
ULBP2 Promotes Tumor Progression by Suppressing NKG2D-Mediated Anti-Tumor Immunity


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
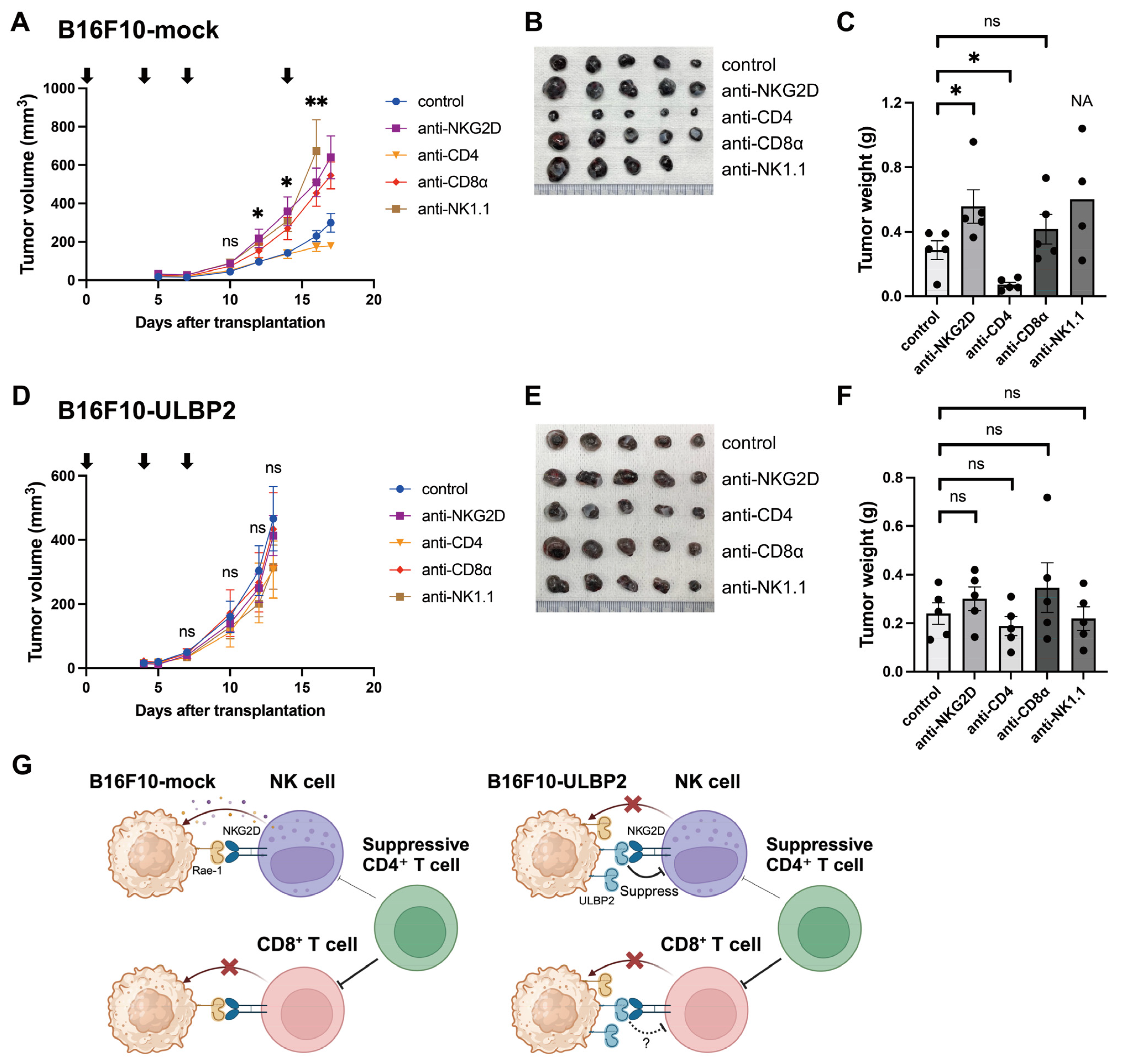
2.1. ULBP2 Suppresses Anti-Tumor Immunity via NKG2D and Promotes Tumor Growth
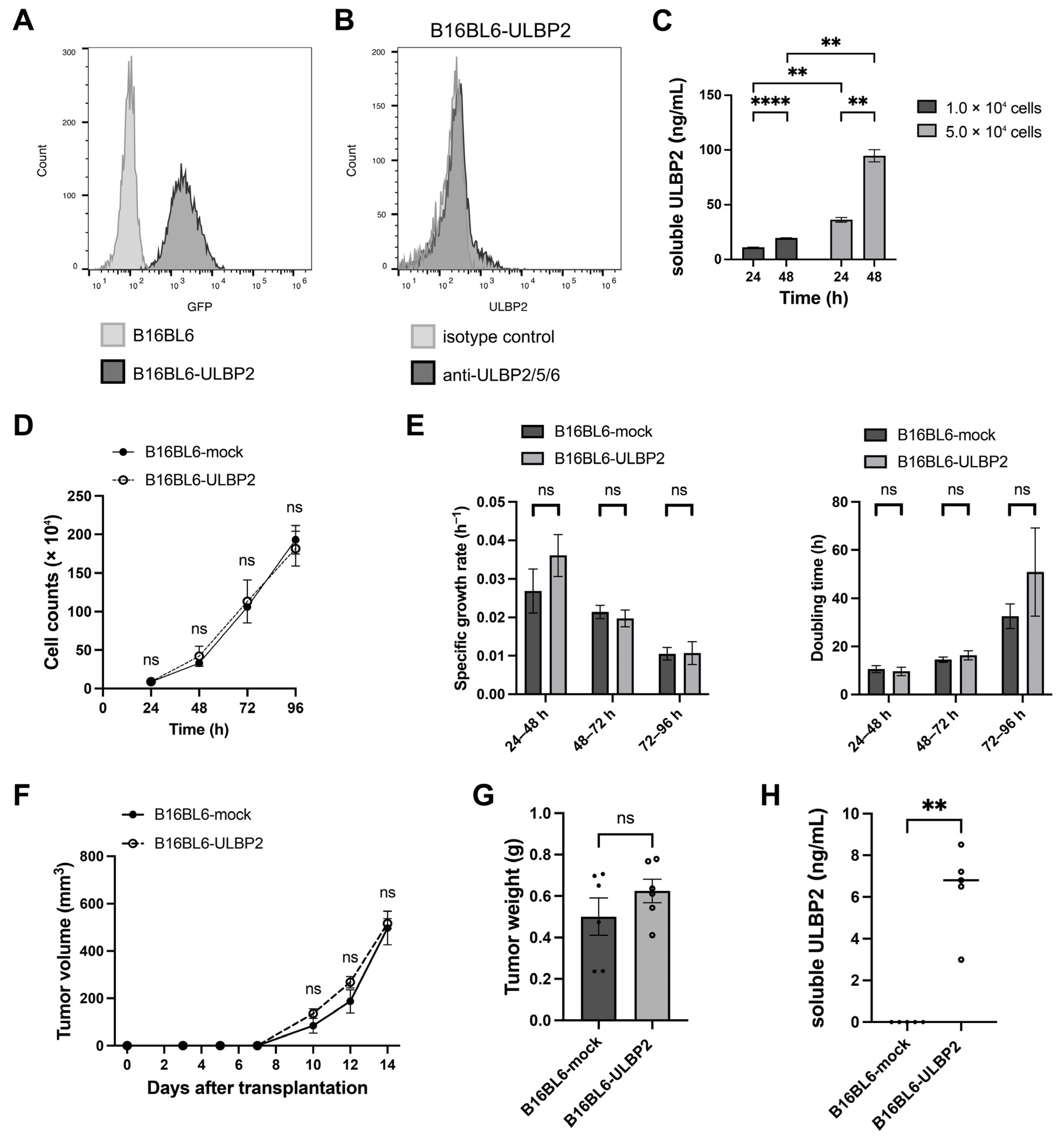
2.2. Soluble ULBP2 Does Not Promote Tumor Growth
2.3. Surface-Expressed ULBP2 Downregulates NKG2D Expression on NK Cells
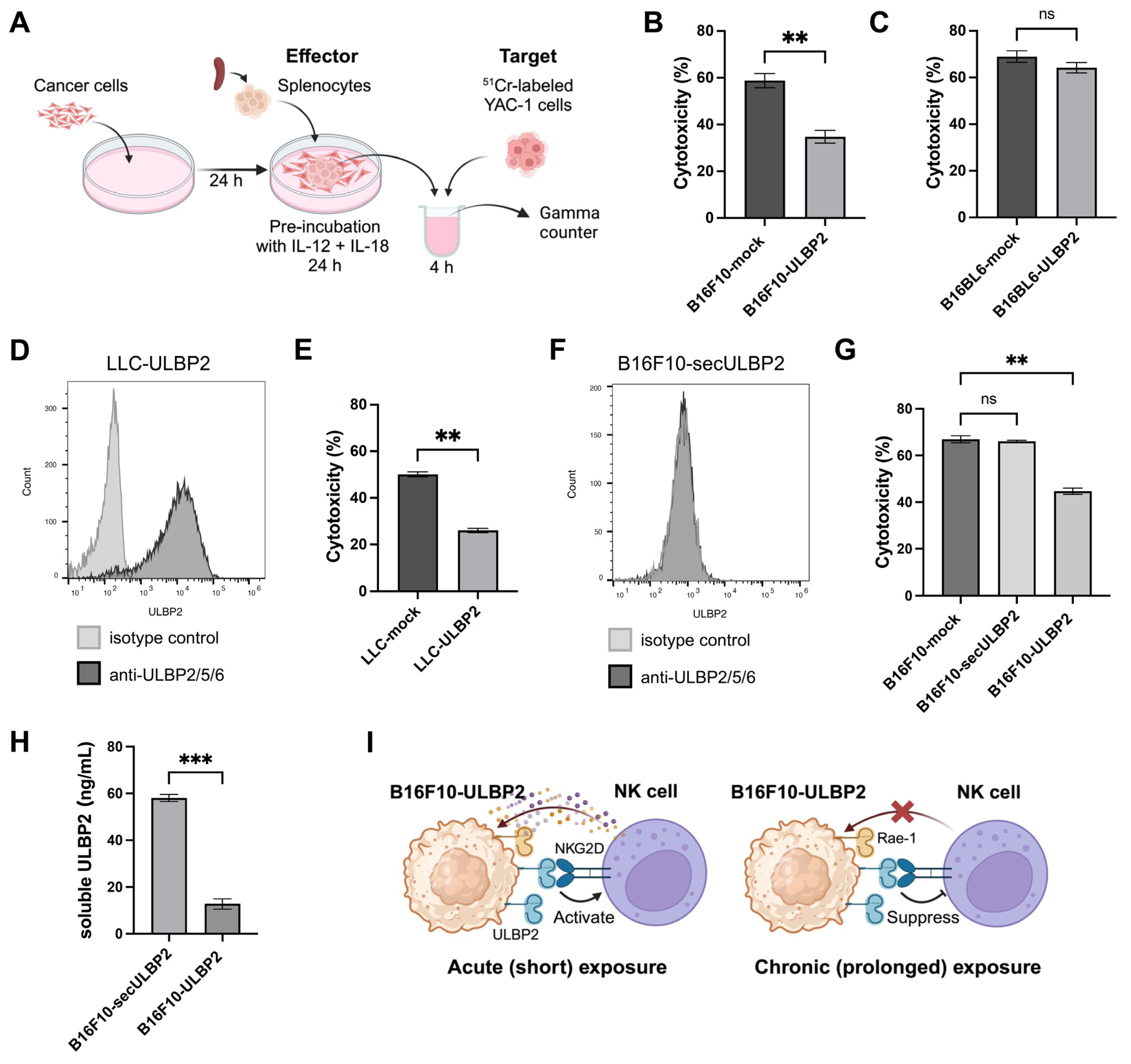
2.4. ULBP2 Inhibits Anti-Tumor Immunity Mediated by NK Cells
2.5. Transient ULBP2 Exposure Enhances the Cytotoxic Activity of Splenocytes
2.6. Sustained ULBP2 Exposure Suppresses the Cytotoxic Activity of Splenocytes
3. Discussion
4. Materials and Methods
4.1. Cell Lines
4.2. Cell Lines Stably Expressing ULBP2
4.3. ELISA
4.4. In Vitro Proliferation Assay
4.5. Flow Cytometry for Cancer Cell Lines
4.6. Mice
4.7. Co-Culture of Cancer Cell Lines and Murine Splenocytes
4.8. Flow Cytometric Analysis for Murine Splenic NK Cells
4.9. Tumor Transplantation and In Vivo Experimental Procedures
4.10. Four-Hour 51Cr Release Assay
4.11. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Bauer, S.; Groh, V.; Wu, J.; Steinle, A.; Phillips, J.H.; Lanier, L.L.; Spies, T. Activation of NK cells and T cells by NKG2D, a receptor for stress-inducible MICA. Science 1999, 285, 727–729. [Google Scholar] [CrossRef] [PubMed]
- Groh, V.; Bruhl, A.; El-Gabalawy, H.; Nelson, J.L.; Spies, T. Stimulation of T cell autoreactivity by anomalous expression of NKG2D and its MIC ligands in rheumatoid arthritis. Proc. Natl. Acad. Sci. USA 2003, 100, 9452–9457. [Google Scholar] [CrossRef]
- Groh, V.; Smythe, K.; Dai, Z.; Spies, T. Fas-ligand-mediated paracrine T cell regulation by the receptor NKG2D in tumor immunity. Nat. Immunol. 2006, 7, 755–762. [Google Scholar] [CrossRef]
- Vivier, E.; Tomasello, E.; Paul, P. Lymphocyte activation via NKG2D: Towards a new paradigm in immune recognition? Curr. Opin. Immunol. 2002, 14, 306–311. [Google Scholar]
- Wu, J.; Song, Y.; Bakker, A.B.; Bauer, S.; Spies, T.; Lanier, L.L.; Phillips, J.H. An activating immunoreceptor complex formed by NKG2D and DAP10. Science 1999, 285, 730–732. [Google Scholar] [CrossRef]
- Cosman, D.; Mullberg, J.; Sutherland, C.L.; Chin, W.; Armitage, R.; Fanslow, W.; Kubin, M.; Chalupny, N.J. ULBPs, novel MHC class I-related molecules, bind to CMV glycoprotein UL16 and stimulate NK cytotoxicity through the NKG2D receptor. Immunity 2001, 14, 123–133. [Google Scholar]
- Onda, H.; Ohkubo, S.; Shintani, Y.; Ogi, K.; Kikuchi, K.; Tanaka, H.; Yamamoto, K.; Tsuji, I.; Ishibashi, Y.; Yamada, T.; et al. A novel secreted tumor antigen with a glycosylphosphatidylinositol-anchored structure ubiquitously expressed in human cancers. Biochem. Biophys. Res. Commun. 2001, 285, 235–243. [Google Scholar] [CrossRef]
- Tan, G.; Spillane, K.M.; Maher, J. The Role and Regulation of the NKG2D/NKG2D Ligand System in Cancer. Biology 2023, 12, 1079. [Google Scholar] [CrossRef]
- Diefenbach, A.; Jensen, E.R.; Jamieson, A.M.; Raulet, D.H. Rae1 and H60 ligands of the NKG2D receptor stimulate tumour immunity. Nature 2001, 413, 165–171. [Google Scholar] [CrossRef]
- Cerwenka, A.; Baron, J.L.; Lanier, L.L. Ectopic expression of retinoic acid early inducible-1 gene (RAE-1) permits natural killer cell-mediated rejection of a MHC class I-bearing tumor in vivo. Proc. Natl. Acad. Sci. USA 2001, 98, 11521–11526. [Google Scholar] [CrossRef]
- Oppenheim, D.E.; Roberts, S.J.; Clarke, S.L.; Filler, R.; Lewis, J.M.; Tigelaar, R.E.; Girardi, M.; Hayday, A.C. Sustained localized expression of ligand for the activating NKG2D receptor impairs natural cytotoxicity in vivo and reduces tumor immunosurveillance. Nat. Immunol. 2005, 6, 928–937. [Google Scholar] [CrossRef] [PubMed]
- Wensveen, F.M.; Jelencic, V.; Polic, B. NKG2D: A Master Regulator of Immune Cell Responsiveness. Front. Immunol. 2018, 9, 441. [Google Scholar] [CrossRef] [PubMed]
- Jinushi, M.; Takehara, T.; Tatsumi, T.; Kanto, T.; Groh, V.; Spies, T.; Kimura, R.; Miyagi, T.; Mochizuki, K.; Sasaki, Y.; et al. Expression and role of MICA and MICB in human hepatocellular carcinomas and their regulation by retinoic acid. Int. J. Cancer 2003, 104, 354–361. [Google Scholar] [CrossRef]
- Sutherland, C.L.; Rabinovich, B.; Chalupny, N.J.; Brawand, P.; Miller, R.; Cosman, D. ULBPs, human ligands of the NKG2D receptor, stimulate tumor immunity with enhancement by IL-15. Blood 2006, 108, 1313–1319. [Google Scholar] [CrossRef]
- Pende, D.; Rivera, P.; Marcenaro, S.; Chang, C.C.; Biassoni, R.; Conte, R.; Kubin, M.; Cosman, D.; Ferrone, S.; Moretta, L.; et al. Major histocompatibility complex class I-related chain A and UL16-binding protein expression on tumor cell lines of different histotypes: Analysis of tumor susceptibility to NKG2D-dependent natural killer cell cytotoxicity. Cancer Res. 2002, 62, 6178–6186. [Google Scholar]
- Waldhauer, I.; Steinle, A. Proteolytic release of soluble UL16-binding protein 2 from tumor cells. Cancer Res. 2006, 66, 2520–2526. [Google Scholar] [CrossRef]
- Groh, V.; Wu, J.; Yee, C.; Spies, T. Tumour-derived soluble MIC ligands impair expression of NKG2D and T-cell activation. Nature 2002, 419, 734–738. [Google Scholar] [CrossRef]
- Song, H.; Kim, J.; Cosman, D.; Choi, I. Soluble ULBP suppresses natural killer cell activity via down-regulating NKG2D expression. Cell. Immunol. 2006, 239, 22–30. [Google Scholar] [CrossRef]
- Yamaguchi, K.; Chikumi, H.; Shimizu, A.; Takata, M.; Kinoshita, N.; Hashimoto, K.; Nakamoto, M.; Matsunaga, S.; Kurai, J.; Miyake, N.; et al. Diagnostic and prognostic impact of serum-soluble UL16-binding protein 2 in lung cancer patients. Cancer Sci. 2012, 103, 1405–1413. [Google Scholar] [CrossRef]
- Paschen, A.; Sucker, A.; Hill, B.; Moll, I.; Zapatka, M.; Nguyen, X.D.; Sim, G.C.; Gutmann, I.; Hassel, J.; Becker, J.C.; et al. Differential clinical significance of individual NKG2D ligands in melanoma: Soluble ULBP2 as an indicator of poor prognosis superior to S100B. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2009, 15, 5208–5215. [Google Scholar] [CrossRef]
- Chen, J.; Zhu, X.X.; Xu, H.; Fang, H.Z.; Zhao, J.Q. Expression and prognostic significance of unique ULBPs in pancreatic cancer. OncoTargets Ther. 2016, 9, 5271–5279. [Google Scholar] [CrossRef] [PubMed]
- Kegasawa, T.; Tatsumi, T.; Yoshioka, T.; Suda, T.; Ikezawa, K.; Nakabori, T.; Yamada, R.; Kodama, T.; Shigekawa, M.; Hikita, H.; et al. Soluble UL16-binding protein 2 is associated with a poor prognosis in pancreatic cancer patients. Biochem. Biophys. Res. Commun. 2019, 517, 84–88. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Su, X.; Wang, Z.; Yu, Y.; Wu, Z.; Zhang, D. ULBP2 is a biomarker related to prognosis and immunity in colon cancer. Mol. Cell. Biochem. 2023, 478, 2207–2219. [Google Scholar] [CrossRef] [PubMed]
- Gao, Z.; Fu, P.; Yu, Z.; Zhen, F.; Gu, Y. Comprehensive Analysis of lncRNA-miRNA-mRNA Network Ascertains Prognostic Factors in Patients with Colon Cancer. Technol. Cancer Res. Treat. 2019, 18, 1533033819853237. [Google Scholar] [CrossRef]
- Ye, Z.; Zhang, H.; Liang, J.; Yi, S.; Zhan, X. Significance of logistic regression scoring model based on natural killer cell-mediated cytotoxic pathway in the diagnosis of colon cancer. Front. Immunol. 2023, 14, 1117908. [Google Scholar] [CrossRef]
- Ruan, G.T.; Wang, S.; Zhu, L.C.; Liao, X.W.; Wang, X.K.; Liao, C.; Yan, L.; Xie, H.L.; Gong, Y.Z.; Gan, J.L.; et al. Investigation and verification of the clinical significance and perspective of natural killer group 2 member D ligands in colon adenocarcinoma. Aging 2021, 13, 12565–12586. [Google Scholar] [CrossRef]
- Zhang, S.; Zhang, W.; Zhang, J. 8-Gene signature related to CD8(+) T cell infiltration by integrating single-cell and bulk RNA-sequencing in head and neck squamous cell carcinoma. Front. Genet. 2022, 13, 938611. [Google Scholar] [CrossRef]
- Sun, Y.; Zhang, Q.; Yao, L.; Wang, S.; Zhang, Z. Comprehensive analysis reveals novel gene signature in head and neck squamous cell carcinoma: Predicting is associated with poor prognosis in patients. Transl. Cancer Res. 2020, 9, 5882–5892. [Google Scholar] [CrossRef]
- Li, L.; Li, L.; Liu, M.; Li, Y.; Sun, Q. Novel immune-related prognostic model and nomogram for breast cancer based on ssGSEA. Front. Genet. 2022, 13, 957675. [Google Scholar] [CrossRef]
- Dong, M.; Cui, X.; Wang, G.; Zhang, Q.; Li, X. Development of a prognostic signature based on immune-related genes and the correlation with immune microenvironment in breast cancer. Aging 2022, 14, 5427–5448. [Google Scholar] [CrossRef]
- D’Angelo, A.; Kilili, H.; Chapman, R.; Generali, D.; Tinhofer, I.; Luminari, S.; Donati, B.; Ciarrocchi, A.; Giannini, R.; Moretto, R.; et al. Immune-related pan-cancer gene expression signatures of patient survival revealed by NanoString-based analyses. PLoS ONE 2023, 18, e0280364. [Google Scholar] [CrossRef]
- Li, Q.; Hirata, Y.; Piao, S.; Minami, M. Immunotoxicity of N,N-diethylaniline in mice: Effect on natural killer activity, cytotoxic T lymphocyte activity, lymphocyte proliferation response and cellular components of the spleen. Toxicology 2000, 150, 179–189. [Google Scholar] [CrossRef] [PubMed]
- Li, Q. Natural Killer (NK) Cell Assays in Immunotoxicity Testing. Methods Mol. Biol. 2018, 1803, 231–241. [Google Scholar] [CrossRef] [PubMed]
- Lauwerys, B.R.; Renauld, J.C.; Houssiau, F.A. Synergistic proliferation and activation of natural killer cells by interleukin 12 and interleukin 18. Cytokine 1999, 11, 822–830. [Google Scholar] [CrossRef]
- Wright, S.C.; Bonavida, B. YAC-1 variant clones selected for resistance to natural killer cytotoxic factors are also resistant to natural killer cell-mediated cytotoxicity. Proc. Natl. Acad. Sci. USA 1983, 80, 1688–1692. [Google Scholar] [CrossRef]
- Coudert, J.D.; Zimmer, J.; Tomasello, E.; Cebecauer, M.; Colonna, M.; Vivier, E.; Held, W. Altered NKG2D function in NK cells induced by chronic exposure to NKG2D ligand-expressing tumor cells. Blood 2005, 106, 1711–1717. [Google Scholar] [CrossRef]
- Duan, S.; Guo, W.; Xu, Z.; He, Y.; Liang, C.; Mo, Y.; Wang, Y.; Xiong, F.; Guo, C.; Li, Y.; et al. Natural killer group 2D receptor and its ligands in cancer immune escape. Mol. Cancer 2019, 18, 29. [Google Scholar] [CrossRef]
- Waldhauer, I.; Steinle, A. NK cells and cancer immunosurveillance. Oncogene 2008, 27, 5932–5943. [Google Scholar] [CrossRef]
- Salih, H.R.; Goehlsdorf, D.; Steinle, A. Release of MICB molecules by tumor cells: Mechanism and soluble MICB in sera of cancer patients. Human Immunol. 2006, 67, 188–195. [Google Scholar] [CrossRef]
- Liu, G.; Lu, S.; Wang, X.; Page, S.T.; Higano, C.S.; Plymate, S.R.; Greenberg, N.M.; Sun, S.; Li, Z.; Wu, J.D. Perturbation of NK cell peripheral homeostasis accelerates prostate carcinoma metastasis. J. Clin. Investig. 2013, 123, 4410–4422. [Google Scholar] [CrossRef]
- Nausch, N.; Cerwenka, A. NKG2D ligands in tumor immunity. Oncogene 2008, 27, 5944–5958. [Google Scholar] [CrossRef] [PubMed]
- Coudert, J.D.; Scarpellino, L.; Gros, F.; Vivier, E.; Held, W. Sustained NKG2D engagement induces cross-tolerance of multiple distinct NK cell activation pathways. Blood 2008, 111, 3571–3578. [Google Scholar] [CrossRef] [PubMed]
- Wiemann, K.; Mittrucker, H.W.; Feger, U.; Welte, S.A.; Yokoyama, W.M.; Spies, T.; Rammensee, H.G.; Steinle, A. Systemic NKG2D down-regulation impairs NK and CD8 T cell responses in vivo. J. Immunol. 2005, 175, 720–729. [Google Scholar] [CrossRef]
- Kim, Y.; Born, C.; Blery, M.; Steinle, A. MICAgen Mice Recapitulate the Highly Restricted but Activation-Inducible Expression of the Paradigmatic Human NKG2D Ligand MICA. Front. Immunol. 2020, 11, 960. [Google Scholar] [CrossRef]
- Basher, F.; Dhar, P.; Wang, X.; Wainwright, D.A.; Zhang, B.; Sosman, J.; Ji, Z.; Wu, J.D. Antibody targeting tumor-derived soluble NKG2D ligand sMIC reprograms NK cell homeostatic survival and function and enhances melanoma response to PDL1 blockade therapy. J. Hematol. Oncol. 2020, 13, 74. [Google Scholar] [CrossRef]
- Lerner, E.C.; Woroniecka, K.I.; D’Anniballe, V.M.; Wilkinson, D.S.; Mohan, A.A.; Lorrey, S.J.; Waibl-Polania, J.; Wachsmuth, L.P.; Miggelbrink, A.M.; Jackson, J.D.; et al. CD8(+) T cells maintain killing of MHC-I-negative tumor cells through the NKG2D-NKG2DL axis. Nat. Cancer 2023, 4, 1258–1272. [Google Scholar] [CrossRef]
- Ueha, S.; Yokochi, S.; Ishiwata, Y.; Ogiwara, H.; Chand, K.; Nakajima, T.; Hachiga, K.; Shichino, S.; Terashima, Y.; Toda, E.; et al. Robust Antitumor Effects of Combined Anti-CD4-Depleting Antibody and Anti-PD-1/PD-L1 Immune Checkpoint Antibody Treatment in Mice. Cancer Immunol. Res. 2015, 3, 631–640. [Google Scholar] [CrossRef]
- Aoki, H.; Ueha, S.; Shichino, S.; Ogiwara, H.; Shitara, K.; Shimomura, M.; Suzuki, T.; Nakatsura, T.; Yamashita, M.; Kitano, S.; et al. Transient Depletion of CD4(+) Cells Induces Remodeling of the TCR Repertoire in Gastrointestinal Cancer. Cancer Immunol. Res. 2021, 9, 624–636. [Google Scholar] [CrossRef]
- Wang, Y.; Kim, M.; Su, S.; Halwatura, L.; You, S.; Kim, H.L. Using major histocompatibility complex (MHC) II expression to predict antitumor response to CD4 + lymphocyte depletion. Sci. Rep. 2025, 15, 5469. [Google Scholar] [CrossRef]
- Dinavahi, S.S.; Chen, Y.C.; Punnath, K.; Berg, A.; Herlyn, M.; Foroutan, M.; Huntington, N.D.; Robertson, G.P. Targeting WEE1/AKT Restores p53-Dependent Natural Killer-Cell Activation to Induce Immune Checkpoint Blockade Responses in “Cold” Melanoma. Cancer Immunol. Res. 2022, 10, 757–769. [Google Scholar] [CrossRef]
- Schaefer, M.R.; Williams, M.; Kulpa, D.A.; Blakely, P.K.; Yaffee, A.Q.; Collins, K.L. A novel trafficking signal within the HLA-C cytoplasmic tail allows regulated expression upon differentiation of macrophages. J. Immunol. 2008, 180, 7804–7817. [Google Scholar] [CrossRef]
- Kurai, J.; Chikumi, H.; Hashimoto, K.; Yamaguchi, K.; Yamasaki, A.; Sako, T.; Touge, H.; Makino, H.; Takata, M.; Miyata, M.; et al. Antibody-dependent cellular cytotoxicity mediated by cetuximab against lung cancer cell lines. Clin. Cancer Res. 2007, 13, 1552–1561. [Google Scholar] [CrossRef] [PubMed]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yamane, K.; Yamaguchi, K.; Teruya, Y.; Miyake, N.; Nakayama, Y.; Nonaka, T.; Chikumi, H.; Yamasaki, A. ULBP2 Promotes Tumor Progression by Suppressing NKG2D-Mediated Anti-Tumor Immunity. Int. J. Mol. Sci. 2025, 26, 2950. https://doi.org/10.3390/ijms26072950
Yamane K, Yamaguchi K, Teruya Y, Miyake N, Nakayama Y, Nonaka T, Chikumi H, Yamasaki A. ULBP2 Promotes Tumor Progression by Suppressing NKG2D-Mediated Anti-Tumor Immunity. International Journal of Molecular Sciences. 2025; 26(7):2950. https://doi.org/10.3390/ijms26072950
Chicago/Turabian StyleYamane, Kohei, Kosuke Yamaguchi, Yasuhiko Teruya, Naomi Miyake, Yuji Nakayama, Takafumi Nonaka, Hiroki Chikumi, and Akira Yamasaki. 2025. "ULBP2 Promotes Tumor Progression by Suppressing NKG2D-Mediated Anti-Tumor Immunity" International Journal of Molecular Sciences 26, no. 7: 2950. https://doi.org/10.3390/ijms26072950
APA StyleYamane, K., Yamaguchi, K., Teruya, Y., Miyake, N., Nakayama, Y., Nonaka, T., Chikumi, H., & Yamasaki, A. (2025). ULBP2 Promotes Tumor Progression by Suppressing NKG2D-Mediated Anti-Tumor Immunity. International Journal of Molecular Sciences, 26(7), 2950. https://doi.org/10.3390/ijms26072950