in cerebral palsy. Ann. Clin. Transl. Neurol. 2022, 9, 193–205. [Google Scholar] [CrossRef]
- Li, N.; Zhou, P.; Tang, H.; He, L.; Fang, X.; Zhao, J.; Wang, X.; Qi, Y.; Sun, C.; Lin, Y. In-depth analysis reveals complex molecular aetiology in a cohort of idiopathic cerebral palsy. Brain 2022, 145, 119–141. [Google Scholar]
- Yechieli, M.; Gulsuner, S.; Ben-Pazi, H.; Fattal, A.; Aran, A.; Kuzminsky, A.; Sagi, L.; Guttman, D.; Schneebaum Sender, N.; Gross-Tsur, V.; et al. Diagnostic yield of chromosomal microarray and trio whole exome sequencing in cryptogenic cerebral palsy. J. Med. Genet. 2022, 59, 759–767. [Google Scholar] [CrossRef]
- Gupta, R.; Appleton, R.E. Cerebral palsy: Not always what it seems. Arch. Dis. Child. 2001, 85, 356–360. [Google Scholar] [CrossRef]
- Pham, R.; Mol, B.W.; Gecz, J.; MacLennan, A.H.; MacLennan, S.C.; Corbett, M.A.; van Eyk, C.L.; Webber, D.L.; Palmer, L.J.; Berry, J.G. Definition and diagnosis of cerebral palsy in genetic studies: A systematic review. Dev. Med. Child Neurol. 2020, 62, 1024–1030. [Google Scholar] [CrossRef]
- Kearney, H.M.; Thorland, E.C.; Brown, K.K.; Quintero-Rivera, F.; South, S.T. American College of Medical Genetics standards and guidelines for interpretation and reporting of postnatal constitutional copy number variants. Genet. Med. 2011, 13, 680–685. [Google Scholar] [CrossRef]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef]
- Pejaver, V.; Byrne, A.B.; Feng, B.J.; Pagel, K.A.; Mooney, S.D.; Karchin, R.; O’Donnell-Luria, A.; Harrison, S.M.; Tavtigian, S.V.; Greenblatt, M.S.; et al. Calibration of computational tools for missense variant pathogenicity classification and ClinGen recommendations for PP3/BP4 criteria. Am. J. Hum. Genet. 2022, 109, 2163–2177. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
| Patient | S/A (y) | GA/BW | CP Types | GMFCS | ID | Epi/OA | Other Comorbidities | Brain MRI |
|---|---|---|---|---|---|---|---|---|
| acp01k | F/13 | 38 wks/3 kg | SQ | V | Profound | +/1 mo | NH, microcephaly | Normal |
| acp02s | M/21 | 29+5 wks/1.42 kg | SQ | V | Severe | +/20 y | None | Periventricular leukomalacia (grade II) |
| acp03k | F/15 | 38 wks/3 kg | SQ | V | Profound | +/12 mo | None | Frontotemporal atrophy with demyelinating changes |
| acp04m | F/39 | 38 wks/3 kg | Athetoid | II | Normal | −/− | None | Normal |
| acp05y | M/9 | 36 wks/2.5 kg | SQ | V | Profound | +/2 mo | None | Schizencephaly with associated callosal agenesis, hydranencephaly |
| acp06l | M/18 | 40 wks/3.36 kg | SD | V | Severe | −/− | NH | Normal |
| acp07l | M/20 | 37 wks/2.9 kg | SQ | V | Profound | +/16 y | NH | Normal |
| acp08p | M/17 | 33 wks/2.56 kg | SQ | V | Profound | −/− | NH, cryptorchidism, facial dysmorphism, thin and long fingers | Widening of the side ventricles |
| acp09k | M/25 | 41 wks/3 kg | SQ | V | Profound | +/25 y | NH, cleft palate | Schizencephaly with cortical dysplasia |
| acp10k | F/8 | 39 wks/3.5 kg | SD | III | Mild | +/7 y | SNHL, ataxia | Normal |
| Patient | Genetic Alteration | Origin/Zygosity | gnomAD | ACMG Class | OMIM Phenotype |
|---|---|---|---|---|---|
| acp01k | SLC2A1, c.277C>T/p.Arg93Trp | De novo/Het | n.f. | PV | GLUT1DS1 (# 606777) |
| acp02s | 15q11.2 microdeletion | Mat/Het | n.f. | PV | CHR15q11.2DS (# 615656) |
| acp03k | PLAA, c.1039+1G>A PLAA, c.1834C>T/p.Pro612Ser | Pat/Het Mat/Het | n.f. | LPV | NDMSBA (# 617527) |
| acp05y | CDC42BPB, c.4049G>A/p.Arg1350Gln | De novo/Het | n.f. | LPV | CHOCNS (# 619841) |
| acp06l | 17p11.2 duplication | Mat/Het | n.f. | PV | PTLS (# 610883) |
| acp07l | CACNA1D, c.1846T>C/p.Cys616Arg | De novo/Het | n.f. | LPV | PASNA (# 615474) |
| acp08p | ALG12, c.437G>A/p.Arg146Gln ALG12, c.788A>G/p.Tyr263Cys | Pat/Het Mat/Het | 0.0113 0.0004 | PV LPV | CDG1G (# 607143) |
| acp09k | 12p13.33p11.23 duplication 18p11.32 microdeletion | De novo/Het De novo/Het | n.f. | LPV LPV | n.a. |
| acp10k | SACS, c.11101T>C/p.Trp3701Arg SACS, c.12973C>T/p.Arg4325Ter | Pat/Het Mat/Het | 0.0008 0.0008 | PV LPV | SACS (# 270550) |
| References | Patients (n) | Diagnostic Yield (%) | Genetic Tests | Patients | Genetic Alterations | Remarks |
|---|---|---|---|---|---|---|
| Moreno-De-Luca et al. (2021) United States of America [36] | 1345 | 32.7% (CNV 4.3%, SNV 94.3%, both 1.4%) | CES (trio and nontrio) | Cryptogenic (referral to genetic tests) | AP4B1, SPAST, ATL1, REEP1, KIF1A, PLP1, RNASEH2B, TREX1, GNB1, GNAO1, PGK1, SPATA5, IFIH1 | Diagnostic yield: health-care-based control 10.5% Specific genomic locations of the identified CNVs were not detailed. |
| Takezawa et al. (2018) Japan [59] | 17 | 52.9% (only SNV) | aCGH, trio-WES | Full-term CP without specific MRI findings | CTNNB1,CYP2U1,SPAST,GNAO1,CACNA1A,AMPD2, STXBP1, SCN2A | |
| Matthews et al. (2019) Canada [60] | 50 | 65% | NGS | Atypical CP | AKT3, ASXL1, ATP1A3, ATP8A2, CHRNA1, CSTB, DGKZ, EHMT1, EPHA4, GCDH, GNAO1, ITPA, KANK1, KCNJ6, KIDINS220, KMT2C, MECP2, NAA10, NBAS, PAK3, PALM, PLP1, PLXNA2, RANBP2, SCN3A, SPAST, TBCK, TCF4, TMEM67, TUBB4A, WDR45 | CMA-negative patients |
| Zouvelou et al. (2019) Greece [61] | 47 | 48.9% | aCGH, MS-PCR, FMR1, TH, MLPA, CES | CP mimics | ACTA1, ACTB, AMPD2, AP4M1, ATL1, ATP1A2, ATP1A3, ATP8A2, BSCL2, C12orf65, CASK, CERS1, CLCN2, COL4A1, COL4A2, CPA6, CPT1A, CPT2, CSF1R, CYP27A1, DAOA, DARS, DDC, DMD, DYNC1H1, EDA, EIF2B5, EIF4A2, ELOVL4, ERCC6, FA2H, FARS2, FBXO7, FGF14, FLNA, FOXP1, GAD1, GAD2, GATM, GBA, GCH1, GLRA1, GLRB, GNAO1, GNB1, GPR56, GRIN1, GRIN2A, GRIN2B, HADHA, HADHB, HARS, HNRNPU, HSPB1, HSPB8, HTRA2, IARS2, IGHMBP2, IKBKG, INF2, ISCU, KCNA1, KCNC3, KCNJ10, KCNQ2, KIF1A, KIF5A, KMT2B, L1CAM, LAMA2, LAMP2, LARGE, LARS2, LMNA, LRRK2, MARS2, MECP2, MFN2, and MOBP | Risk factor: epilepsy |
| Nejabat et al. (2021) Iran [62] | 66 | 45.2% (only SNV) | WES | Atypical | HACE1, SPEG, SLC13A5, TRAPPC4, FBXL4, TDP2, GAMT, LAMB1, OCLN, WWOX, TREX1, SURF1, WDR45, LAMA2, MTHFR, MOCS1, DOM1, SEPSECS, GABRB1, KCNT1, AP4M1, PDHX, FOXG1, ATP6V1A, SPR, PIGG, ATL1, SNX14 | |
| Rosello et al. (2021) Spain [63] | 20 | 55% (only SNV) | CMA, trio-CES | Idiopathic CP | AP4B1, IFIH1, SPAST, ATL1, PGK1, SPATA5, GNAO1, GNB1, RNASEH2B | Sporadic 64% Risk factors: spastic quadriplegia, epilepsy, gross motor impairment |
| Chopra et al. (2022) United States of America [64] | 24 | 29% (only SNV) | WES (trio and nontrio) | Cryptogenic | ECHS1, SATB2, ZMYM2, ADAT3, COL4A1, THOC2, SLC16A2, SPAST, POLR2A, GNAO1, PDHX, ACADM, ATL1 | Diagnostic yield: CP masquerader 60% (3/5) |
| Li et al. (2022) [65] | 66 | 45% (only SNV) | WES | Idiopathic CP | SPAST, KIF1A, COL4A1, BCL11A, BCL11B, TUBA1A, TUBB2B, ATL1, RARS2, PTK7, TARS, TYW1, GPAM | |
| Yechieli et al. (2022) Israel [66] | 45 | 58% (CNV 18%, SNV 40%) | CMA, trio-WES | Cryptogenic CP | 15q11.2 microdeletion, 17p11.2 duplication, 12p13.33-p11.23 duplication, and 18p11.32 deletion AIFM1, ELOVL1, VPS11, SYNE1, MAPK8IP3, PCDH19, AP2M1, PHF8, ARHGEF10 | Sporadic 62% Risk factors: associated comorbidities |
| Our study | 10 | 90% (CNV 30%, SNV 60%) | Karyotyping, CMA, WES, WGS (trio and nontrio) | Atypical CP | 15p11.2 microdeletion, 17p11.2 duplication, 12p13.33p11.23 duplication, and 18p11.32 microdeletion SLC2A1, PLAA, CDC42BPB, CACNA1D, ALG12, SACS | Sporadic 40% |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Han, J.Y.; Gwack, J.; Kim, J.H.; Park, M.K.; Park, J. Genetic Alterations in Atypical Cerebral Palsy Identified Through Chromosomal Microarray and Exome Sequencing. Int. J. Mol. Sci. 2025, 26, 2929. https://doi.org/10.3390/ijms26072929
Han JY, Gwack J, Kim JH, Park MK, Park J. Genetic Alterations in Atypical Cerebral Palsy Identified Through Chromosomal Microarray and Exome Sequencing. International Journal of Molecular Sciences. 2025; 26(7):2929. https://doi.org/10.3390/ijms26072929
Chicago/Turabian StyleHan, Ji Yoon, Jin Gwack, Jong Hun Kim, Min Kyu Park, and Joonhong Park. 2025. "Genetic Alterations in Atypical Cerebral Palsy Identified Through Chromosomal Microarray and Exome Sequencing" International Journal of Molecular Sciences 26, no. 7: 2929. https://doi.org/10.3390/ijms26072929
APA StyleHan, J. Y., Gwack, J., Kim, J. H., Park, M. K., & Park, J. (2025). Genetic Alterations in Atypical Cerebral Palsy Identified Through Chromosomal Microarray and Exome Sequencing. International Journal of Molecular Sciences, 26(7), 2929. https://doi.org/10.3390/ijms26072929