

Unraveling Molecular Targets for Neurodegenerative Diseases Through Caenorhabditis elegans Models

Abstract

{kind=link}
{kind=link}
{kind=link}
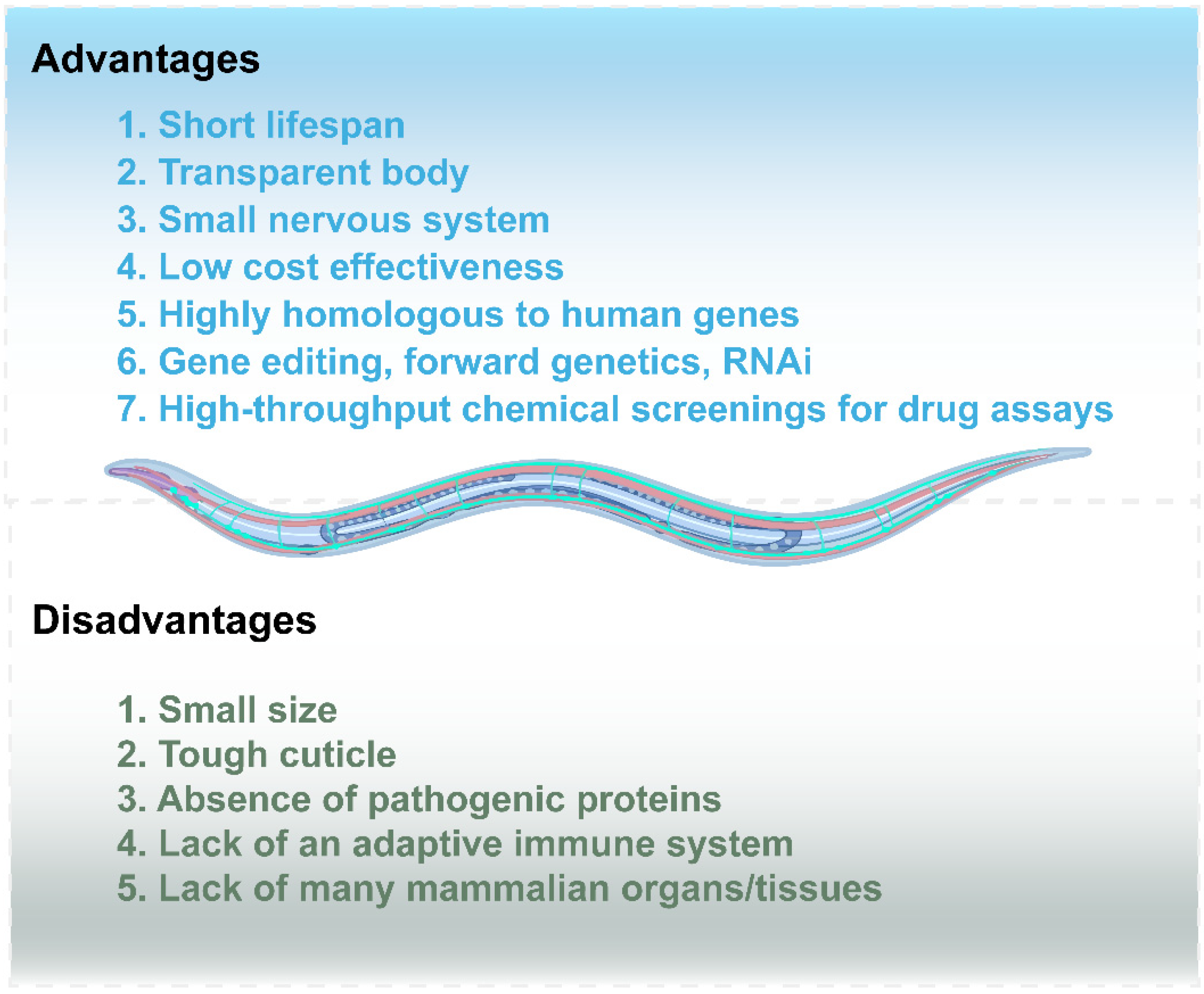
1. Introduction
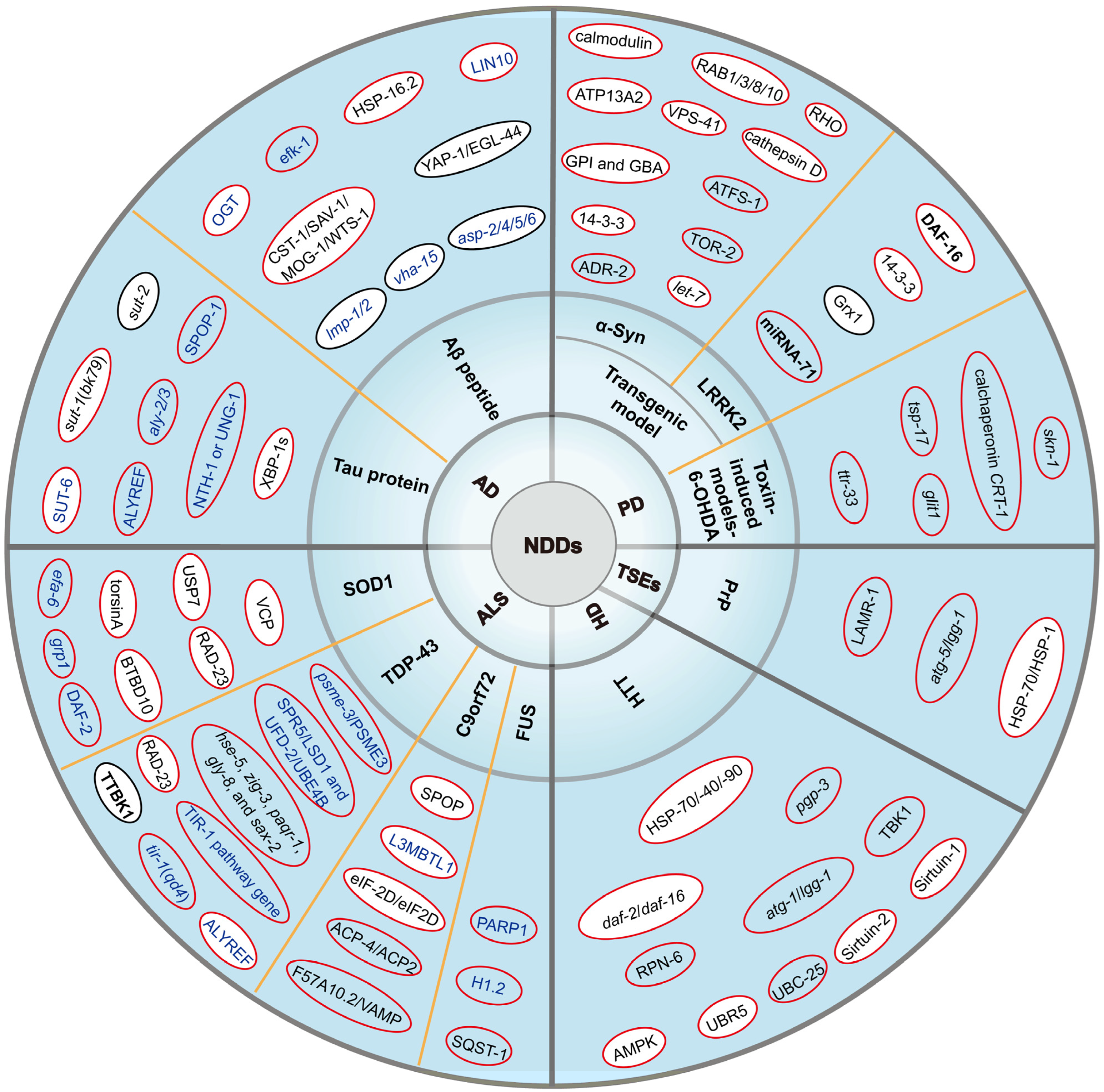
2. Alzheimer’s Disease (AD)
2.1. Amyloid-Beta Peptide (Aβ Peptide)
2.2. Tau Protein
3. Parkinson’s Disease (PD)
3.1. Transgenic Model
3.1.1. Alpha-Synuclein (Alpha-Syn)
3.1.2. Leucine-Rich Repeat Kinase 2 (LRRK2)
3.2. Toxin-Induced Models-6-Hydroxydopamine (6-OHDA)
4. Amyotrophic Lateral Sclerosis (ALS)
4.1. Cu/Zn Superoxide Dismutase 1(SOD1)
4.2. TAR DNA-Binding Protein 43 (TDP-43)
4.3. Chromosome 9 Open Reading Frame 72 (C9orf72)
4.4. Fusion Sarcoma (FUS)
5. Huntington’s Disease (HD)
6. Prion Diseases
7. Discussion and Future Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| NDDs | Neurodegenerative diseases |
| AD | Alzheimer’s Disease |
| PD | Parkinson’s Disease |
| ALS | amyotrophic lateral sclerosis |
| HD | Huntington’s Disease |
| C. elegans | Caenorhabditis elegans |
| KO | knockout |
| EOAD | early-onset AD |
| LOAD | late-onset AD |
| NFTs | neurofibrillary tangles |
| OE | overexpressing |
| Aβ peptide | amyloid-beta peptide |
| OGT | O-GlcNAc transferase |
| MAP2 | microtubule-associated protein 2 |
| UPRER | endoplasmic reticulum unfolded protein response |
| alpha-Syn | alpha Synuclein |
| DA | dopamine |
| GFP | green fluorescent protein |
| BiFC | bimolecular fluorescence complementation |
| miRNAs | microRNAs |
| LRRK2 | Leucine-rich repeat kinase 2 |
| Grx1 | Glutaredoxin 1 |
| 6-OHDA | 6-hydroxydopamine |
| DAT | dopamine transporters |
| PQ | Paraquat |
| sALS | sporadic ALS |
| fALS | familial ALS |
| SOD1 | Cu/Zn superoxide dismutase 1 |
| TDP-43 | TAR DNA-binding protein 43 |
| C9orf72 | Chromosome 9 Open Reading Frame 72 |
| FUS | Fusion sarcoma |
| hmSOD1 | human SOD1 |
| VCP | valine-containing protein |
| MPS | α-methyl-α-phenylsuccinimide |
| DPRs | dipeptide repeat protein |
| L3MBTL1 | Lethal (3) malignant brain tumor-like protein 1 |
| G4C2 | GGGGCC |
| NLS | nuclear localization signal |
| HTT | huntingtin |
| polyQ | polyglutamine |
| TSEs | transmissible spongiform encephalopathies |
| PrP | prion protein |
References
- Wu, Y.; Chen, Y.; Yu, X.; Zhang, M.; Li, Z. Towards Understanding Neurodegenerative Diseases: Insights from Caenorhabditis elegans. Int. J. Mol. Sci. 2023, 25, 443. [Google Scholar] [CrossRef] [PubMed]
- Cheslow, L.; Snook, A.E.; Waldman, S.A. Biomarkers for Managing Neurodegenerative Diseases. Biomolecules 2024, 14, 398. [Google Scholar] [CrossRef]
- Tenchov, R.; Sasso, J.M.; Zhou, Q.A. Polyglutamine (PolyQ) Diseases: Navigating the Landscape of Neurodegeneration. ACS Chem. Neurosci. 2024, 15, 2665–2694. [Google Scholar] [CrossRef] [PubMed]
- Baiardi, S.; Mammana, A.; Capellari, S.; Parchi, P. Human prion disease: Molecular pathogenesis, and possible therapeutic targets and strategies. Expert. Opin. Ther. Targets 2023, 27, 1271–1284. [Google Scholar] [CrossRef] [PubMed]
- Sundaramoorthy, T.H.; Castanho, I. The Neuroepigenetic Landscape of Vertebrate and Invertebrate Models of Neurodegenerative Diseases. Epigenet. Insights 2022, 15, 25168657221135848. [Google Scholar] [CrossRef] [PubMed]
- Rani, N.; Alam, M.M.; Jamal, A.; Bin Ghaffar, U.; Parvez, S. Caenorhabditis elegans: A transgenic model for studying age-associated neurodegenerative diseases. Ageing Res. Rev. 2023, 91, 102036. [Google Scholar] [CrossRef]
- Brenner, S. The genetics of Caenorhabditis elegans. Genetics 1974, 77, 71–94. [Google Scholar] [CrossRef] [PubMed]
- Lai, C.H.; Chou, C.Y.; Ch’ang, L.Y.; Liu, C.S.; Lin, W. Identification of novel human genes evolutionarily conserved in Caenorhabditis elegans by comparative proteomics. Genome Res. 2000, 10, 703–713. [Google Scholar] [CrossRef]
- Cook, S.J.; Jarrell, T.A.; Brittin, C.A.; Wang, Y.; Bloniarz, A.E.; Yakovlev, M.A.; Nguyen, K.C.Q.; Tang, L.T.; Bayer, E.A.; Duerr, J.S.; et al. Whole-animal connectomes of both Caenorhabditis elegans sexes. Nature 2019, 571, 63–71. [Google Scholar] [CrossRef]
- White, J.G.; Southgate, E.; Thomson, J.N.; Brenner, S. The structure of the nervous system of the nematode Caenorhabditis elegans. Philos. Trans. R. Soc. Lond. B Biol. Sci. 1986, 314, 1–340. [Google Scholar] [CrossRef]
- Roussos, A.; Kitopoulou, K.; Borbolis, F.; Palikaras, K. Caenorhabditis elegans as a Model System to Study Human Neurodegenerative Disorders. Biomolecules 2023, 13, 478. [Google Scholar] [CrossRef] [PubMed]
- Flavell, S.W.; Pokala, N.; Macosko, E.Z.; Albrecht, D.R.; Larsch, J.; Bargmann, C.I. Serotonin and the neuropeptide PDF initiate and extend opposing behavioral states in C. elegans. Cell 2013, 154, 1023–1035. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Barclay, J.W.; Burgoyne, R.D.; Morgan, A. Using C. elegans to discover therapeutic compounds for ageing-associated neurodegenerative diseases. Chem. Cent. J. 2015, 9, 65. [Google Scholar] [CrossRef]
- Ikenaka, K.; Tsukada, Y.; Giles, A.C.; Arai, T.; Nakadera, Y.; Nakano, S.; Kawai, K.; Mochizuki, H.; Katsuno, M.; Sobue, G.; et al. A behavior-based drug screening system using a Caenorhabditis elegans model of motor neuron disease. Sci. Rep. 2019, 9, 10104. [Google Scholar] [CrossRef]
- Rajendran, K.; Krishnan, U.M. Biomarkers in Alzheimer’s disease. Clin. Chim. Acta 2024, 562, 119857. [Google Scholar] [CrossRef] [PubMed]
- Better, M.A. Alzheimer’s disease facts and figures. Alzheimer’s Dement. 2023, 19, 1598–1695. [Google Scholar] [CrossRef]
- Andrade-Guerrero, J.; Santiago-Balmaseda, A.; Jeronimo-Aguilar, P.; Vargas-Rodriguez, I.; Cadena-Suarez, A.R.; Sanchez-Garibay, C.; Pozo-Molina, G.; Mendez-Catala, C.F.; Cardenas-Aguayo, M.D.; Diaz-Cintra, S.; et al. Alzheimer’s Disease: An Updated Overview of Its Genetics. Int. J. Mol. Sci. 2023, 24, 3754. [Google Scholar] [CrossRef] [PubMed]
- Ashrafian, H.; Zadeh, E.H.; Khan, R.H. Review on Alzheimer’s disease: Inhibition of amyloid beta and tau tangle formation. Int. J. Biol. Macromol. 2021, 167, 382–394. [Google Scholar] [CrossRef]
- Alvarez, J.; Alvarez-Illera, P.; Santo-Domingo, J.; Fonteriz, R.I.; Montero, M. Modeling Alzheimer’s Disease in Caenorhabditis elegans. Biomedicines 2022, 10, 288. [Google Scholar] [CrossRef]
- Ortega, F.; Stott, J.; Visser, S.A.; Bendtsen, C. Interplay between alpha-, beta-, and gamma-secretases determines biphasic amyloid-beta protein level in the presence of a gamma-secretase inhibitor. J. Biol. Chem. 2013, 288, 785–792. [Google Scholar] [CrossRef]
- Hata, S. Molecular Pathogenesis of Sporadic Alzheimer’s Disease (AD) and Pharmaceutical Research to Develop a Biomarker for AD Diagnosis. Yakugaku Zasshi 2015, 135, 1023–1027. [Google Scholar] [CrossRef] [PubMed]
- Chang, Y.J.; Chen, Y.R. The coexistence of an equal amount of Alzheimer’s amyloid-beta 40 and 42 forms structurally stable and toxic oligomers through a distinct pathway. FEBS J. 2014, 281, 2674–2687. [Google Scholar] [CrossRef]
- Link, C.D. Expression of human beta-amyloid peptide in transgenic Caenorhabditis elegans. Proc. Natl. Acad. Sci. USA 1995, 92, 9368–9372. [Google Scholar] [CrossRef] [PubMed]
- Dinda, B.; Dinda, M.; Kulsi, G.; Chakraborty, A.; Dinda, S. Therapeutic potentials of plant iridoids in Alzheimer’s and Parkinson’s diseases: A review. Eur. J. Med. Chem. 2019, 169, 185–199. [Google Scholar] [CrossRef]
- Wang, P.; Lazarus, B.D.; Forsythe, M.E.; Love, D.C.; Krause, M.W.; Hanover, J.A. O-GlcNAc cycling mutants modulate proteotoxicity in Caenorhabditis elegans models of human neurodegenerative diseases. Proc. Natl. Acad. Sci. USA 2012, 109, 17669–17674. [Google Scholar] [CrossRef] [PubMed]
- Jan, A.; Jansonius, B.; Delaidelli, A.; Somasekharan, S.P.; Bhanshali, F.; Vandal, M.; Negri, G.L.; Moerman, D.; MacKenzie, I.; Calon, F.; et al. eEF2K inhibition blocks Abeta42 neurotoxicity by promoting an NRF2 antioxidant response. Acta Neuropathol. 2017, 133, 101–119. [Google Scholar] [CrossRef] [PubMed]
- Fonte, V.; Kipp, D.R.; Yerg, J., 3rd; Merin, D.; Forrestal, M.; Wagner, E.; Roberts, C.M.; Link, C.D. Suppression of in vivo beta-amyloid peptide toxicity by overexpression of the HSP-16.2 small chaperone protein. J. Biol. Chem. 2008, 283, 784–791. [Google Scholar] [CrossRef] [PubMed]
- Link, C.D.; Fonte, V.; Hiester, B.; Yerg, J.; Ferguson, J.; Csontos, S.; Silverman, M.A.; Stein, G.H. Conversion of green fluorescent protein into a toxic, aggregation-prone protein by C-terminal addition of a short peptide. J. Biol. Chem. 2006, 281, 1808–1816. [Google Scholar] [CrossRef] [PubMed]
- Zhu, M.; Gu, H.; Bai, H.; Li, Y.; Zhong, C.; Huang, X. Role and molecular regulatory mechanisms of Hippo signaling pathway in Caenorhabditis elegans and mammalian cell models of Alzheimer’s disease. Neurobiol. Aging 2024, 134, 9–20. [Google Scholar] [CrossRef]
- Florez-McClure, M.L.; Hohsfield, L.A.; Fonte, G.; Bealor, M.T.; Link, C.D. Decreased insulin-receptor signaling promotes the autophagic degradation of beta-amyloid peptide in C. elegans. Autophagy 2007, 3, 569–580. [Google Scholar] [CrossRef]
- McDermott, J.B.; Aamodt, S.; Aamodt, E. ptl-1, a Caenorhabditis elegans gene whose products are homologous to the tau microtubule-associated proteins. Biochemistry 1996, 35, 9415–9423. [Google Scholar] [CrossRef] [PubMed]
- Chew, Y.L.; Fan, X.; Gotz, J.; Nicholas, H.R. PTL-1 regulates neuronal integrity and lifespan in C. elegans. J. Cell Sci. 2013, 126, 2079–2091. [Google Scholar] [CrossRef] [PubMed]
- Natale, C.; Barzago, M.M.; Diomede, L. Caenorhabditis elegans Models to Investigate the Mechanisms Underlying Tau Toxicity in Tauopathies. Brain Sci. 2020, 10, 838. [Google Scholar] [CrossRef]
- Kraemer, B.C.; Zhang, B.; Leverenz, J.B.; Thomas, J.H.; Trojanowski, J.Q.; Schellenberg, G.D. Neurodegeneration and defective neurotransmission in a Caenorhabditis elegans model of tauopathy. Proc. Natl. Acad. Sci. USA 2003, 100, 9980–9985. [Google Scholar] [CrossRef] [PubMed]
- Miyasaka, T.; Ding, Z.; Gengyo-Ando, K.; Oue, M.; Yamaguchi, H.; Mitani, S.; Ihara, Y. Progressive neurodegeneration in C. elegans model of tauopathy. Neurobiol. Dis. 2005, 20, 372–383. [Google Scholar] [CrossRef]
- Brandt, R.; Gergou, A.; Wacker, I.; Fath, T.; Hutter, H. A Caenorhabditis elegans model of tau hyperphosphorylation: Induction of developmental defects by transgenic overexpression of Alzheimer’s disease-like modified tau. Neurobiol. Aging 2009, 30, 22–33. [Google Scholar] [CrossRef]
- Mi, K.; Johnson, G.V. The role of tau phosphorylation in the pathogenesis of Alzheimer’s disease. Curr. Alzheimer Res. 2006, 3, 449–463. [Google Scholar] [CrossRef]
- Cohen, T.J.; Guo, J.L.; Hurtado, D.E.; Kwong, L.K.; Mills, I.P.; Trojanowski, J.Q.; Lee, V.M. The acetylation of tau inhibits its function and promotes pathological tau aggregation. Nat. Commun. 2011, 2, 252. [Google Scholar] [CrossRef]
- Min, S.W.; Chen, X.; Tracy, T.E.; Li, Y.; Zhou, Y.; Wang, C.; Shirakawa, K.; Minami, S.S.; Defensor, E.; Mok, S.A.; et al. Critical role of acetylation in tau-mediated neurodegeneration and cognitive deficits. Nat. Med. 2015, 21, 1154–1162. [Google Scholar] [CrossRef]
- Augustinack, J.C.; Schneider, A.; Mandelkow, E.M.; Hyman, B.T. Specific tau phosphorylation sites correlate with severity of neuronal cytopathology in Alzheimer’s disease. Acta Neuropathol. 2002, 103, 26–35. [Google Scholar] [CrossRef]
- Guha, S.; Fischer, S.; Johnson, G.V.W.; Nehrke, K. Tauopathy-associated tau modifications selectively impact neurodegeneration and mitophagy in a novel C. elegans single-copy transgenic model. Mol. Neurodegener. 2020, 15, 65. [Google Scholar] [CrossRef] [PubMed]
- Han, M.; Saxton, A.; Currey, H.; Waldherr, S.M.; Liachko, N.F.; Kraemer, B.C. Transgenic Dendra2::tau expression allows in vivo monitoring of tau proteostasis in Caenorhabditis elegans. Dis. Model. Mech. 2024, 17, dmm050473. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Saar, V.; Leung, K.L.; Chen, L.; Wong, G. Human amyloid beta peptide and tau co-expression impairs behavior and causes specific gene expression changes in Caenorhabditis elegans. Neurobiol. Dis. 2018, 109, 88–101. [Google Scholar] [CrossRef] [PubMed]
- Kraemer, B.C.; Schellenberg, G.D. SUT-1 enables tau-induced neurotoxicity in C. elegans. Hum. Mol. Genet. 2007, 16, 1959–1971. [Google Scholar] [CrossRef] [PubMed]
- Currey, H.N.; Kraemer, B.C.; Liachko, N.F. sut-2 loss of function mutants protect against tau-driven shortened lifespan and hyperactive pharyngeal pumping in a C. elegans model of tau toxicity. MicroPubl. Biol. 2023, 2023, 10.17912. [Google Scholar] [CrossRef]
- Guthrie, C.R.; Schellenberg, G.D.; Kraemer, B.C. SUT-2 potentiates tau-induced neurotoxicity in Caenorhabditis elegans. Hum. Mol. Genet. 2009, 18, 1825–1838. [Google Scholar] [CrossRef]
- Kow, R.L.; Black, A.H.; Henderson, B.P.; Kraemer, B.C. Sut-6/NIPP1 modulates tau toxicity. Hum. Mol. Genet. 2023, 32, 2292–2306. [Google Scholar] [CrossRef]
- Kow, R.L.; Black, A.H.; Saxton, A.D.; Liachko, N.F.; Kraemer, B.C. Loss of aly/ALYREF suppresses toxicity in both tau and TDP-43 models of neurodegeneration. Geroscience 2022, 44, 747–761. [Google Scholar] [CrossRef]
- Eck, R.J.; Kow, R.L.; Black, A.H.; Liachko, N.F.; Kraemer, B.C. SPOP loss of function protects against tauopathy. Proc. Natl. Acad. Sci. USA 2023, 120, e2207250120. [Google Scholar] [CrossRef]
- Tiwari, V.; Buvarp, E.; Borbolis, F.; Puligilla, C.; Croteau, D.L.; Palikaras, K.; Bohr, V.A. Loss of DNA glycosylases improves health and cognitive function in a C. elegans model of human tauopathy. Nucleic. Acids Res. 2024, 52, 10965–10985. [Google Scholar] [CrossRef]
- Vidovic, M.; Rikalovic, M.G. Alpha-Synuclein Aggregation Pathway in Parkinson’s Disease: Current Status and Novel Therapeutic Approaches. Cells 2022, 11, 1732. [Google Scholar] [CrossRef] [PubMed]
- Ye, H.; Robak, L.A.; Yu, M.; Cykowski, M.; Shulman, J.M. Genetics and Pathogenesis of Parkinson’s Syndrome. Annu. Rev. Pathol. 2023, 18, 95–121. [Google Scholar] [CrossRef] [PubMed]
- Munhoz, R.P.; Tumas, V.; Pedroso, J.L.; Silveira-Moriyama, L. The clinical diagnosis of Parkinson’s disease. Arq. Neuropsiquiatr. 2024, 82, 1–10. [Google Scholar] [CrossRef] [PubMed]
- da Silva, L.P.D.; da Cruz Guedes, E.; Fernandes, I.C.O.; Pedroza, L.A.L.; da Silva Pereira, G.J.; Gubert, P. Exploring Caenorhabditis elegans as Parkinson’s Disease Model: Neurotoxins and Genetic Implications. Neurotox. Res. 2024, 42, 11. [Google Scholar] [CrossRef] [PubMed]
- Ma, L.; Li, X.; Liu, C.; Yan, W.; Ma, J.; Petersen, R.B.; Peng, A.; Huang, K. Modelling Parkinson’s Disease in C. elegans: Strengths and Limitations. Curr. Pharm. Des. 2022, 28, 3033–3048. [Google Scholar] [CrossRef]
- Cooper, J.F.; Van Raamsdonk, J.M. Modeling Parkinson’s Disease in C. elegans. J. Park. Dis. 2018, 8, 17–32. [Google Scholar] [CrossRef]
- Kawahata, I.; Finkelstein, D.I.; Fukunaga, K. Pathogenic Impact of alpha-Synuclein Phosphorylation and Its Kinases in alpha-Synucleinopathies. Int. J. Mol. Sci. 2022, 23, 6216. [Google Scholar] [CrossRef]
- Perni, M.; Flagmeier, P.; Limbocker, R.; Cascella, R.; Aprile, F.A.; Galvagnion, C.; Heller, G.T.; Meisl, G.; Chen, S.W.; Kumita, J.R.; et al. Multistep Inhibition of alpha-Synuclein Aggregation and Toxicity in Vitro and in Vivo by Trodusquemine. ACS Chem. Biol. 2018, 13, 2308–2319. [Google Scholar] [CrossRef] [PubMed]
- Vicario, M.; Cieri, D.; Vallese, F.; Catoni, C.; Barazzuol, L.; Berto, P.; Grinzato, A.; Barbieri, L.; Brini, M.; Cali, T. A split-GFP tool reveals differences in the sub-mitochondrial distribution of wt and mutant alpha-synuclein. Cell Death Dis. 2019, 10, 857. [Google Scholar] [CrossRef]
- Asthana, J.; Shravage, B.V. Exploring therapeutic potential of mitophagy modulators using Drosophila models of Parkinson’s disease. Front. Aging Neurosci. 2022, 14, 986849. [Google Scholar] [CrossRef]
- Lakso, M.; Vartiainen, S.; Moilanen, A.M.; Sirvio, J.; Thomas, J.H.; Nass, R.; Blakely, R.D.; Wong, G. Dopaminergic neuronal loss and motor deficits in Caenorhabditis elegans overexpressing human alpha-synuclein. J. Neurochem. 2003, 86, 165–172. [Google Scholar] [CrossRef] [PubMed]
- Hamamichi, S.; Rivas, R.N.; Knight, A.L.; Cao, S.; Caldwell, K.A.; Caldwell, G.A. Hypothesis-based RNAi screening identifies neuroprotective genes in a Parkinson’s disease model. Proc. Natl. Acad. Sci. USA 2008, 105, 728–733. [Google Scholar] [CrossRef] [PubMed]
- Cooper, A.A.; Gitler, A.D.; Cashikar, A.; Haynes, C.M.; Hill, K.J.; Bhullar, B.; Liu, K.; Xu, K.; Strathearn, K.E.; Liu, F.; et al. Alpha-synuclein blocks ER-Golgi traffic and Rab1 rescues neuron loss in Parkinson’s models. Science 2006, 313, 324–328. [Google Scholar] [CrossRef] [PubMed]
- Qiao, L.; Hamamichi, S.; Caldwell, K.A.; Caldwell, G.A.; Yacoubian, T.A.; Wilson, S.; Xie, Z.L.; Speake, L.D.; Parks, R.; Crabtree, D.; et al. Lysosomal enzyme cathepsin D protects against alpha-synuclein aggregation and toxicity. Mol. Brain 2008, 1, 17. [Google Scholar] [CrossRef] [PubMed]
- Yacoubian, T.A.; Slone, S.R.; Harrington, A.J.; Hamamichi, S.; Schieltz, J.M.; Caldwell, K.A.; Caldwell, G.A.; Standaert, D.G. Differential neuroprotective effects of 14-3-3 proteins in models of Parkinson’s disease. Cell Death Dis. 2010, 1, e2. [Google Scholar] [CrossRef]
- Kim, H.; Calatayud, C.; Guha, S.; Fernandez-Carasa, I.; Berkowitz, L.; Carballo-Carbajal, I.; Ezquerra, M.; Fernandez-Santiago, R.; Kapahi, P.; Raya, A.; et al. The Small GTPase RAC1/CED-10 Is Essential in Maintaining Dopaminergic Neuron Function and Survival Against alpha-Synuclein-Induced Toxicity. Mol. Neurobiol. 2018, 55, 7533–7552. [Google Scholar] [CrossRef] [PubMed]
- Knight, A.L.; Yan, X.; Hamamichi, S.; Ajjuri, R.R.; Mazzulli, J.R.; Zhang, M.W.; Daigle, J.G.; Zhang, S.; Borom, A.R.; Roberts, L.R.; et al. The glycolytic enzyme, GPI, is a functionally conserved modifier of dopaminergic neurodegeneration in Parkinson’s models. Cell Metab. 2014, 20, 145–157. [Google Scholar] [CrossRef]
- Cooper, J.F.; Machiela, E.; Dues, D.J.; Spielbauer, K.K.; Senchuk, M.M.; Van Raamsdonk, J.M. Activation of the mitochondrial unfolded protein response promotes longevity and dopamine neuron survival in Parkinson’s disease models. Sci. Rep. 2017, 7, 16441. [Google Scholar] [CrossRef]
- Martinez, B.A.; Petersen, D.A.; Gaeta, A.L.; Stanley, S.P.; Caldwell, G.A.; Caldwell, K.A. Dysregulation of the Mitochondrial Unfolded Protein Response Induces Non-Apoptotic Dopaminergic Neurodegeneration in C. elegans Models of Parkinson’s Disease. J. Neurosci. 2017, 37, 11085–11100. [Google Scholar] [CrossRef]
- Gaeta, A.L.; Caldwell, K.A.; Caldwell, G.A. Found in Translation: The Utility of C. elegans Alpha-Synuclein Models of Parkinson’s Disease. Brain Sci. 2019, 9, 73. [Google Scholar] [CrossRef]
- Sharma, N.; Hewett, J.; Ozelius, L.J.; Ramesh, V.; McLean, P.J.; Breakefield, X.O.; Hyman, B.T. A close association of torsinA and alpha-synuclein in Lewy bodies: A fluorescence resonance energy transfer study. Am. J. Pathol. 2001, 159, 339–344. [Google Scholar] [CrossRef] [PubMed]
- Tyson, T.; Senchuk, M.; Cooper, J.F.; George, S.; Van Raamsdonk, J.M.; Brundin, P. Novel animal model defines genetic contributions for neuron-to-neuron transfer of alpha-synuclein. Sci. Rep. 2017, 7, 7506. [Google Scholar] [CrossRef] [PubMed]
- Ray, A.; Zhang, S.; Rentas, C.; Caldwell, K.A.; Caldwell, G.A. RTCB-1 mediates neuroprotection via XBP-1 mRNA splicing in the unfolded protein response pathway. J. Neurosci. 2014, 34, 16076–16085. [Google Scholar] [CrossRef] [PubMed]
- Starr, L.A.; McKay, L.E.; Peter, K.N.; Seyfarth, L.M.; Berkowitz, L.A.; Caldwell, K.A.; Caldwell, G.A. Attenuation of Dopaminergic Neurodegeneration in a C. elegans Parkinson’s Model through Regulation of Xanthine Dehydrogenase (XDH-1) Expression by the RNA Editase, ADR-2. J. Dev. Biol. 2023, 11, 20. [Google Scholar] [CrossRef] [PubMed]
- Shamsuzzama; Kumar, L.; Nazir, A. Modulation of Alpha-synuclein Expression and Associated Effects by MicroRNA Let-7 in Transgenic C. elegans. Front. Mol. Neurosci. 2017, 10, 328. [Google Scholar] [CrossRef]
- Bae, J.R.; Lee, B.D. Function and dysfunction of leucine-rich repeat kinase 2 (LRRK2): Parkinson’s disease and beyond. BMB Rep. 2015, 48, 243–248. [Google Scholar] [CrossRef]
- Seegobin, S.P.; Heaton, G.R.; Liang, D.; Choi, I.; Blanca Ramirez, M.; Tang, B.; Yue, Z. Progress in LRRK2-Associated Parkinson’s Disease Animal Models. Front. Neurosci. 2020, 14, 674. [Google Scholar] [CrossRef]
- Sakaguchi-Nakashima, A.; Meir, J.Y.; Jin, Y.; Matsumoto, K.; Hisamoto, N. LRK-1, a C. elegans PARK8-related kinase, regulates axonal-dendritic polarity of SV proteins. Curr. Biol. 2007, 17, 592–598. [Google Scholar] [CrossRef]
- Mata, I.; Salles, P.; Cornejo-Olivas, M.; Saffie, P.; Ross, O.A.; Reed, X.; Bandres-Ciga, S. LRRK2: Genetic mechanisms vs genetic subtypes. Handb. Clin. Neurol. 2023, 193, 133–154. [Google Scholar] [CrossRef]
- Samann, J.; Hegermann, J.; von Gromoff, E.; Eimer, S.; Baumeister, R.; Schmidt, E. Caenorhabditits elegans LRK-1 and PINK-1 act antagonistically in stress response and neurite outgrowth. J. Biol. Chem. 2009, 284, 16482–16491. [Google Scholar] [CrossRef]
- Yao, C.; El Khoury, R.; Wang, W.; Byrd, T.A.; Pehek, E.A.; Thacker, C.; Zhu, X.; Smith, M.A.; Wilson-Delfosse, A.L.; Chen, S.G. LRRK2-mediated neurodegeneration and dysfunction of dopaminergic neurons in a Caenorhabditis elegans model of Parkinson’s disease. Neurobiol. Dis. 2010, 40, 73–81. [Google Scholar] [CrossRef] [PubMed]
- Yao, C.; Johnson, W.M.; Gao, Y.; Wang, W.; Zhang, J.; Deak, M.; Alessi, D.R.; Zhu, X.; Mieyal, J.J.; Roder, H.; et al. Kinase inhibitors arrest neurodegeneration in cell and C. elegans models of LRRK2 toxicity. Hum. Mol. Genet. 2013, 22, 328–344. [Google Scholar] [CrossRef]
- Saha, S.; Ash, P.E.; Gowda, V.; Liu, L.; Shirihai, O.; Wolozin, B. Mutations in LRRK2 potentiate age-related impairment of autophagic flux. Mol. Neurodegener. 2015, 10, 26. [Google Scholar] [CrossRef] [PubMed]
- Chandler, R.J.; Cogo, S.; Lewis, P.A.; Kevei, E. Modelling the functional genomics of Parkinson’s disease in Caenorhabditis elegans: LRRK2 and beyond. Biosci. Rep. 2021, 41, BSR20203672. [Google Scholar] [CrossRef] [PubMed]
- Senchuk, M.M.; Van Raamsdonk, J.M.; Moore, D.J. Multiple genetic pathways regulating lifespan extension are neuroprotective in a G2019S LRRK2 nematode model of Parkinson’s disease. Neurobiol. Dis. 2021, 151, 105267. [Google Scholar] [CrossRef]
- Bae, E.J.; Kim, D.K.; Kim, C.; Mante, M.; Adame, A.; Rockenstein, E.; Ulusoy, A.; Klinkenberg, M.; Jeong, G.R.; Bae, J.R.; et al. LRRK2 kinase regulates alpha-synuclein propagation via RAB35 phosphorylation. Nat. Commun. 2018, 9, 3465. [Google Scholar] [CrossRef] [PubMed]
- Ray, A.; Martinez, B.A.; Berkowitz, L.A.; Caldwell, G.A.; Caldwell, K.A. Mitochondrial dysfunction, oxidative stress, and neurodegeneration elicited by a bacterial metabolite in a C. elegans Parkinson’s model. Cell Death Dis. 2014, 5, e984. [Google Scholar] [CrossRef]
- Antunes, A.; Saia-Cereda, V.M.; Crunfli, F.; Martins-de-Souza, D. 14-3-3 proteins at the crossroads of neurodevelopment and schizophrenia. World J. Biol. Psychiatry 2022, 23, 14–32. [Google Scholar] [CrossRef]
- Morrison, D.K. The 14-3-3 proteins: Integrators of diverse signaling cues that impact cell fate and cancer development. Trends Cell Biol. 2009, 19, 16–23. [Google Scholar] [CrossRef]
- Fan, X.; Cui, L.; Zeng, Y.; Song, W.; Gaur, U.; Yang, M. 14-3-3 Proteins Are on the Crossroads of Cancer, Aging, and Age-Related Neurodegenerative Disease. Int. J. Mol. Sci. 2019, 20, 3518. [Google Scholar] [CrossRef]
- Long, S.; Guo, W.; Hu, S.; Su, F.; Zeng, Y.; Zeng, J.; Tan, E.K.; Ross, C.A.; Pei, Z. G2019S LRRK2 Increases Stress Susceptibility Through Inhibition of DAF-16 Nuclear Translocation in a 14-3-3 Associated-Manner in Caenorhabditis elegans. Front. Neurosci. 2018, 12, 782. [Google Scholar] [CrossRef]
- Johnson, W.M.; Yao, C.; Siedlak, S.L.; Wang, W.; Zhu, X.; Caldwell, G.A.; Wilson-Delfosse, A.L.; Mieyal, J.J.; Chen, S.G. Glutaredoxin deficiency exacerbates neurodegeneration in C. elegans models of Parkinson’s disease. Hum. Mol. Genet. 2015, 24, 1322–1335. [Google Scholar] [CrossRef]
- Grecco, A.; Macchiaroli, N.; Perez, M.G.; Casulli, A.; Cucher, M.A.; Rosenzvit, M.C. microRNA silencing in a whole worm cestode model provides insight into miR-71 function. Int. J. Parasitol. 2023, 53, 699–710. [Google Scholar] [CrossRef] [PubMed]
- Naidoo, D.; de Lencastre, A. Regulation of TIR-1/SARM-1 by miR-71 Protects Dopaminergic Neurons in a C. elegans Model of LRRK2-Induced Parkinson’s Disease. Int. J. Mol. Sci. 2024, 25, 8795. [Google Scholar] [CrossRef]
- Hernandez-Baltazar, D.; Zavala-Flores, L.M.; Villanueva-Olivo, A. The 6-hydroxydopamine model and parkinsonian pathophysiology: Novel findings in an older model. Neurologia 2017, 32, 533–539. [Google Scholar] [CrossRef]
- Chia, S.J.; Tan, E.K.; Chao, Y.X. Historical Perspective: Models of Parkinson’s Disease. Int. J. Mol. Sci. 2020, 21, 2464. [Google Scholar] [CrossRef] [PubMed]
- Harrington, A.J.; Hamamichi, S.; Caldwell, G.A.; Caldwell, K.A. C. elegans as a model organism to investigate molecular pathways involved with Parkinson’s disease. Dev. Dyn. 2010, 239, 1282–1295. [Google Scholar] [CrossRef] [PubMed]
- Hsu, Y.L.; Hung, H.S.; Tsai, C.W.; Liu, S.P.; Chiang, Y.T.; Kuo, Y.H.; Shyu, W.C.; Lin, S.Z.; Fu, R.H. Peiminine Reduces ARTS-Mediated Degradation of XIAP by Modulating the PINK1/Parkin Pathway to Ameliorate 6-Hydroxydopamine Toxicity and alpha-Synuclein Accumulation in Parkinson’s Disease Models In Vivo and In Vitro. Int. J. Mol. Sci. 2021, 22, 10240. [Google Scholar] [CrossRef]
- Offenburger, S.L.; Gartner, A. 6-hydroxydopamine (6-OHDA) Oxidative Stress Assay for Observing Dopaminergic Neuron Loss in Caenorhabditis elegans. Bio-Protocol 2018, 8, e3025. [Google Scholar] [CrossRef]
- Lal, R.; Singh, A.; Watts, S.; Chopra, K. Experimental models of Parkinson’s disease: Challenges and Opportunities. Eur. J. Pharmacol. 2024, 980, 176819. [Google Scholar] [CrossRef]
- Boos, J.; Shubbar, A.; Geldenhuys, W.J. Dual monoamine oxidase B and acetylcholine esterase inhibitors for treating movement and cognition deficits in a C. elegans model of Parkinson’s disease. Med. Chem. Res. 2021, 30, 1166–1174. [Google Scholar] [CrossRef]
- Offenburger, S.L.; Jongsma, E.; Gartner, A. Mutations in Caenorhabditis elegans neuroligin-like glit-1, the apoptosis pathway and the calcium chaperone crt-1 increase dopaminergic neurodegeneration after 6-OHDA treatment. PLoS Genet. 2018, 14, e1007106. [Google Scholar] [CrossRef]
- Offenburger, S.L.; Ho, X.Y.; Tachie-Menson, T.; Coakley, S.; Hilliard, M.A.; Gartner, A. 6-OHDA-induced dopaminergic neurodegeneration in Caenorhabditis elegans is promoted by the engulfment pathway and inhibited by the transthyretin-related protein TTR-33. PLoS Genet. 2018, 14, e1007125. [Google Scholar] [CrossRef]
- Hu, K.; Zhu, S.; Wu, F.; Zhang, Y.; Li, M.; Yuan, L.; Huang, W.; Zhang, Y.; Wang, J.; Ren, J.; et al. Aureusidin ameliorates 6-OHDA-induced neurotoxicity via activating Nrf2/HO-1 signaling pathway and preventing mitochondria-dependent apoptosis pathway in SH-SY5Y cells and Caenorhabditis elegans. Chem. Biol. Interact. 2024, 387, 110824. [Google Scholar] [CrossRef]
- Chen, Q.X.; Zhou, L.; Long, T.; Qin, D.L.; Wang, Y.L.; Ye, Y.; Zhou, X.G.; Wu, J.M.; Wu, A.G. Galangin Exhibits Neuroprotective Effects in 6-OHDA-Induced Models of Parkinson’s Disease via the Nrf2/Keap1 Pathway. Pharmaceuticals 2022, 15, 1014. [Google Scholar] [CrossRef]
- Ren, J.; Yuan, L.; Wang, W.; Zhang, M.; Wang, Q.; Li, S.; Zhang, L.; Hu, K. Tricetin protects against 6-OHDA-induced neurotoxicity in Parkinson’s disease model by activating Nrf2/HO-1 signaling pathway and preventing mitochondria-dependent apoptosis pathway. Toxicol. Appl. Pharmacol. 2019, 378, 114617. [Google Scholar] [CrossRef]
- Morton, K.S.; Hartman, J.H.; Heffernan, N.; Ryde, I.T.; Kenny-Ganzert, I.W.; Meng, L.; Sherwood, D.R.; Meyer, J.N. Chronic high-sugar diet in adulthood protects Caenorhabditis elegans from 6-OHDA-induced dopaminergic neurodegeneration. BMC Biol. 2023, 21, 252. [Google Scholar] [CrossRef]
- Tai, H.; Cui, L.; Shen, D.; Li, D.; Cui, B.; Fang, J. Military service and the risk of amyotrophic lateral sclerosis: A meta-analysis. J. Clin. Neurosci. 2017, 45, 337–342. [Google Scholar] [CrossRef]
- van Es, M.A.; Hardiman, O.; Chio, A.; Al-Chalabi, A.; Pasterkamp, R.J.; Veldink, J.H.; van den Berg, L.H. Amyotrophic lateral sclerosis. Lancet 2017, 390, 2084–2098. [Google Scholar] [CrossRef]
- Eck, R.J.; Stair, J.G.; Kraemer, B.C.; Liachko, N.F. Simple models to understand complex disease: 10 years of progress from Caenorhabditis elegans models of amyotrophic lateral sclerosis and frontotemporal lobar degeneration. Front. Neurosci. 2023, 17, 1300705. [Google Scholar] [CrossRef]
- Smukowski, S.N.; Maioli, H.; Latimer, C.S.; Bird, T.D.; Jayadev, S.; Valdmanis, P.N. Progress in Amyotrophic Lateral Sclerosis Gene Discovery: Reflecting on Classic Approaches and Leveraging Emerging Technologies. Neurol. Genet. 2022, 8, e669. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.; Xu, R. Invertebrate genetic models of amyotrophic lateral sclerosis. Front. Mol. Neurosci. 2024, 17, 1328578. [Google Scholar] [CrossRef]
- Rosen, D.R.; Siddique, T.; Patterson, D.; Figlewicz, D.A.; Sapp, P.; Hentati, A.; Donaldson, D.; Goto, J.; O’Regan, J.P.; Deng, H.X.; et al. Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature 1993, 362, 59–62. [Google Scholar] [CrossRef]
- Bernard, E.; Pegat, A.; Svahn, J.; Bouhour, F.; Leblanc, P.; Millecamps, S.; Thobois, S.; Guissart, C.; Lumbroso, S.; Mouzat, K. Clinical and Molecular Landscape of ALS Patients with SOD1 Mutations: Novel Pathogenic Variants and Novel Phenotypes. A Single ALS Center Study. Int. J. Mol. Sci. 2020, 21, 6807. [Google Scholar] [CrossRef]
- Wei, Q.; Zhou, Q.; Chen, Y.; Ou, R.; Cao, B.; Xu, Y.; Yang, J.; Shang, H.F. Analysis of SOD1 mutations in a Chinese population with amyotrophic lateral sclerosis: A case-control study and literature review. Sci. Rep. 2017, 7, 44606. [Google Scholar] [CrossRef]
- Chen, Y.P.; Yu, S.H.; Wei, Q.Q.; Cao, B.; Gu, X.J.; Chen, X.P.; Song, W.; Zhao, B.; Wu, Y.; Sun, M.M.; et al. Role of genetics in amyotrophic lateral sclerosis: A large cohort study in Chinese mainland population. J. Med. Genet. 2022, 59, 840–849. [Google Scholar] [CrossRef] [PubMed]
- Yamashita, S.; Ando, Y. Genotype-phenotype relationship in hereditary amyotrophic lateral sclerosis. Transl. Neurodegener. 2015, 4, 13. [Google Scholar] [CrossRef] [PubMed]
- Ogawa, M.; Shidara, H.; Oka, K.; Kurosawa, M.; Nukina, N.; Furukawa, Y. Cysteine residues in Cu,Zn-superoxide dismutase are essential to toxicity in Caenorhabditis elegans model of amyotrophic lateral sclerosis. Biochem. Biophys. Res. Commun. 2015, 463, 1196–1202. [Google Scholar] [CrossRef]
- Oeda, T.; Shimohama, S.; Kitagawa, N.; Kohno, R.; Imura, T.; Shibasaki, H.; Ishii, N. Oxidative stress causes abnormal accumulation of familial amyotrophic lateral sclerosis-related mutant SOD1 in transgenic Caenorhabditis elegans. Hum. Mol. Genet. 2001, 10, 2013–2023. [Google Scholar] [CrossRef]
- Xu, H.; Jia, C.; Cheng, C.; Wu, H.; Cai, H.; Le, W. Activation of autophagy attenuates motor deficits and extends lifespan in a C. elegans model of ALS. Free. Radic. Biol. Med. 2022, 181, 52–61. [Google Scholar] [CrossRef]
- Boccitto, M.; Lamitina, T.; Kalb, R.G. Daf-2 signaling modifies mutant SOD1 toxicity in C. elegans. PLoS ONE 2012, 7, e33494. [Google Scholar] [CrossRef]
- Li, J.; Huang, K.X.; Le, W.D. Establishing a novel C. elegans model to investigate the role of autophagy in amyotrophic lateral sclerosis. Acta Pharmacol. Sin. 2013, 34, 644–650. [Google Scholar] [CrossRef] [PubMed]
- Nawa, M.; Kage-Nakadai, E.; Aiso, S.; Okamoto, K.; Mitani, S.; Matsuoka, M. Reduced expression of BTBD10, an Akt activator, leads to motor neuron death. Cell Death Differ. 2012, 19, 1398–1407. [Google Scholar] [CrossRef] [PubMed]
- Jablonski, A.M.; Lamitina, T.; Liachko, N.F.; Sabatella, M.; Lu, J.; Zhang, L.; Ostrow, L.W.; Gupta, P.; Wu, C.Y.; Doshi, S.; et al. Loss of RAD-23 Protects Against Models of Motor Neuron Disease by Enhancing Mutant Protein Clearance. J. Neurosci. 2015, 35, 14286–14306. [Google Scholar] [CrossRef] [PubMed]
- Neumann, M.; Sampathu, D.M.; Kwong, L.K.; Truax, A.C.; Micsenyi, M.C.; Chou, T.T.; Bruce, J.; Schuck, T.; Grossman, M.; Clark, C.M.; et al. Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science 2006, 314, 130–133. [Google Scholar] [CrossRef]
- Arai, T.; Hasegawa, M.; Akiyama, H.; Ikeda, K.; Nonaka, T.; Mori, H.; Mann, D.; Tsuchiya, K.; Yoshida, M.; Hashizume, Y.; et al. TDP-43 is a component of ubiquitin-positive tau-negative inclusions in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Biochem. Biophys. Res. Commun. 2006, 351, 602–611. [Google Scholar] [CrossRef]
- Saldi, T.K.; Ash, P.E.; Wilson, G.; Gonzales, P.; Garrido-Lecca, A.; Roberts, C.M.; Dostal, V.; Gendron, T.F.; Stein, L.D.; Blumenthal, T.; et al. TDP-1, the Caenorhabditis elegans ortholog of TDP-43, limits the accumulation of double-stranded RNA. EMBO J. 2014, 33, 2947–2966. [Google Scholar] [CrossRef]
- Mitra, J.; Guerrero, E.N.; Hegde, P.M.; Liachko, N.F.; Wang, H.; Vasquez, V.; Gao, J.; Pandey, A.; Taylor, J.P.; Kraemer, B.C.; et al. Motor neuron disease-associated loss of nuclear TDP-43 is linked to DNA double-strand break repair defects. Proc. Natl. Acad. Sci. USA 2019, 116, 4696–4705. [Google Scholar] [CrossRef]
- Koopman, M.; Gungordu, L.; Seinstra, R.I.; Nollen, E.A.A. Neuronal overexpression of hTDP-43 in Caenorhabditis elegans impairs motor function. MicroPubl. Biol. 2023, 2023. [Google Scholar] [CrossRef]
- Liachko, N.F.; Saxton, A.D.; McMillan, P.J.; Strovas, T.J.; Keene, C.D.; Bird, T.D.; Kraemer, B.C. Genome wide analysis reveals heparan sulfate epimerase modulates TDP-43 proteinopathy. PLoS Genet. 2019, 15, e1008526. [Google Scholar] [CrossRef]
- Zhang, T.; Hwang, H.Y.; Hao, H.; Talbot, C., Jr.; Wang, J. Caenorhabditis elegans RNA-processing protein TDP-1 regulates protein homeostasis and life span. J. Biol. Chem. 2012, 287, 8371–8382. [Google Scholar] [CrossRef] [PubMed]
- Veriepe, J.; Fossouo, L.; Parker, J.A. Neurodegeneration in C. elegans models of ALS requires TIR-1/Sarm1 immune pathway activation in neurons. Nat. Commun. 2015, 6, 7319. [Google Scholar] [CrossRef] [PubMed]
- Aggad, D.; Veriepe, J.; Tauffenberger, A.; Parker, J.A. TDP-43 toxicity proceeds via calcium dysregulation and necrosis in aging Caenorhabditis elegans motor neurons. J. Neurosci. 2014, 34, 12093–12103. [Google Scholar] [CrossRef] [PubMed]
- Liachko, N.F.; McMillan, P.J.; Guthrie, C.R.; Bird, T.D.; Leverenz, J.B.; Kraemer, B.C. CDC7 inhibition blocks pathological TDP-43 phosphorylation and neurodegeneration. Ann. Neurol. 2013, 74, 39–52. [Google Scholar] [CrossRef] [PubMed]
- Rojas-Prats, E.; Martinez-Gonzalez, L.; Gonzalo-Consuegra, C.; Liachko, N.F.; Perez, C.; Ramirez, D.; Kraemer, B.C.; Martin-Requero, A.; Perez, D.I.; Gil, C.; et al. Targeting nuclear protein TDP-43 by cell division cycle kinase 7 inhibitors: A new therapeutic approach for amyotrophic lateral sclerosis. Eur. J. Med. Chem. 2021, 210, 112968. [Google Scholar] [CrossRef]
- Taylor, L.M.; McMillan, P.J.; Kraemer, B.C.; Liachko, N.F. Tau tubulin kinases in proteinopathy. FEBS J. 2019, 286, 2434–2446. [Google Scholar] [CrossRef] [PubMed]
- Wong, S.Q.; Pontifex, M.G.; Phelan, M.M.; Pidathala, C.; Kraemer, B.C.; Barclay, J.W.; Berry, N.G.; O’Neill, P.M.; Burgoyne, R.D.; Morgan, A. alpha-Methyl-alpha-phenylsuccinimide ameliorates neurodegeneration in a C. elegans model of TDP-43 proteinopathy. Neurobiol. Dis. 2018, 118, 40–54. [Google Scholar] [CrossRef]
- Sin, O.; Michels, H.; Nollen, E.A. Genetic screens in Caenorhabditis elegans models for neurodegenerative diseases. Biochim. Biophys. Acta. 2014, 1842, 1951–1959. [Google Scholar] [CrossRef]
- Lee, H.J.; Alirzayeva, H.; Koyuncu, S.; Rueber, A.; Noormohammadi, A.; Vilchez, D. Cold temperature extends longevity and prevents disease-related protein aggregation through PA28gamma-induced proteasomes. Nat. Aging 2023, 3, 546–566. [Google Scholar] [CrossRef]
- Zhang, T.; Mullane, P.C.; Periz, G.; Wang, J. TDP-43 neurotoxicity and protein aggregation modulated by heat shock factor and insulin/IGF-1 signaling. Hum. Mol. Genet. 2011, 20, 1952–1965. [Google Scholar] [CrossRef]
- Tseng, Y.L.; Lu, P.C.; Lee, C.C.; He, R.Y.; Huang, Y.A.; Tseng, Y.C.; Cheng, T.R.; Huang, J.J.; Fang, J.M. Degradation of neurodegenerative disease-associated TDP-43 aggregates and oligomers via a proteolysis-targeting chimera. J. Biomed. Sci. 2023, 30, 27. [Google Scholar] [CrossRef] [PubMed]
- Bose, P.; Tremblay, E.; Maios, C.; Narasimhan, V.; Armstrong, G.A.B.; Liao, M.; Parker, J.A.; Robitaille, R.; Wen, X.Y.; Barden, C.; et al. Correction to: The Novel Small Molecule TRVA242 Stabilizes Neuromuscular Junction Defects in Multiple Animal Models of Amyotrophic Lateral Sclerosis. Neurotherapeutics 2021, 18, 2128. [Google Scholar] [CrossRef] [PubMed]
- Webster, C.P.; Smith, E.F.; Bauer, C.S.; Moller, A.; Hautbergue, G.M.; Ferraiuolo, L.; Myszczynska, M.A.; Higginbottom, A.; Walsh, M.J.; Whitworth, A.J.; et al. The C9orf72 protein interacts with Rab1a and the ULK1 complex to regulate initiation of autophagy. EMBO J. 2016, 35, 1656–1676. [Google Scholar] [CrossRef] [PubMed]
- Webster, C.P.; Smith, E.F.; Grierson, A.J.; De Vos, K.J. C9orf72 plays a central role in Rab GTPase-dependent regulation of autophagy. Small GTPases 2018, 9, 399–408. [Google Scholar] [CrossRef]
- Aoki, Y.; Manzano, R.; Lee, Y.; Dafinca, R.; Aoki, M.; Douglas, A.G.L.; Varela, M.A.; Sathyaprakash, C.; Scaber, J.; Barbagallo, P.; et al. C9orf72 and RAB7L1 regulate vesicle trafficking in amyotrophic lateral sclerosis and frontotemporal dementia. Brain 2017, 140, 887–897. [Google Scholar] [CrossRef]
- DeJesus-Hernandez, M.; Mackenzie, I.R.; Boeve, B.F.; Boxer, A.L.; Baker, M.; Rutherford, N.J.; Nicholson, A.M.; Finch, N.A.; Flynn, H.; Adamson, J.; et al. Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron 2011, 72, 245–256. [Google Scholar] [CrossRef] [PubMed]
- Ratti, A.; Corrado, L.; Castellotti, B.; Del Bo, R.; Fogh, I.; Cereda, C.; Tiloca, C.; D’Ascenzo, C.; Bagarotti, A.; Pensato, V.; et al. C9ORF72 repeat expansion in a large Italian ALS cohort: Evidence of a founder effect. Neurobiol. Aging 2012, 33, 2528.e7–2528.e14. [Google Scholar] [CrossRef]
- Renton, A.E.; Majounie, E.; Waite, A.; Simon-Sanchez, J.; Rollinson, S.; Gibbs, J.R.; Schymick, J.C.; Laaksovirta, H.; van Swieten, J.C.; Myllykangas, L.; et al. A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron 2011, 72, 257–268. [Google Scholar] [CrossRef]
- Wang, X.; Hao, L.; Saur, T.; Joyal, K.; Zhao, Y.; Zhai, D.; Li, J.; Pribadi, M.; Coppola, G.; Cohen, B.M.; et al. Forward Genetic Screen in Caenorhabditis elegans Suggests F57A10.2 and acp-4 As Suppressors of C9ORF72 Related Phenotypes. Front. Mol. Neurosci. 2016, 9, 113. [Google Scholar] [CrossRef]
- Corrionero, A.; Horvitz, H.R. A C9orf72 ALS/FTD Ortholog Acts in Endolysosomal Degradation and Lysosomal Homeostasis. Curr. Biol. 2018, 28, 1522–1535.e5. [Google Scholar] [CrossRef]
- Therrien, M.; Rouleau, G.A.; Dion, P.A.; Parker, J.A. Deletion of C9ORF72 results in motor neuron degeneration and stress sensitivity in C. elegans. PLoS ONE 2013, 8, e83450. [Google Scholar] [CrossRef] [PubMed]
- Ji, Y.J.; Ugolino, J.; Zhang, T.; Lu, J.; Kim, D.; Wang, J. C9orf72/ALFA-1 controls TFEB/HLH-30-dependent metabolism through dynamic regulation of Rag GTPases. PLoS Genet. 2020, 16, e1008738. [Google Scholar] [CrossRef] [PubMed]
- Beckers, J.; Van Damme, P. Toxic gain-of-function mechanisms in C9orf72 ALS-FTD neurons drive autophagy and lysosome dysfunction. Autophagy 2024, 20, 2102–2104. [Google Scholar] [CrossRef] [PubMed]
- Burberry, A.; Wells, M.F.; Limone, F.; Couto, A.; Smith, K.S.; Keaney, J.; Gillet, G.; van Gastel, N.; Wang, J.Y.; Pietilainen, O.; et al. C9orf72 suppresses systemic and neural inflammation induced by gut bacteria. Nature 2020, 582, 89–94. [Google Scholar] [CrossRef]
- Snoznik, C.; Medvedeva, V.; Mojsilovic-Petrovic, J.; Rudich, P.; Oosten, J.; Kalb, R.G.; Lamitina, T. The nuclear ubiquitin ligase adaptor SPOP is a conserved regulator of C9orf72 dipeptide toxicity. Proc. Natl. Acad. Sci. USA 2021, 118, e2104664118. [Google Scholar] [CrossRef] [PubMed]
- Sonobe, Y.; Aburas, J.; Krishnan, G.; Fleming, A.C.; Ghadge, G.; Islam, P.; Warren, E.C.; Gu, Y.; Kankel, M.W.; Brown, A.E.X.; et al. A C. elegans model of C9orf72-associated ALS/FTD uncovers a conserved role for eIF2D in RAN translation. Nat. Commun. 2021, 12, 6025. [Google Scholar] [CrossRef]
- Lu, J.; Periz, G.; Lu, Y.N.; Tang, Q.; Liu, Y.; Zhang, T.; Shah, Y.; Thombre, R.; Aljumaah, R.; Li, W.; et al. L3MBTL1 regulates ALS/FTD-associated proteotoxicity and quality control. Nat. Neurosci. 2019, 22, 875–886. [Google Scholar] [CrossRef]
- Lagier-Tourenne, C.; Cleveland, D.W. Rethinking ALS: The FUS about TDP-43. Cell 2009, 136, 1001–1004. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, J.C.; Ebmeier, C.C.; Podell, E.R.; Heimiller, J.; Taatjes, D.J.; Cech, T.R. FUS binds the CTD of RNA polymerase II and regulates its phosphorylation at Ser2. Genes Dev. 2012, 26, 2690–2695. [Google Scholar] [CrossRef]
- Tan, A.Y.; Riley, T.R.; Coady, T.; Bussemaker, H.J.; Manley, J.L. TLS/FUS (translocated in liposarcoma/fused in sarcoma) regulates target gene transcription via single-stranded DNA response elements. Proc. Natl. Acad. Sci. USA 2012, 109, 6030–6035. [Google Scholar] [CrossRef]
- Murakami, T.; Yang, S.P.; Xie, L.; Kawano, T.; Fu, D.; Mukai, A.; Bohm, C.; Chen, F.; Robertson, J.; Suzuki, H.; et al. ALS mutations in FUS cause neuronal dysfunction and death in Caenorhabditis elegans by a dominant gain-of-function mechanism. Hum. Mol. Genet. 2012, 21, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Kino, Y.; Washizu, C.; Aquilanti, E.; Okuno, M.; Kurosawa, M.; Yamada, M.; Doi, H.; Nukina, N. Intracellular localization and splicing regulation of FUS/TLS are variably affected by amyotrophic lateral sclerosis-linked mutations. Nucleic Acids Res. 2011, 39, 2781–2798. [Google Scholar] [CrossRef] [PubMed]
- Lorenzo-Betancor, O.; Ogaki, K.; Soto-Ortolaza, A.; Labbe, C.; Vilarino-Guell, C.; Rajput, A.; Rajput, A.H.; Pastor, P.; Ortega, S.; Lorenzo, E.; et al. Analysis of nuclear export sequence regions of FUS-Related RNA-binding proteins in essential tremor. PLoS ONE 2014, 9, e111989. [Google Scholar] [CrossRef] [PubMed]
- Tyzack, G.E.; Luisier, R.; Taha, D.M.; Neeves, J.; Modic, M.; Mitchell, J.S.; Meyer, I.; Greensmith, L.; Newcombe, J.; Ule, J.; et al. Widespread FUS mislocalization is a molecular hallmark of amyotrophic lateral sclerosis. Brain 2019, 142, 2572–2580. [Google Scholar] [CrossRef]
- Ishigaki, S.; Sobue, G. Importance of Functional Loss of FUS in FTLD/ALS. Front. Mol. Biosci. 2018, 5, 44. [Google Scholar] [CrossRef] [PubMed]
- Birsa, N.; Bentham, M.P.; Fratta, P. Cytoplasmic functions of TDP-43 and FUS and their role in ALS. Semin. Cell Dev. Biol. 2020, 99, 193–201. [Google Scholar] [CrossRef] [PubMed]
- Xiao, X.; Li, M.; Ye, Z.; He, X.; Wei, J.; Zha, Y. FUS gene mutation in amyotrophic lateral sclerosis: A new case report and systematic review. Amyotroph. Lateral Scler. Front. Degener. 2024, 25, 1–15. [Google Scholar] [CrossRef]
- Labarre, A.; Tossing, G.; Maios, C.; Doyle, J.J.; Parker, J.A. A single copy transgenic mutant FUS strain reproduces age-dependent ALS phenotypes in C. elegans. MicroPubl. Biol. 2021, 2021. [Google Scholar] [CrossRef]
- Therrien, M.; Dion, P.A.; Rouleau, G.A. ALS: Recent Developments from Genetics Studies. Curr. Neurol. Neurosci. Rep. 2016, 16, 59. [Google Scholar] [CrossRef]
- Zhang, T.; Wu, Y.C.; Mullane, P.; Ji, Y.J.; Liu, H.; He, L.; Arora, A.; Hwang, H.Y.; Alessi, A.F.; Niaki, A.G.; et al. FUS Regulates Activity of MicroRNA-Mediated Gene Silencing. Mol. Cell 2018, 69, 787–801.e8. [Google Scholar] [CrossRef]
- Taylor, M.; Marx, O.; Norris, A. TDP-1 and FUST-1 co-inhibit exon inclusion and control fertility together with transcriptional regulation. bioRxiv 2023, 51, 9610–9628. [Google Scholar] [CrossRef] [PubMed]
- Baskoylu, S.N.; Chapkis, N.; Unsal, B.; Lins, J.; Schuch, K.; Simon, J.; Hart, A.C. Disrupted autophagy and neuronal dysfunction in C. elegans knockin models of FUS amyotrophic lateral sclerosis. Cell Rep. 2022, 38, 110195. [Google Scholar] [CrossRef] [PubMed]
- Wong, C.H.; Rahat, A.; Chang, H.C. Fused in sarcoma regulates glutamate signaling and oxidative stress response. Free. Radic. Biol. Med. 2024, 210, 172–182. [Google Scholar] [CrossRef] [PubMed]
- Markert, S.M.; Skoruppa, M.; Yu, B.; Mulcahy, B.; Zhen, M.; Gao, S.; Sendtner, M.; Stigloher, C. Overexpression of an ALS-associated FUS mutation in C. elegans disrupts NMJ morphology and leads to defective neuromuscular transmission. Biol. Open 2020, 9, bio055129. [Google Scholar] [CrossRef] [PubMed]
- Alirzayeva, H.; Loureiro, R.; Koyuncu, S.; Hommen, F.; Nabawi, Y.; Zhang, W.H.; Dao, T.T.P.; Wehrmann, M.; Lee, H.J.; Vilchez, D. ALS-FUS mutations cause abnormal PARylation and histone H1.2 interaction, leading to pathological changes. Cell Rep. 2024, 43, 114626. [Google Scholar] [CrossRef] [PubMed]
- Wexler, N.S.; Collett, L.; Wexler, A.R.; Rawlins, M.D.; Tabrizi, S.J.; Douglas, I.; Smeeth, L.; Evans, S.J. Incidence of adult Huntington’s disease in the UK: A UK-based primary care study and a systematic review. BMJ Open 2016, 6, e009070. [Google Scholar] [CrossRef]
- Borrell-Pages, M.; Zala, D.; Humbert, S.; Saudou, F. Huntington’s disease: From huntingtin function and dysfunction to therapeutic strategies. Cell. Mol. Life Sci. 2006, 63, 2642–2660. [Google Scholar] [CrossRef]
- Ajitkumar, A.; De Jesus, O. Huntington Disease. In StatPearls; StatPearls: Treasure Island, FL, USA, 2024. [Google Scholar]
- Jiang, A.; Handley, R.R.; Lehnert, K.; Snell, R.G. From Pathogenesis to Therapeutics: A Review of 150 Years of Huntington’s Disease Research. Int. J. Mol. Sci. 2023, 24, 13021. [Google Scholar] [CrossRef]
- Rudich, P.; Watkins, S.; Lamitina, T. PolyQ-independent toxicity associated with novel translational products from CAG repeat expansions. PLoS ONE 2020, 15, e0227464. [Google Scholar] [CrossRef]
- Faber, P.W.; Alter, J.R.; MacDonald, M.E.; Hart, A.C. Polyglutamine-mediated dysfunction and apoptotic death of a Caenorhabditis elegans sensory neuron. Proc. Natl. Acad. Sci. USA 1999, 96, 179–184. [Google Scholar] [CrossRef]
- Koyuncu, S.; Fatima, A.; Gutierrez-Garcia, R.; Vilchez, D. Proteostasis of Huntingtin in Health and Disease. Int. J. Mol. Sci. 2017, 18, 1568. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Gomez-Pastor, R. HSF1 and Its Role in Huntington’s Disease Pathology. Adv. Exp. Med. Biol. 2023, 1410, 35–95. [Google Scholar] [CrossRef] [PubMed]
- Sinnige, T.; Yu, A.; Morimoto, R.I. Challenging Proteostasis: Role of the Chaperone Network to Control Aggregation-Prone Proteins in Human Disease. Adv. Exp. Med. Biol. 2020, 1243, 53–68. [Google Scholar] [CrossRef] [PubMed]
- Jia, K.; Hart, A.C.; Levine, B. Autophagy genes protect against disease caused by polyglutamine expansion proteins in Caenorhabditis elegans. Autophagy 2007, 3, 21–25. [Google Scholar] [CrossRef] [PubMed]
- Farina, F.; Lambert, E.; Commeau, L.; Lejeune, F.X.; Roudier, N.; Fonte, C.; Parker, J.A.; Boddaert, J.; Verny, M.; Baulieu, E.E.; et al. The stress response factor daf-16/FOXO is required for multiple compound families to prolong the function of neurons with Huntington’s disease. Sci. Rep. 2017, 7, 4014. [Google Scholar] [CrossRef]
- Luthi-Carter, R.; Taylor, D.M.; Pallos, J.; Lambert, E.; Amore, A.; Parker, A.; Moffitt, H.; Smith, D.L.; Runne, H.; Gokce, O.; et al. SIRT2 inhibition achieves neuroprotection by decreasing sterol biosynthesis. Proc. Natl. Acad. Sci. USA 2010, 107, 7927–7932. [Google Scholar] [CrossRef]
- Duan, W. Targeting sirtuin-1 in Huntington’s disease: Rationale and current status. CNS Drugs 2013, 27, 345–352. [Google Scholar] [CrossRef]
- Chondrogianni, N.; Georgila, K.; Kourtis, N.; Tavernarakis, N.; Gonos, E.S. 20S proteasome activation promotes life span extension and resistance to proteotoxicity in Caenorhabditis elegans. FASEB J. 2015, 29, 611–622. [Google Scholar] [CrossRef]
- Koyuncu, S.; Saez, I.; Lee, H.J.; Gutierrez-Garcia, R.; Pokrzywa, W.; Fatima, A.; Hoppe, T.; Vilchez, D. The ubiquitin ligase UBR5 suppresses proteostasis collapse in pluripotent stem cells from Huntington’s disease patients. Nat. Commun. 2018, 9, 2886. [Google Scholar] [CrossRef]
- Vazquez-Manrique, R.P.; Farina, F.; Cambon, K.; Dolores Sequedo, M.; Parker, A.J.; Millan, J.M.; Weiss, A.; Deglon, N.; Neri, C. AMPK activation protects from neuronal dysfunction and vulnerability across nematode, cellular and mouse models of Huntington’s disease. Hum. Mol. Genet. 2016, 25, 1043–1058. [Google Scholar] [CrossRef]
- Hegde, R.N.; Chiki, A.; Petricca, L.; Martufi, P.; Arbez, N.; Mouchiroud, L.; Auwerx, J.; Landles, C.; Bates, G.P.; Singh-Bains, M.K.; et al. TBK1 phosphorylates mutant Huntingtin and suppresses its aggregation and toxicity in Huntington’s disease models. EMBO J. 2020, 39, e104671. [Google Scholar] [CrossRef] [PubMed]
- Machiela, E.; Rudich, P.D.; Traa, A.; Anglas, U.; Soo, S.K.; Senchuk, M.M.; Van Raamsdonk, J.M. Targeting Mitochondrial Network Disorganization is Protective in C. elegans Models of Huntington’s Disease. Aging Dis. 2021, 12, 1753–1772. [Google Scholar] [CrossRef] [PubMed]
- Traa, A.; Machiela, E.; Rudich, P.D.; Soo, S.K.; Senchuk, M.M.; Van Raamsdonk, J.M. Identification of Novel Therapeutic Targets for Polyglutamine Diseases That Target Mitochondrial Fragmentation. Int. J. Mol. Sci. 2021, 22, 13447. [Google Scholar] [CrossRef]
- Querfurth, H.; Lee, H.K. Mammalian/mechanistic target of rapamycin (mTOR) complexes in neurodegeneration. Mol. Neurodegener. 2021, 16, 44. [Google Scholar] [CrossRef] [PubMed]
- Gidalevitz, T.; Ben-Zvi, A.; Ho, K.H.; Brignull, H.R.; Morimoto, R.I. Progressive disruption of cellular protein folding in models of polyglutamine diseases. Science 2006, 311, 1471–1474. [Google Scholar] [CrossRef] [PubMed]
- Auluck, P.K.; Chan, H.Y.; Trojanowski, J.Q.; Lee, V.M.; Bonini, N.M. Chaperone suppression of alpha-synuclein toxicity in a Drosophila model for Parkinson’s disease. Science 2002, 295, 865–868. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, R.; Tabrizi, S.J. Huntington disease. Handb. Clin. Neurol. 2018, 147, 255–278. [Google Scholar] [CrossRef] [PubMed]
- Ravikumar, B.; Vacher, C.; Berger, Z.; Davies, J.E.; Luo, S.; Oroz, L.G.; Scaravilli, F.; Easton, D.F.; Duden, R.; O’Kane, C.J.; et al. Inhibition of mTOR induces autophagy and reduces toxicity of polyglutamine expansions in fly and mouse models of Huntington disease. Nat. Genet. 2004, 36, 585–595. [Google Scholar] [CrossRef]
- Park, K.W.; Li, L. Cytoplasmic expression of mouse prion protein causes severe toxicity in Caenorhabditis elegans. Biochem. Biophys. Res. Commun. 2008, 372, 697–702. [Google Scholar] [CrossRef]
- Sandhof, C.A.; Hoppe, S.O.; Tittelmeier, J.; Nussbaum-Krammer, C. C. elegans Models to Study the Propagation of Prions and Prion-Like Proteins. Biomolecules 2020, 10, 1188. [Google Scholar] [CrossRef]
- Nussbaum-Krammer, C.I.; Park, K.W.; Li, L.; Melki, R.; Morimoto, R.I. Spreading of a prion domain from cell-to-cell by vesicular transport in Caenorhabditis elegans. PLoS Genet. 2013, 9, e1003351. [Google Scholar] [CrossRef] [PubMed]
- Lackie, R.E.; Maciejewski, A.; Ostapchenko, V.G.; Marques-Lopes, J.; Choy, W.Y.; Duennwald, M.L.; Prado, V.F.; Prado, M.A.M. The Hsp70/Hsp90 Chaperone Machinery in Neurodegenerative Diseases. Front. Neurosci. 2017, 11, 254. [Google Scholar] [CrossRef]
- Abdulrahman, B.A.; Tahir, W.; Doh-Ura, K.; Gilch, S.; Schatzl, H.M. Combining autophagy stimulators and cellulose ethers for therapy against prion disease. Prion 2019, 13, 185–196. [Google Scholar] [CrossRef] [PubMed]
- Donnelly, K.M.; Coleman, C.M.; Fuller, M.L.; Reed, V.L.; Smerina, D.; Tomlinson, D.S.; Pearce, M.M.P. Hunting for the cause: Evidence for prion-like mechanisms in Huntington’s disease. Front. Neurosci. 2022, 16, 946822. [Google Scholar] [CrossRef]
- Vana, K.; Zuber, C.; Pflanz, H.; Kolodziejczak, D.; Zemora, G.; Bergmann, A.K.; Weiss, S. LRP/LR as an alternative promising target in therapy of prion diseases, Alzheimer’s disease and cancer. Infect. Disord. Drug Targets 2009, 9, 69–80. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xu, R.; Kang, Q.; Yang, X.; Yi, P.; Zhang, R. Unraveling Molecular Targets for Neurodegenerative Diseases Through Caenorhabditis elegans Models. Int. J. Mol. Sci. 2025, 26, 3030. https://doi.org/10.3390/ijms26073030
Xu R, Kang Q, Yang X, Yi P, Zhang R. Unraveling Molecular Targets for Neurodegenerative Diseases Through Caenorhabditis elegans Models. International Journal of Molecular Sciences. 2025; 26(7):3030. https://doi.org/10.3390/ijms26073030
Chicago/Turabian StyleXu, Rongmei, Qiaoju Kang, Xuefei Yang, Ping Yi, and Rongying Zhang. 2025. "Unraveling Molecular Targets for Neurodegenerative Diseases Through Caenorhabditis elegans Models" International Journal of Molecular Sciences 26, no. 7: 3030. https://doi.org/10.3390/ijms26073030
APA StyleXu, R., Kang, Q., Yang, X., Yi, P., & Zhang, R. (2025). Unraveling Molecular Targets for Neurodegenerative Diseases Through Caenorhabditis elegans Models. International Journal of Molecular Sciences, 26(7), 3030. https://doi.org/10.3390/ijms26073030