Abstract
The inner ear and/or lateral line are responsible for hearing and balance of vertebrate. The otic sensory hair cells (HCs) employ cilium organelles, namely stereocilia and/or kinocilia, to mediate mechanical stimuli to electrical signal transition. Tektins (Tekts) are known as the cilium microtubule stabilizer and inner-space filler, and four Tekt(1-4)-encoding genes are identified in zebrafish HCs, but the subcellular location of Tekts in HCs remains unknown. In the present study, we first found that tekt3 is expressed in the inner ear and lateral line neuromast. Antibody staining revealed that Tekt3 is present in neuromast and utricular HCs. It is absent in the saccule, the authentic hearing end-organ of zebrafish and the crista of semi-circular canals. Furthermore, Tekt3 were enriched at the apical side of neuromast and utricular HCs, mainly in the cytosol. Similar subcellular distribution of Tekt3 was also evident in the outer HCs of mature mouse cochlea, which are not directly linked to the hearing sense. However, only neuromast HCs exerted morphological defect of kinocilia in tekt3 mutant. The disrupted or distorted HC kinocilia of mutant neuromast ultimately resulted in slower vital dye intake, delayed HC regeneration after neomycin treatment, and reduced startle response to vibration stimulation. All functional defects of tekt3 mutant were largely rescued by wild-type tekt3 mRNA. Our study thus suggests that zebrafish Tekt3 maintains the integrity and function of neuromast kinocilia to against surrounding and persistent low-frequency noises, perhaps via the intracellular distribution of Tekt3. Nevertheless, TEKT3/Tekt3 could be used to clarify HC sub-types in both zebrafish and mice, to highlight the non-hearing HCs.
1. Introduction
Vertebrates’ sense of hearing and balance rely on the function of mechanosensory receptors (hair cells, HCs) in the inner ear and/or lateral line. HCs employ the actin-filled stereocilia and/or kinocilia to detect physical stimuli such as soundwaves and vibration and transduce them into nerve signals via opening mechanoelectrical transducer (MET) channels. HC kinocilium is a single non-motile cilium, also called the true cilium. The kinocilia are different from non-motile primary cilia and motile cilia/flagella in that each kinocilium possesses the 9+2 axonemal MTs yet it is non-motile, due to a lack of inner dynein arms and nexin links [1,2]. Among vertebrate HCs, there is a great diversity in kinocilial morphology—from the transient kinocilia of cochlear HCs, the bulbed kinocilia of amphibian HCs, to the towering kinocilia of lateral line neuromasts [2,3]. However, little is known about the molecular bases of this morphological diversity and if functional diversity of these different shaped kinocilia also exists. Furthermore, since the mature vestibular HCs of all vertebrates possess both kinocilia and stereocilia, while mature cochlear HCs do not have any kinocilia, the roles of kinocilia played in the cochlear vs. vestibular HC differentiation and during the gain of hearing warrant in depth exploration.
Tektins (Tekts) are a highly conserved family of coiled-coil domain-containing proteins. They are known to play a role in the structure, stability, and function of cilia and flagella [4]. Tekts form filaments to run the length of the axoneme along the inner surface of the A tubule of each ‘9+2’ microtubule (MT) doublet. For motile cilia and flagella of both unicellular and multicellular organisms, enabling the movement of axoneme is one of the common features of Tekts. The members of Tekt family, ranging from one to up to five in each species, are all derived from an ancestral Tekt member. Evolutionarily, tektin1, 2 and 4 encoding genes are usually co-regulated to show similar spatial and temporal expression patterns. The expression of Tekt3 or 5 encoding (3/5) gene appears in species-specific cell types including the sensory cells. For instance, sea urchin tektin filaments are composed of hetero- and homodimers of Tekt1, 2 and 4. Recent structural analyses reveal a complex arrangement of filaments made of all tektins, including Tekt3/5 in bovine respiratory cilia and mammalian and sea urchin sperm flagella [5,6,7,8]. In general, Tekt1, 2 and 4 appear to form a conserved core of filaments while species-specific Tekt3/5 are added depending on species and cell types. Furthermore, the presence or absence of Tekt3/5 is likely a key factor in differentiating cilia type and function, between sensory cell (with multiciliary arrays) and motile cilia of the other ciliated structures. The complex composition and decoration of tektin filaments within the axoneme may contribute to cilia diversity and function, most likely by fine-tuning the stability of the axoneme to withstand mechanical and beating forces. However, little is known about the role of tektins, particularly the Tekt3 that is specifically expressed in zebrafish HCs [9,10,11] and associated with nodal and olfactory cilia in functional knockdown experiment [12].
Previous studies indicate that mutations of individual tektins lead to structural and functional defects of cilia and flagella in green algae, zebrafish, rodents, and humans [13,14,15,16,17,18,19,20]. In humans, all tektins, including TEKT3, are found in the spermatozoa, mainly localized to the flagella, and present in the respiratory doublet microtubules. In rodent species, TEKT3 is found in the sperm/spermatozoa flagella, head, and acrosome membrane, and Tekt3 KO mouse sperms exert reduced motility and forward progression and increased flagellar bending. Point mutations in patients with a complete loss of TEKT3 expression causes the acrosomal hypoplasia with no apparent defects in sperm flagellar but decreased progressive motility of their sperms [14]. However, all these studies focus on sperm and male infertility, whether these mammalian mutants have any hearing and vestibular defects is yet to be reported.
The zebrafish genome contains four tektin-coding genes (1–4), and these genes, from tekt1 to tekt4, are expressed in otic and/or neuromast HCs [9,10,11,21]. Interestingly, we have found that during HC development and regeneration of larval zebrafish, only tekt3 expression was kept in mature HCs. To understand the role of tekt3 in HC development and function, the present study obtained a loss-of-function zebrafish mutant, via CRISPER/Cas9 technique, that developed normally and was fertile. Tekt3 was inclusively expressed in utricular and neuromast, present and enriched in the cytosol at the apical end of HCs. Such a TEKT3 distribution was also found in the outer HCs of mouse cochlea. However, only neuromast HCs of tekt3 mutant exerted clustered, bent and/or bubbled kinocilia. Functional defects included vital dye intake, delayed HC regeneration after neomycin was used to kill lateral line HCs, and reduced startle response to vibration stimulation. On the other hand, the normal VOR score of tekt3 mutant indicated that utricular HC development and function was not affected, suggesting that zebrafish Tekt3 is involved in a protective mechanism to enable structural stability of HC kinocilia that are constantly in action.
2. Results
2.1. Zebrafish Tekt3 Is Specifically Expressed in the Ear and Neuromast
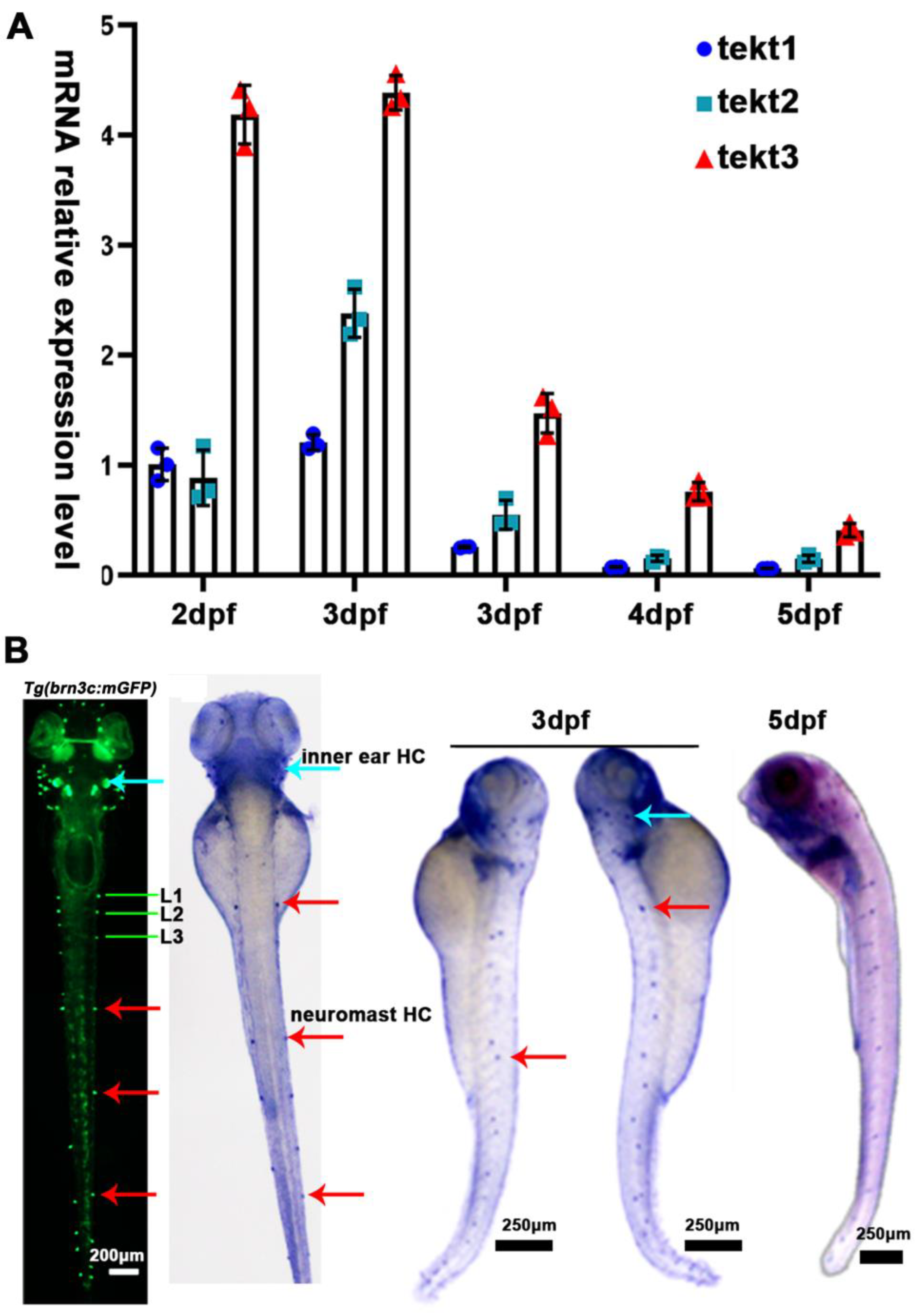
Using Neighbor-Joining method, we compared the amino acid sequences of zebrafish and mammalian tektin3-coding genes to indicate the conservation of tekt3 (Figure 1A,B). Whole body qPCR analyses revealed that the tekt3 mRNA level was much higher than tekt1 and tekt2 mRNA between 2 and 6 dpf (Figure 2A). Whole-mount in situ hybridization revealed that tekt3 was exclusively expressed in the inner ear and lateral line neuromast at 3 and 5 dpf (Figure 2B). By analyzing the available single-cell RNA sequencing data [21], tekt3 mRNA was indeed found to be only expressed in mature neuromast HCs, while tekt1 and tekt2 were less abundant (Figure S1).

Figure 1.
TEKT3/Tekt3 is evolutionarily conserved. (A) Amino acid sequences of zebrafish and a few mammalian TEKT3/Tekt3 are aligned, and the resulted phylogenetic tree (B) reveals that zebrafish Tekt3 is older than human TEKT3 in evolution. Zebrafish (Danio rerio) is highlighted in red color in (A,B).
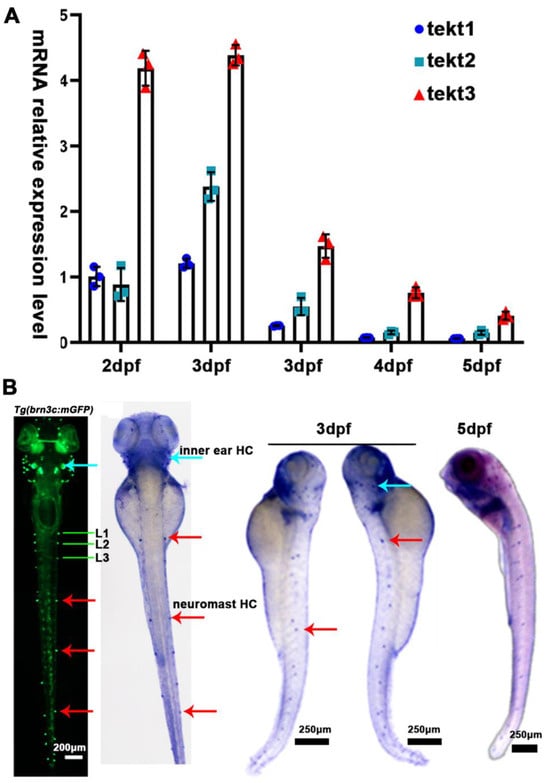
Figure 2.
tekt3 mRNA is specifically present in zebrafish HC-bearing organs. (A) The expression profiles of zebrafish tekt1-3 between 2 and 6 dpf. (B) Expression patterns of tekt3 were detected by the whole-mount in situ hybridization at 3 and 5 dpf. Red arrows point to neuromasts and light blue arrows point to the ear. HC: the hair cell.
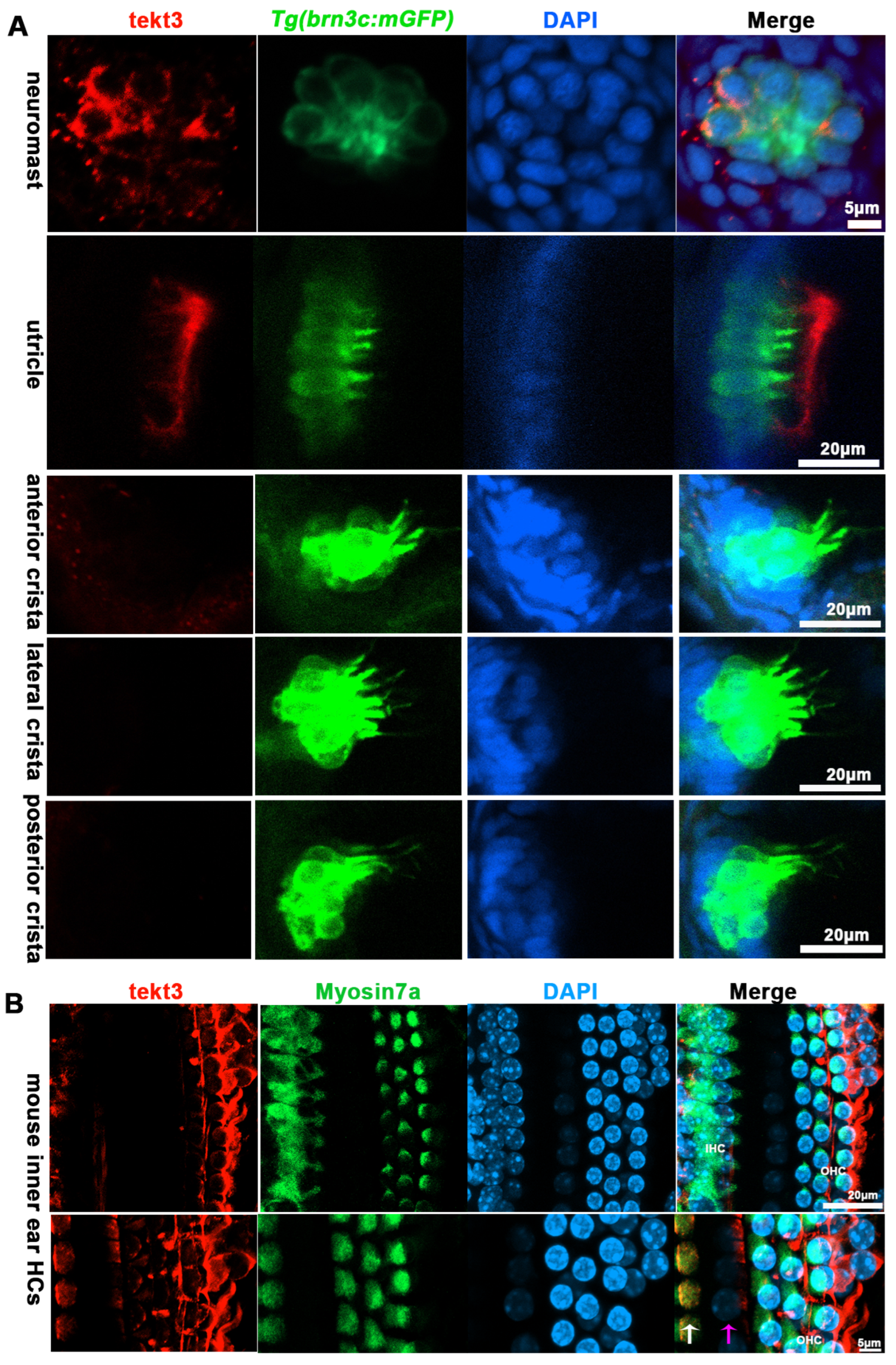
Using a tektin3-specific antibody, the immunostaining showed that tekt3 was present in the cytosol of neuromast HCs and the main signal was enriched apically in utricular HCs at 5 dpf (Figure 3A). Interestingly, a barrier plate-like structure, presumably the otolithic membrane (OTM) of the zebrafish’s inner ear, was intensively stained. Furthermore, the OTM-like structure staining did not overlap with the kinocilium-GFP cells but did with some cytosol GFP signals, suggesting the absence of tektin3 in utricular kinocilia and stereocilia. When the utricle HC image was blown up, the cytosolic co-localization of both green and red signals was clearly evident at the apical surface (Figure 4). In mouse cochlea, tektin3 was present in the cytosol and apically enriched in outer HCs (Figure 3B), indicating the conserved subcellular localization. Tektin3 was absent in saccular and crista HCs (Figure 3A). Therefore, tekt3 expression is quite specific at the young larval stage of zebrafish and tekt3 is mainly present in the cytosol of HCs, and could therefore be used to distinguish sub-types of HCs in both zebrafish and mice.
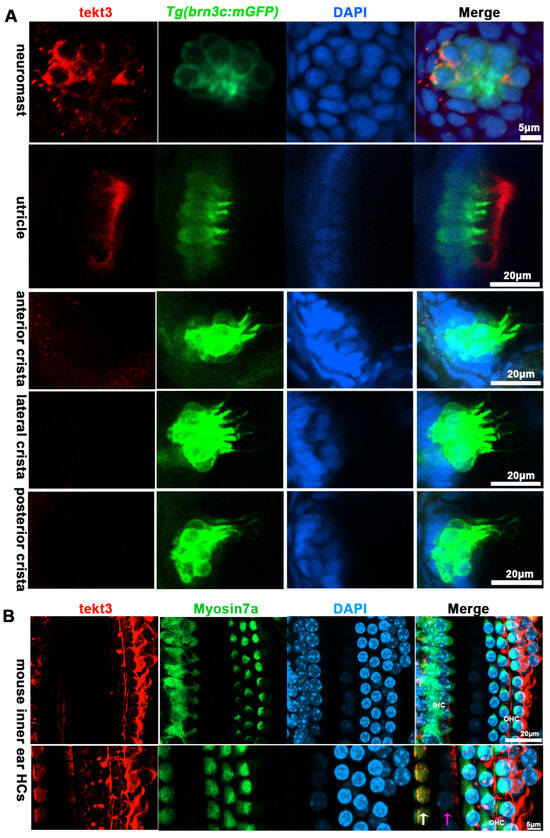
Figure 3.
TEKT3/Tekt3 is present in some HCs. (A) Neuromast and utricular HCs pf zebrafish larvae at 5 dpf are positive for anti-Tekt3 antibody staining while the crista HCs are negative. (B) TEKT3 is present in mature mouse cochlear HCs. The left row of cells are inner HCs (IHC, pointed by a white arrow) and the right three rows are outer HCs (pillar cell, pointed by a pink arrow). Note: The saccular and crista HCs are always negative.
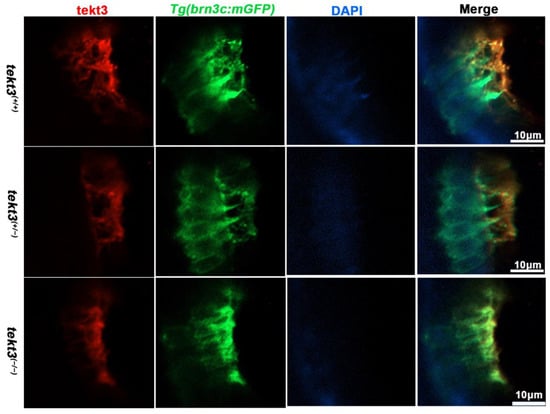
Figure 4.
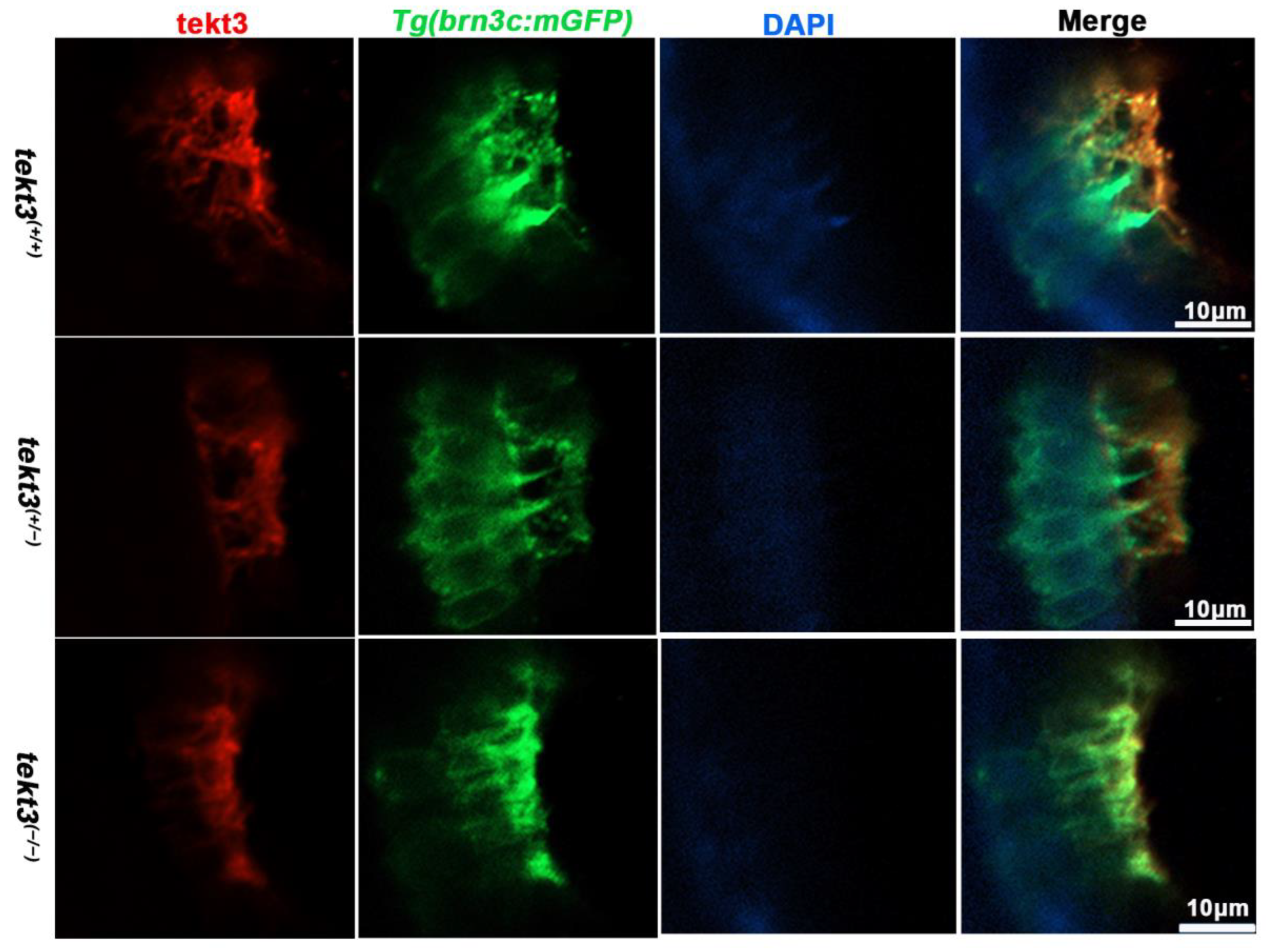
The subcellular location of Tekt3 in zebrafish utricular HCs. Crossed with Tg(brn3c:mGFP), both homozygous and heterozygous tekt3 mutants showed weaker α-Tektin3 staining at the utricle (apical portion of HCs). Note: two colors overlap at the very tip portion of hair bundles, yet at the root portion, they never overlap.
2.2. Disruption of Tekt3 Triggers Genetic Compensation to Boost Total Tektins
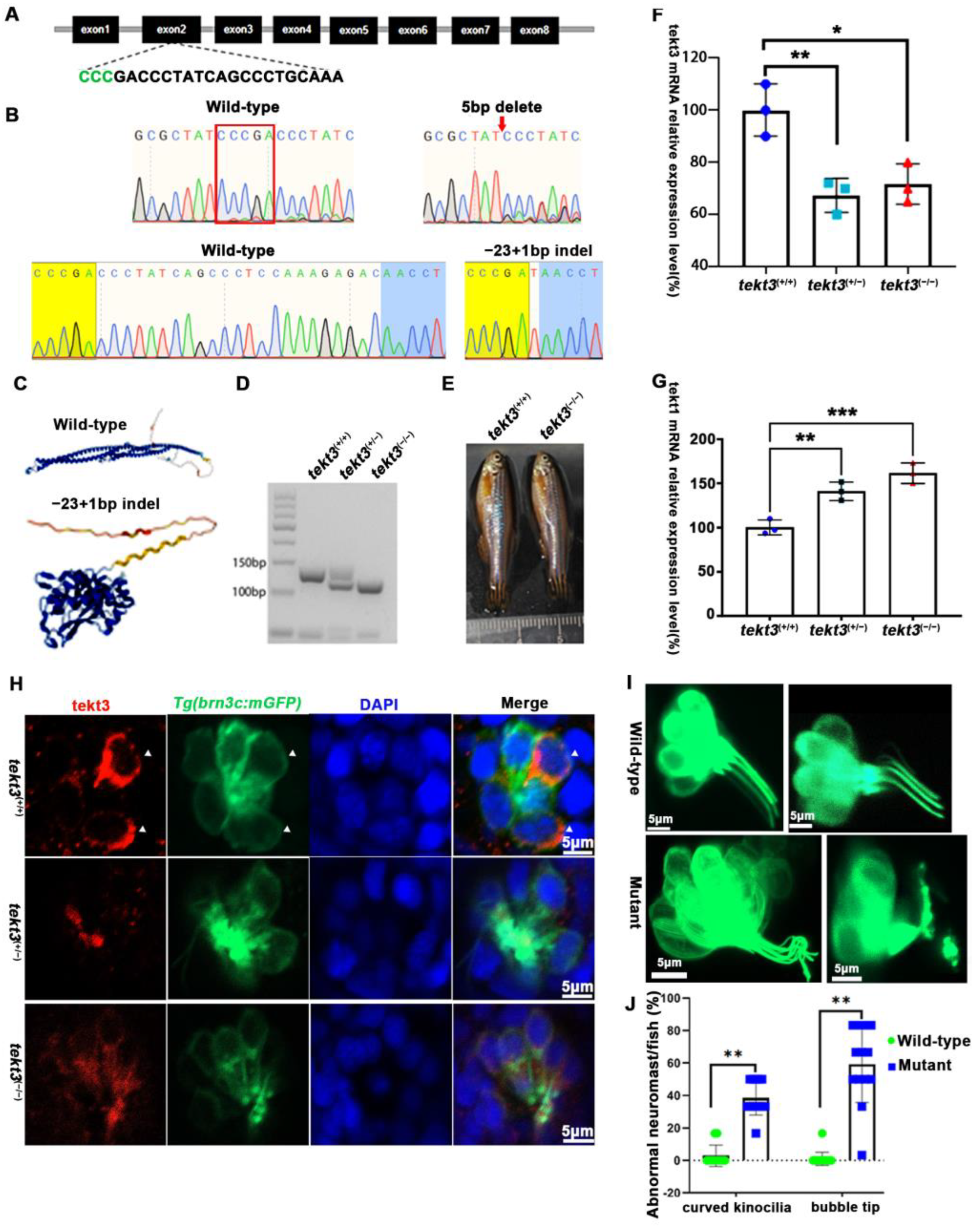
To better understand the role of tektin3, two tekt3 mutant lines were obtained (Figure 5A,B). One (indel, +1–23 bp) was most likely a loss-of-function mutant allele (Figure 5C) while the other one (indel, −5 bp) was not. tekt3 mutant (+1−23) could be easily genotyped via agarose gel electrophoresis (Figure 5D). And the homozygous mutant was vital and fertile (Figure 5E). By qPCR analyses, tekt1 was found to be upregulated in both heterozygous and homozygous mutants, largely due to some genetic compensation mechanisms, whereas tekt3 expression was drastically reduced (Figure 5F,G). The homozygous mutant possessed stronger genetic compensation than that of heterozygous mutant. Genetic compensation is crucial for the genetic robustness and could be achieved by functionally redundant genes [22]. The elevation of tekt1 expression might contribute to the normal fertility of the tekt3 mutant.
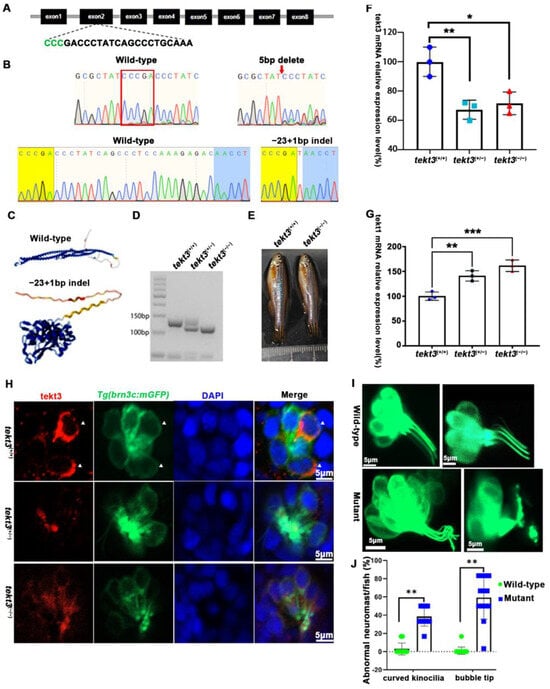
Figure 5.
tekt3 mutant exhibited only neuromast HC kinocilium defect. (A) The CRISPR/Cas9-mediated genome editing targeted the exon 2 of tekt3 locus; the gRNA core sequence is shown. (B) Two tekt3 mutant alleles were verified by sequencing. (C) The (Alphfold2.0) predicted protein structure of one mutated Tekt3 is shown side by side with a normal protein. (D) A typical genotyping result of tekt3 mutant. (E) A 6 mpf homozygous tekt3 mutant looked normal. (F) Significant reduction in tekt3 mRNA level can be seen in both homozygous and heterozygous tekt3 mutants. (G) The tekt1 mRNA level in 5 dpf fish were significantly up in both homozygous and heterozygous tekt3 mutants. (H) Tekt3 was decreased in neuromast HCs of both homozygous and heterozygous tekt3 mutants (5 dpf). The arrowheads are pointed to tekt3 positive cells. (I) The abnormal neuromast HC hair bundle or kinocilia of the 5 dpf tekt3 mutant. (J) Almost all mutant neuromasts (n = 78) showed abnormal kinoclium tips while wild-type (n = 112) rarely possessed the defect. * p < 0.05; ** p < 0.01; and *** p < 0.001.
To learn if the mutant is truly a loss-of-function allele, the tekt3 mutant was crossed into transgenic Tg(brn3c:mGFP) fish, in which both otic and neuromast HCs can be visualized under a fluorescence microscope. No obvious defect was evident in saccular and crista HCs (Figure S2). In contrast, utricular HCs of the tekt3 mutant lacked distinct knicilium structure and lower Tekt3 level (Figure 4). Mutant neuromast HCs also produced much less Tekt3 and their kinocilia were curved/bent with or without a bubbled tip (Figure 5H,I). To quantify the mutant phenotype, we counted the number of abnormal neuromasts (L1–L3/fish) at 5 dpf. Among 17 wild-type fish and 112 neuromasts, only two neuromasts showed slightly bent hair bundles and one carried a tiny bubble at the tip of the kinocilia (Figure S3). The curved kinocilium tips, kinocilia with bubbled tips, and hair bundles mixed with both types of kinocilia were almost exclusively present in 78 tekt3 mutant neuromasts (Figure 5J). Moreover, Tekt3 was largely enriched apically in neuromast HCs (Figure 5H).
2.3. A Lack of Tekt3 Leads to Neuromast Function Defects
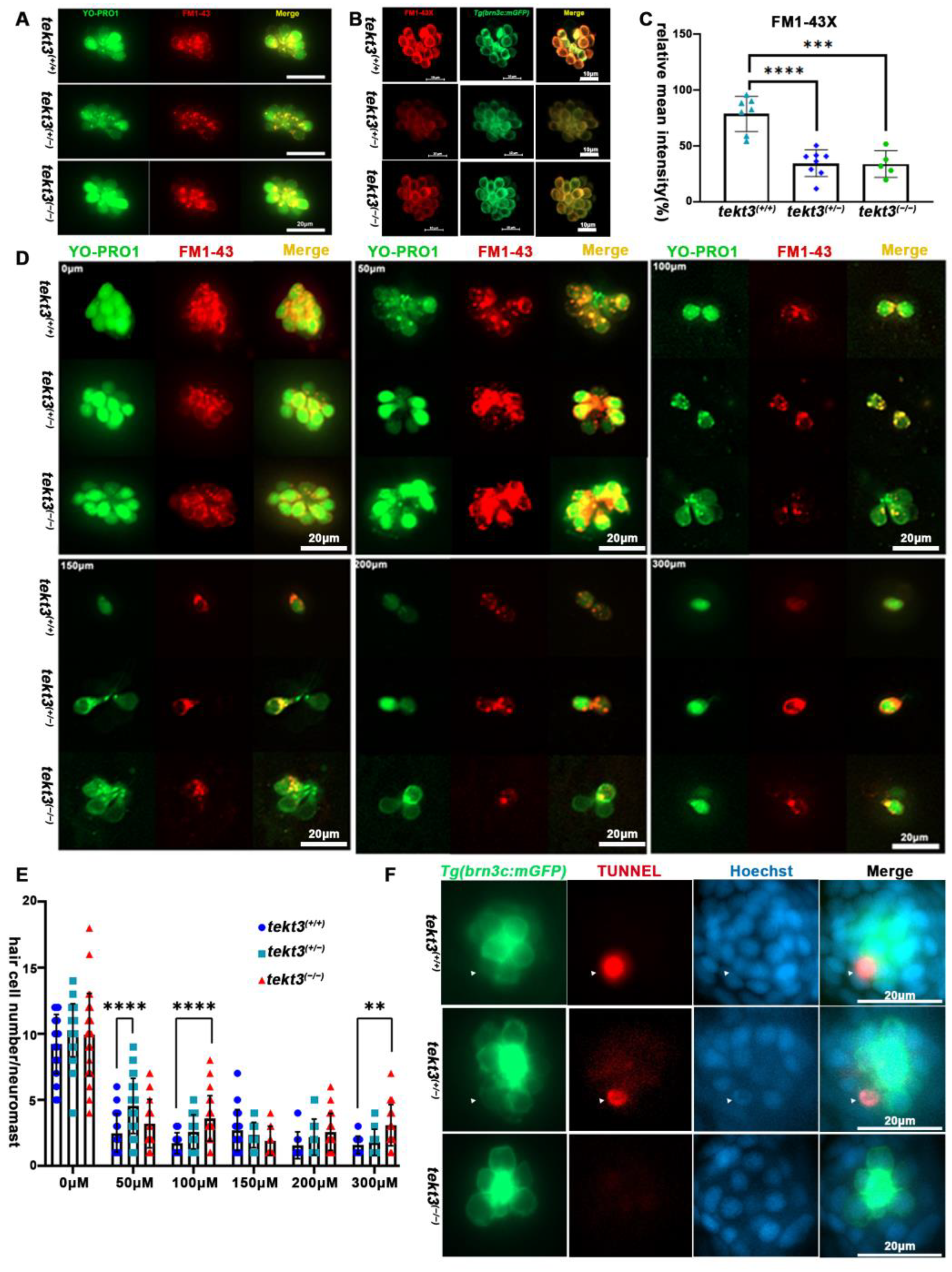
To distinguish if the morphological defect of HC kinocilia is due to developmental defect or function-triggered collapse of kinocilia upon the lack of Tekt3, we first assessed HC functions. From YO-PRO1 and FM1-43 or FM1-43X staining, the heterozygous mutant showed a significantly slower intake of the vital dyes (Figure 6A–C), indicating a possible MET blockage in neuromast HCs. To verify the possibility, neomycin was used to kill pre-existing neuromast HCs for 0.5 h, and the remaining neuromast HCs were counted, revealing that the tekt3 mutant is indeed partially resistant to neomycin-induced HC death (Figure 6D,E). The lower TUNNEL signal was evident in both homozygous and heterozygous tekt3 mutant neuromasts, likely supporting a Tekt3-enabled protective effect on HCs upon a 5 min treatment of 300 μM neomycin (Figure 6F).
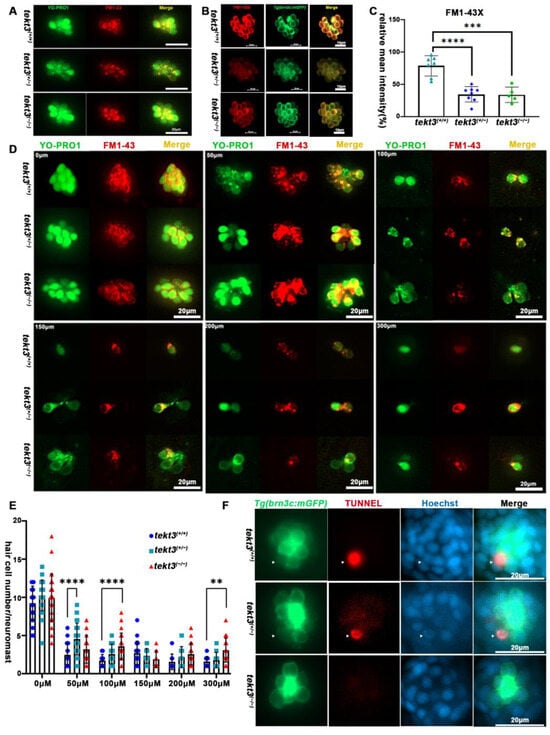
Figure 6.
Neuromast HCs of tekt3 mutant are neomycin resistant. (A) Live dye (YO-PRO1 and FM1-43) intake by neuromast HCs in 5 dpf WT and tekt3 mutant. (B) YO-PRO1 and FM1-43X staining. (C) Weaker dye-intaking of tekt3 mutant neuromast. (D) Resistance to neomycin treatment. (E) Neomycin dose-dependency. (F) Weaker TUNNEL signals of mutant neuromast after neomycin treatment. The arrowheads are pointed to apoptotic neuromast hair cells. ** p < 0.01; *** p < 0.001; and **** p < 0.0001.
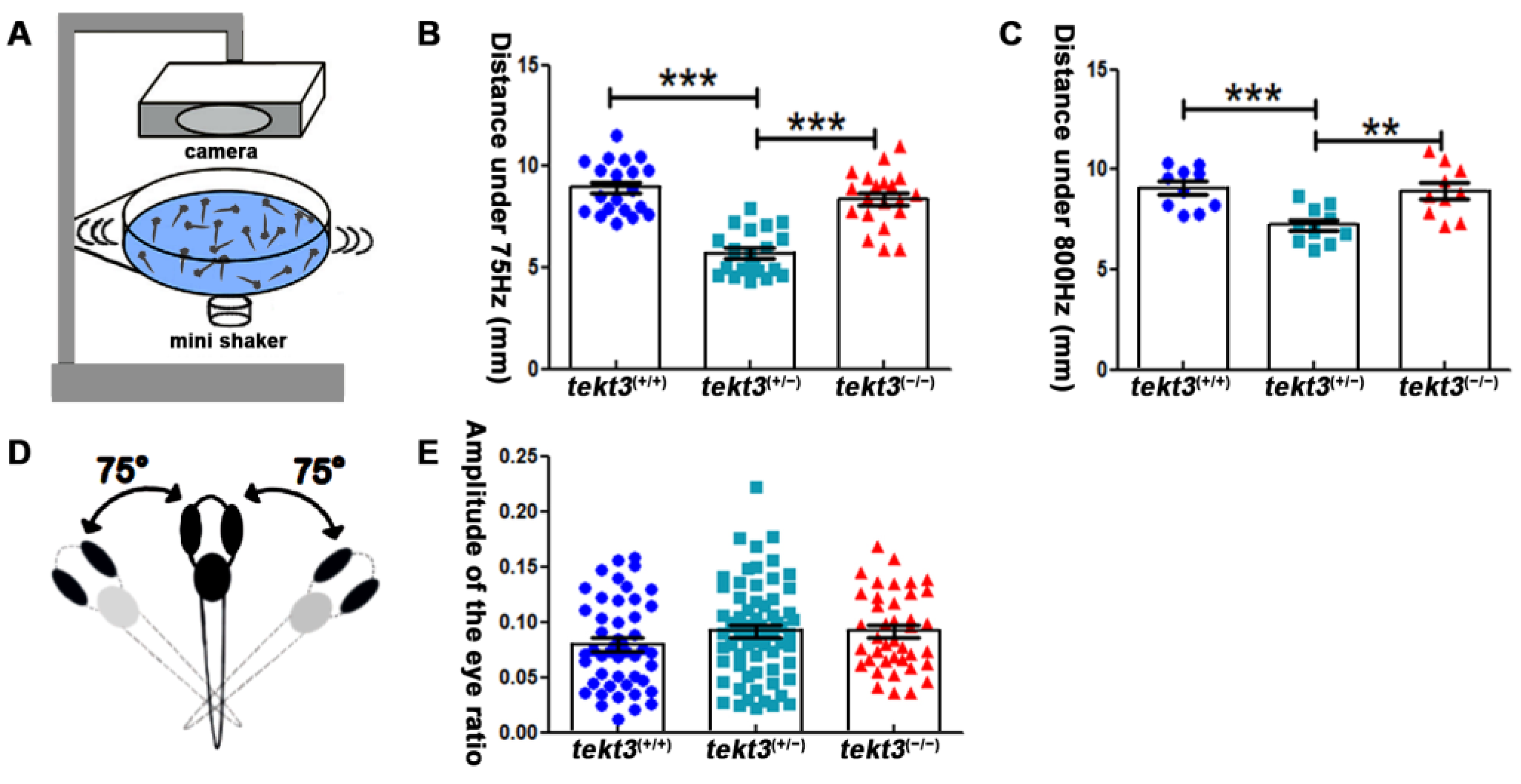
Zebrafish neuromasts can partially mediate vibrational stimuli triggered startle response that can be captured by a self-made equipment (Figure 7A). By measuring the moving distance upon 75 Hz and 800 Hz stimuli (Figure 7B,C), the heterozygous tekt3 mutant was more affected, perhaps also due to the genetic compensation via elevated tekt1 expression. However, the linear VOR test showed no obvious defect of the tekt3 mutant utricle (Figure 7D,E). These results indicated that only neuromast function was negatively affected in the absence of Tekt3, although tekt3 is expressed in both utricle and neuromast HCs. These experiments suggest a critical role of Tekt3 to safeguard functional integrity rather than to affect HC development.
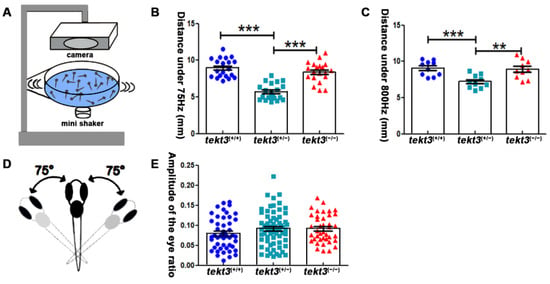
Figure 7.
Heterozygous tekt3 mutant has weaker response to soundwave stimuli. (A) Self-made equipment to measure neuromast function (B,C) showed that the 6 dpf heterozygous tekt3 mutant exerted weaker response upon 75 Hz (B) or 800 Hz (C) stimuli. (D) A diagram of a larval zebrafish in linear VOR test. (E) The 5 dpf tekt3 mutant had a normal VOR score. ** p < 0.01; and *** p < 0.001.
2.4. Lacking Tektin3 Delays HC Regeneration of Neuromast After Neomycin Treatment
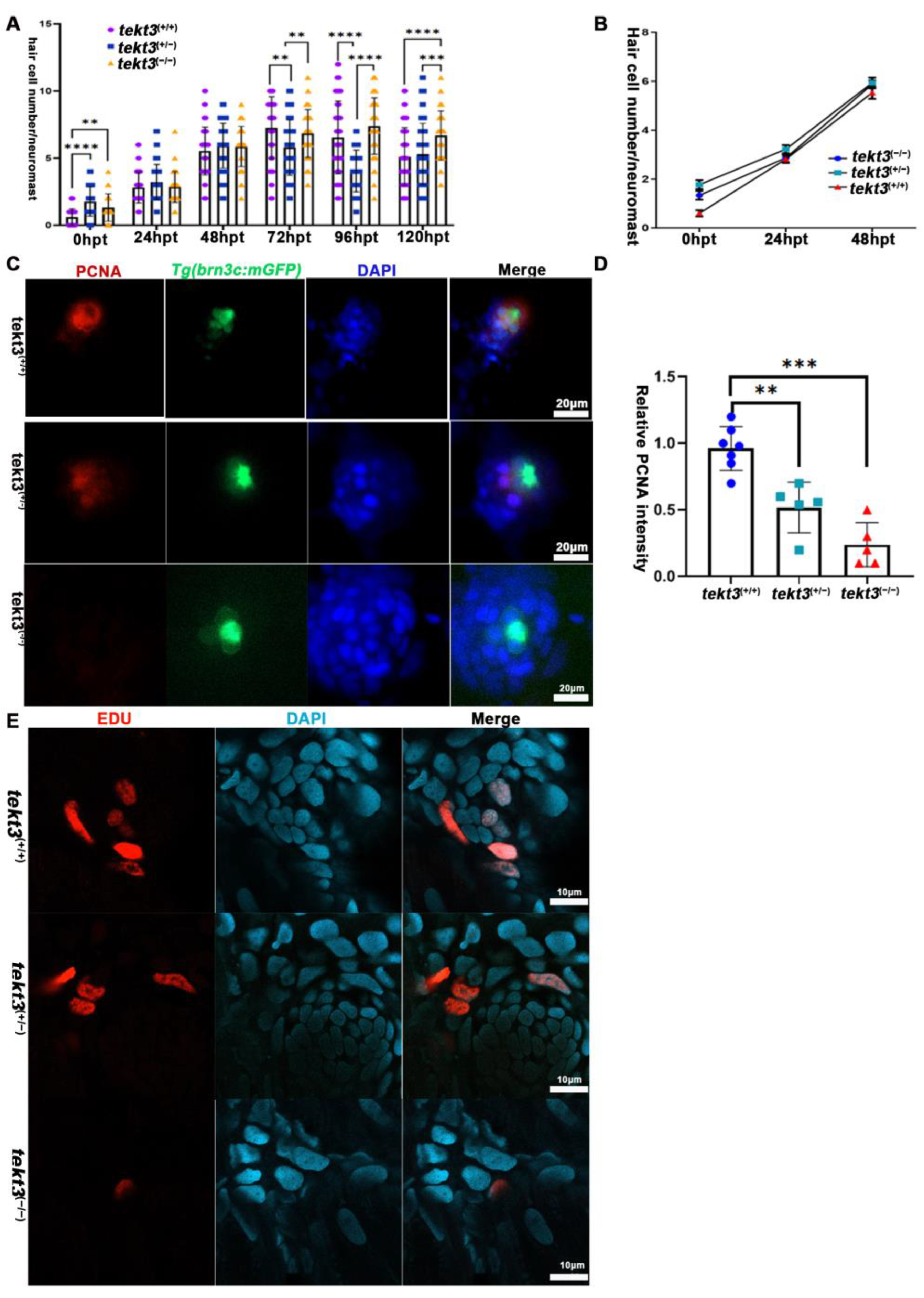
Upon neomycin treatment to kill pre-existing HCs, neuromast HC regeneration program is triggered. If neomycin intake is slower in tekt3 mutant HCs, neuromast HC regeneration, in theory, should be slower than that of wild-type neuromast. We treated the larvae with 300 μM neomycin at RT for 30 min and showed that tekt3 mutant was indeed late in regenerating HCs (Figure 8A,B). This negative effect is due to slower cell proliferation of non-HCs, as demonstrated by PCNA and EDU staining experiments (Figure 8C–E).
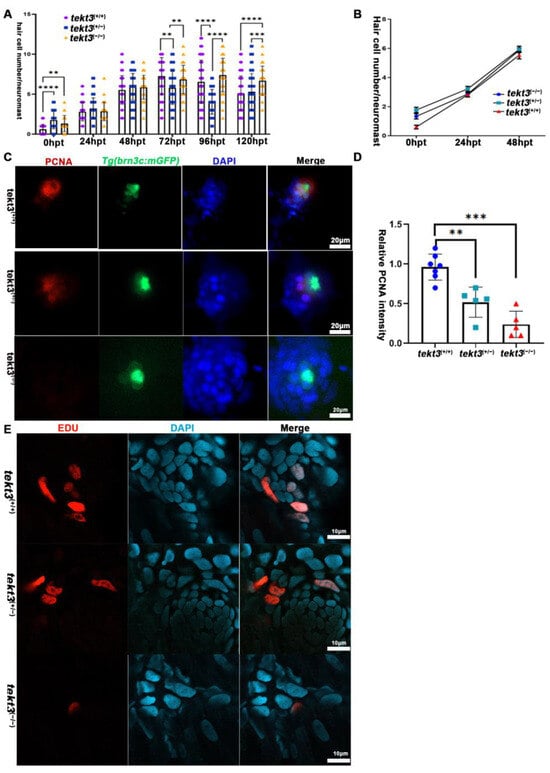
Figure 8.
Neuromast HC regeneration is delayed in tekt3 mutant. (A) The summary of HC numbers in L1-L3 neuromasts, days after neomycin administration. (B) The HC regeneration. (C,D) PCNA staining shows neuromast cell proliferation defect of tekt3 mutant. (E) EDU staining shows neuromast cell proliferation defect of tekt3 mutant 6 h after neomycin treatment. ** p < 0.01; *** p < 0.001; and **** p < 0.0001.
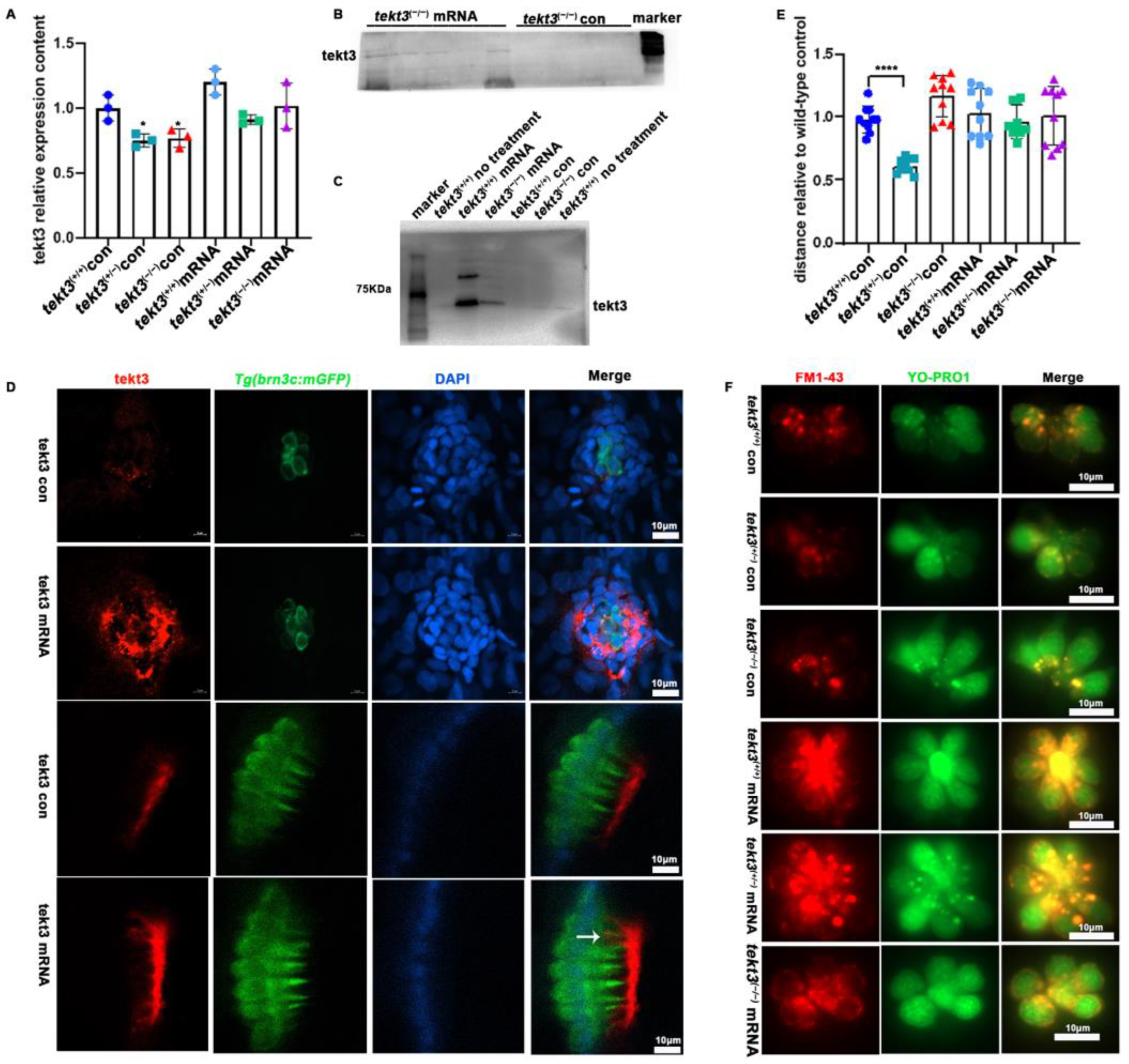
To ultimately ascertain how tektin3 is related to the phenotypes observed above, we constructed a tekt3-expressing vector by inserting the full-length CDS of wild-type tekt3 into pblk-CMV-SV40. The wild-type and mutant fish, co-injected with TP mRNA and pblk-CMV-tekt3-CDS-SV40 vector, were evaluated by PCR, antibody staining and Western blot, and successful rescues were scored (Figure 9A–D). The rescued mutant fish showed better response to the soundwave stimuli (Figure 9E) and improved uptake of YO-PRO1 and FM1-43 (Figure 9F). Interestingly, the presence of Tekt3 was observed in a few kinocilia of the injected wild-type fish (Figure 9D). The fact that the tekt3 mutant possesses functional defects in its neuromasts, which were largely rescued by mis-expressing wild-type tekt3, indicated that Tekt3 is responsible for the mutant phenotypes.
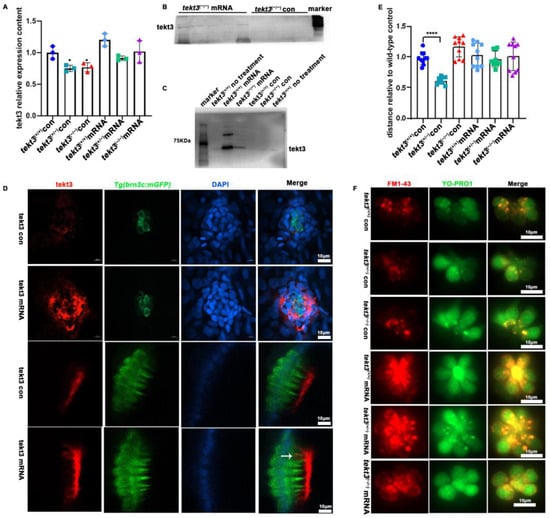
Figure 9.
The rescue of tekt3 mutant. (A) Comparison of tekt3 mRNA levels between control and experiment groups is shown. (B,C) The protein level could be brought back a bit in the injected groups. (D) Rescued neuromast and utricle of tekt3 mutant. (E) The rescued startle response in mRNA injection groups. (F) The YO-PRO1 and FM1-43 intakes become normal in mRNA injected groups. The white arrow is pointed to a kinocilium with positive tekt3 signals. * p < 0.05; and **** p < 0.0001.
3. Discussion
The present study was initially designed to provide supporting evidence of a hypothesis that microtubule-associated Tekt3 is a component of HCs to stabilize structure and safeguard function of kinocilia. After checking the specificity of Tekt3/tekt3 expression in zebrafish larvae, we have discovered that Tekt3/tekt3 is only present in utricular and neuromast HCs; it is absent in HCs of saccule and ampulla crista. We have additionally unveiled that Tekt3 is largely present in the cytosol of neuromast and utricular HCs in zebrafish, and TEKT3 is found in the cytosol of cochlear outer HCs in mature mice. We then bred a loss-of-function tekt3 mutant that develops normally and is fertile, i.e., no obvious sperm defect.
All HCs of the zebrafish inner ear and lateral line possess kinocilium-containing hair bundles. Since tekt3 is not present in all HCs of these organs, one may predict that zebrafish tekt3 is not an essential kinocilium component, regarding the structure and/or function. Given that kinocilia should act similarly in mechanoelectrical transition, Tekt3 may play a specific, balance-related role unique to utricular and neuromast HCs. To make the situation more complex, however, the loss-of-tekt3-function mutant has morphological and function defects of kinocilia only in neuromast HCs.
It is possible that the subcellular location of Tekt3 matters. In addition to the apical cytosol-enriched Tekt3, the immunochemical signal is also present in OTM at the utricle. OTM is composed of a dozen proteins and helps the tips of HC hair bundles to avoid direct contact with the otolith and mediates the movement of otoliths in response to soundwave/vibration triggered mechanical stimulation. Occasionally, Tekt3 was detected in a mesh-like structure at the top of neuromast hair bundles (Figure S4). In a 5–6 dpf neuromast, HCs stick out their apical portion of kinocilia that are embedded in the cupula. After PFA fixation, ~30% of the sampled cupula adopted a mesh-like structure (unpublished observation). The fact that both utricular OTM and neuromast cupula are completely outside the cell body of HCs suggests that Tekt3 might be present at the tip portion of a kinocilium in these HCs. The compartmentation of primary cilia, including zebrafish crista, and neuromast kinocilia, along the proximal-distal axis, could be visualized using labeled/fusion kinocilium-specific proteins [2,23,24]. It is thus possible that PFA fixation breaks kinocilia to allow OTM to retain tip-located Tekt3. The jelly cupula of neuromast, however, may not be able to firmly retain Tekt3 during PFA fixation. It is also possible that kinocilia are extremely fragile [25] and PFA fixation helps to quick lock Tekt3 at kinocilium tip. If so, the strong intensity of Tekt3 in OTM, much higher than that in neuromast cupula, means a higher Tekt3 level in utricular HCs. Nevertheless, live visualizing Tekt3 would ultimately help to resolve the question of whether Tekt3 is truly located in the kinocilium tip.
Alternatively, if Tekt3 is not permanently located in neuromast and utricular kinocilia, the large amount of Tekt3 present in OTM may suggest an active transportation of Tekt3 intracellularly in utricular HCs. When tekt3 is mis-expressed, few kinocilia are tekt3+, also suggesting a traffic of Tekt3 between kinocilia and cytosol of utricular HCs. Perhaps Tekt3 is actively transported in neuromast HCs too (Figure S4) if appropriate fixation is employed. Thus, what type(s) of subcellular event involves Tekt3 may warrant further investigation. As a hint for future research, mammalian TEKTs are found to be not restricted to microtubule-associated structures. In rat spermatozoa, TEKT3 is not directly associated with the flagellar axoneme. Instead, TEKT3 is a component of the peri-axonemal complex and predominantly associated with the surface of mitochondria [16]. But what exactly TEKT3 does in these locations remains unknown.
Zebrafish neuromast HCs are constantly challenged by various low frequency mechanical and vibrational stimulation in a laboratory setting, thus requiring constant recharge or repair. That Neuromast HCs are more active in terms of metabolism than inner ear HCs [11,26] appears to support such a notion. While the chemical composition of neuromast cupula remains unidentified, its protective strength may be much weaker than that of inner ear OTM. If so, less protected neuromast HCs need a quick turnover of Tekt3 for some reason. Otherwise, the mutant kinocilia damage shows up, exhibiting the bent or bulbed kinocilium tips. A better protection mediated by OTM may account for normal HC structure and function of mutant utricular HCs.
The genetic compensation caused by enhanced tket1 expression in the tekt3 mutant is evident (Figure 5G). However, defects of vital dye intake and vibration-triggered startle response are only evidenced in the heterozygous mutant (Figure 6C and Figure 7), suggesting that elevated tekt1 can partially replace the role of tekt3 in fine-tuning neuromast function in homozygous mutants. In contrast, elevated tekt4 expression fails to compensate tekt3 at all in the mutant (unpublished observation). Nonetheless, injected normal tekt3 mRNA can rescue mutant abnormalities, indicating that Tek3 is hard to be replaced and may help to achieve kinocilia diversity during the evolution of tektin family in teleost.
In parallel, we observed that mouse TEKT3 is apically enriched in the cytosol of outer HCs of mature mouse cochlea but is absent in cilia (Figure 3B). Therefore, HCs of mouse cochlea and zebrafish utricle express Tekt3/tekt3 and distribution of TEKT3/Tekt3 exerts somewhat similar subcellular distribution. However, zebrafish saccular HCs and mouse cochlear inner HCs, the true sound sensor cells in both animals, both lack the protein, suggesting less relevance of TEKT3 to hearing loss disease of humans. Defects in the sensory function or structure of primary cilia are known to be associated with diseases termed ciliopathies, and hearing loss (HL) in relation to HCs’ ciliary defects is also considered to be one such kind. Studies on cilia-related HL have largely been focused on stereocilia, since mature cochlear HCs lack any kinocilia [27]. Cochlear stereocilia are responsible for the conversion of mechanical energy into electrical signals in mammalian ears. Kinocilia have drawn a growing interest due to their important roles in the development of hair bundle polarity and the maintenance of normal hearing. For instance, the loss of kinociliary links leads to abnormal polarity of hair bundles. Genetic defects result in the absence of cilia, morphologically defective cilia, or abnormally elongated cilia, which result in hearing loss in mice [28,29,30,31]. The present study suggests that TEKT3 can be listed as a risk factor of noise-triggered and cochlear outer HC-related hearing loss/deafness in humans.
4. Materials and Methods
4.1. Zebrafish Husbandry and Care
The wild-type AB strain and transgenic zebrafish line Tg(Brn3c:mGFP) were raised as previously described [32]. Briefly, all strains were maintained at 28 °C and a density of about 10 fishes/L under a 14 h light/10 h dark cycle. Embryos and larvae were raised in the E3 medium under 28 °C conditions. All animal experiments were approved by the Institutional Animal Care and Use Committee of Southern University of Science and Technology.
4.2. Quantitative Real-Time PCR
Total RNA was extracted from 30 wild-type or mutant larvae at 5 dpf using TRIzol reagent (Invitrogen, Waltham, MA, USA). To be more specific, 1 mL TRIzol was added into the larvae and ground with a TGrinder (TIANGEN, Beijing, China, OSE-Y30) for about 2 min. Then, 200 μL chloroform was added and mixed by vigorously shaking the tubes for 30 s and they were then incubated for 5 min at RT. After centrifuging, the upper 200 μL aqueous phase was carefully transferred into a fresh tube. Then, the same volume of isopropanol was added and gently mixed by inverting the tube. Then, they were incubated at RT for 5 min. After centrifuging, the RNA was washed with 75% cold ethanol. The RNA pellet was air-dried at RT and dissolved in nuclease-free water. The concentration and quality were measured with a Nanodrop and Agilent 2100 bioanalyzer (Thermo Fisher Scientific, Waltham, MA, USA). A total of 1μg RNA was reverse transcribed to the first strand of complementary DNA with the random primer using a complementary DNA synthesis kit (Promega, Madison, WI, USA, A5003). Quantitative real-time PCR primers for tekt gene members and gapdh are listed in Table S1. The real-time PCR reaction was set up with a SYBR® qPCR Master Mix (Vazyme, Nanjing, China). The PCR was performed in an ABI 7500 Real-Time PCR instrument (Applied Biosystems, Waltham, MA, USA) with the SYBR green detection system, and results were normalized with gapdh expression using ΔΔCt method.
4.3. Neomycin Treatment
The 5 dpf embryos were rinsed in culture medium containing different concentrations of neomycin (Merck Millipore, Burlington, MA, USA, 4801-25GM) at 28 °C for 30 min, then the embryos were gently washed with culture medium without neomycin 3 times and used for other detections.
4.4. Immunofluorescence Staining
For immunofluorescence, the zebrafish larvae were maintained with 0.2 mM PTU (1-phenyl-2-thiourea, Sigma, Saint Louis, MO, USA) solution after 24 hpf. Then, the 5 dpf zebrafish larvae were anesthetized with 0.016% (w/v) anesthetic tricaine/E2 medium (MS-222, pH = 7.0, Sigma, Shanghai, China, E10521) on ice for 3 min. After removing most of the fluid, the larvae were fixed with 4% paraformaldehyde (PFA)/PBS at 4 °C overnight. After three washes with 0.2% Tween-20/PBS, the fixed larvae were incubated with 0.1% Triton X-100/PBS at RT for 30 min. Then, the larvae were blocked in a blocking buffer containing 5% donkey serum (v/v), 3% BSA (w/v), and 0.1% Trixon X-100 (v/v) at RT for 1 h. The larvae were incubated with diluted primary antibody at 4 °C overnight. After three times of PBST wash, each for 5 min, the larvae were incubated with diluted secondary antibody at RT in dark for 2 h. Then, larvae were washed and stained with a 1:1000 dilution of DAPI in dark for 5 min. Finally, the larvae were imaged with a Zeiss LSM780 confocal microscope (Oberkochen, Germany) using 20× object lens.
The wild-type C57BL/6 mice were transcardially perfused with 4% (w/v) PFA/PBS after anesthesia with isoflurane. The cochlea was collected and fixed in 4% PFA at 4 °C overnight and then decalcified with JYBL-I decalcifying fluid (Solarbio, Beijing, China, G2470) at 4 °C for 2 days. Then, the tissues around HCs were cut off and the HCs were used for staining. Similarly, the tissues were penetrated with 0.1% Triton X-100/PBS at RT for 30 min, blocked with 5% donkey serum (v/v), 3% BSA (w/v), and 0.1% Trixon X-100 (v/v)/PBS at RT for 1 h. Then, the HCs were incubated with primary antibodies at 4 °C overnight and secondary antibodies at RT in dark for 2 h. Finally, the HCs were mounted with DAPI Fluoromount-GTM mounting medium (20 μL/slice, Yeasen, Shanghai, China, 36308ES11) and imaged with a Zeiss LSM780 confocal microscope using Z-stack projections.
The primary antibodies used were rabbit anti-Tekt3 (1:100, Proteintech, Rosemont, IL, USA, 12959-1-AP-50UL), anti-PCNA (1:100, Proteintech, 10205-2-AP-50UL), and Anti-MYO7A (1:50, DSHB, Iowa, USA, 138-1). The secondary antibodies used are donkey anti-rabbit Alexa Fluor-568 (1:1000, Invitrogen, A10042) and donkey anti-mouse Alexa Fluor-488 (1:1000, Invitrogen, A21206).
As for FM™1-43FX staining, FM™1-43FX dye (300 μM, Thermo Fisher, F35355) was used for incubating the living embryos for 15 s in dark at RT. Then, the embryos were fixed in 4% PFA at RT for 15 min, and the embryos were imaged with a Zeiss LSM780 confocal microscope.
4.5. FM1-43 Intake Assay and YO-PRO1 Staining
The fluorescent vital dye FM1-43 can rapidly and specifically label active hair cells by entering through open mechanotransduction channels [33,34]. The intake of FM 1-43 is commonly used as a hair cell mechanotransduction marker in many animal models, including zebrafish [35]. YO-PRO-1, a cyanine dye, which can entry into hair cells through non-selective mechanotransduction channels and bind to the DNA, is also a reliable indicator of hair cell viability and has been used in hair cell studies [35]. To recognize the functional HCs in neuromasts, a FM1-43 staining experiment was conducted as follows. The free-swimming larvae were incubated in 3μM FM1-43 vital dye (Thermo Fisher, T3163) for 25 s at room temperature (RT) in the dark. Then, the larvae were gently washed with E2 buffer twice. Similarly, 200 μM YO-PRO1 (Thermo Scientific, Y3601-4) was stained in dark at RT for 1 min and washed with culture medium. Finally, the larvae were imaged after being gently mounted in 1% low melted agarose containing 0.016% (w/v) anesthetic tricaine.
4.6. Whole-Mount in Situ Hybridization
The whole-mount in situ hybridization (WISH) of zebrafish was performed according to the following standard procedures. The cDNA fragment of zebrafish tekt3 was amplified via PCR using designed primers—tekt3-ISH-F, 5′AGATTTCAGCGCTGTCCGAT′ and tekt3-ISH-R 5′ AAGCAGCACGTTCACTCTGA3′—and was cloned into an EZ-T cloning vector with an EZ-T Fast Ligation Kit (GenStar, #T168-10, Beijing, China). After linearization of the EZ-T-inserting tekt3 fragment, the DIG RNA Labeling Kit (T3 & T7) (Roche, #11175025910, Indianapolis, IL, USA) was used to prepare digoxigenin-labeled tekt3 sense and antisense mRNA probes through transcription in vitro. The 3 dpf and 5 dpf embryos were anesthetized with 0.016% (w/v) anesthetic tricaine/E2 medium on ice for 3 min and then fixed in 4%PFA at 4 °C overnight, digested in proteinase K at RT for 10 min, and incubated with a pre-hybridized mix at RT for 1 h. After that, the embryos were hybridized with tekt3 sense or antisense probes overnight at 65 °C. After incubation with alkaline phosphatase (AP)-conjugated antibody against digoxigenin (Roche, #11093274910) at 4 °C overnight, the NBT/BCIP solution (Roche, #11681451001) was used to detect the tekt3 mRNA expression.
4.7. CRISPR/Cas9 Generated tekt3 Mutant
The tekt3 mutant zebrafish were generated via CRISPR/Cas9-mediated gene editing technology. The single guide RNA (sgRNA), specifically targeting exon 2 of tekt3 (5′-TTT GCA GGG CTG ATA GGG TC-3′), was synthesized (Genescript, Nanjing, China). Then, the sgRNA was dissolved in nuclease-free water. A mixture of 300 ng/μL Cas9 protein (NEB, Ipswich, MA, USA, M0646T) and 100 ng/μL tekt3 sgRNA was co-injected into one-cell stage zebrafish embryos. Then, those injected embryos (designated as F0) were raised and outcrossed with wild-type zebrafish to achieve F1 fish. F1 fish were raised to adulthood. A small piece of caudal fin of these adult F1 fish was cut and used for genotyping. The DNA was extracted with 30μl 50 mM NaOH at 97 °C for 30 min and adjusted the pH with 1 mM Tris-HCl (pH 7.2). The genotype of F1 fish was first examined with PCR. Then, the genotype of the F1 fish was further identified with Sanger sequencing. Finally, the heterozygous F1 fish were kept and incrossed to obtain a stable tekt3 mutant line. A pair of primers, at both ends of the tekt3 target site, were designed and synthesized for genotyping. The used genotyping primers were: tekt3-gF: 5′-CAGAACTCCAATATGCCCTGG-3′ and tekt3-gR: 5′-CTGTCCTGTTCGAGGGGTAGA-3′.
4.8. Vestibulo-Ocular Reflex (VOR) Assay
A customized VOR testing system was obtained from the Southern University of Science and Technology and used to quantify linear VOR in zebrafish larva, evoked by the head motion to the earth horizontal axis. The detailed procedure for linear VOR testing are as follows. The zebrafish larva was gently mounted in the larva-shaped chamber in a dorsal-up position with the tail glued by 5% methylcellulose and covered with a piece of glass coverslip on the chamber. After adding E3 embryo media in the head region, the chamber unit was then mounted on a device for quantifying linear VOR. After aligning the larval eyes to the center of the infrared camera, the platform started to rotate back and forth around a horizontal axis at a speed of 30 rpm, and the VOR was recorded by the camera [36].
4.9. Startle Response Assay
The sensory function of zebrafish was tested by the startle response assay. In brief, a plastic plate attached to a mini vibrator was used to place 20 normal larvae at 6 dpf, while an infrared digital video tracking system was used to monitor their swimming behavior. The zebrafish lateral line can sense low-frequency stimulation up to 200 Hz, and the inner ear can detect stimulation higher than 100 Hz [37,38]. For indirect stimulation, the zebrafish frequency response curve studies show that best response frequency of lateral line distributes at 74 ± 28 Hz [39]. After removing neuromasts, the hearing is not affected at about 800 Hz in zebrafish [40]. In order to detect the sensory functions of both neuromasts and ear, the 75 or 800 Hz with 32 dB re.1 ms−2 tone bursts were applied to the amplifier to drive the vibrator. The conditions of acoustic vibration stimuli lasting for 30 ms with a 180 s inter-stimulus interval were set and applied. Each sound vibration stimulus level was repeated for 20 times and the locomotion behavior of the larvae with C-shape motion to this stimulus was recorded. Finally, the movement typical parameters of mean distance and peak velocity were analyzed to assess the startle response of larvae to sound vibration stimuli.
4.10. Western Blot Analysis
The 5 dpf zebrafish were homogenized in a cold RIPA lysis buffer (GLPBIO, Shanghai, China) with a protease inhibitor cocktail (CST, Danvers, MA, USA, #5871S). Protein concentration was determined using the BCA protein assay kit (Biomed, Guangzhou, China). Proteins were separated with 10% SDS-PAGE (EpiZyme, Shanghai, China, PG112 ) and transferred to PVDF membranes (Beyotime, FFP39, Shanghai, China). The membranes were blocked with 5% skimmed milk dissolved in TBST buffer at RT for 1 h and then incubated with the Anti-TEKT3 antibody (1:1000, Proteintech) and Anti-GAPDH (1:000, SAB) at 4 °C overnight with gentle shaking. After washing in 1×TBST buffer (20 mM Tris–HCl, 150 mM NaCl, 0.05% Tween 20, and pH 7.6) 3 times, the membranes were incubated with HRP-conjugated secondary antibodies (1:1000; Beyotime) for 2 h at RT. The membranes were then developed using TanonTM High-sig ECL Western Blotting Substrate (Tanon, Shanghai, China, 130-301) and Tanon6100C imaging system (Tanon). Quantitative analysis of protein bands was performed by the Tanon 6100C software.
4.11. Construction of pblk-CMV-tekt3-CDS-SV40 Vector and Microinjection
A vector containing full-length tekt3 CDS was constructed. The full-length tekt3 CDS was amplified from 6 dpf wild-type AB strain cDNA with the following primers: tekt3-csdF: 5′-CCATCGATTCGAATTCAAGGATGGAGCTCATCGGATCCACCC-3′ and tekt3-csdR: 5′-CTTATCATGTCTGGATCTACCTATGTAAATCCAACCAGGCGA-3′. Then, the PCR product was inserted into a pblk-CMV-SV40 vector via a Gibson Assembly HiFi Kit (Invitrogen, A46624EA). The vector was examined by Sanger sequencing. Then, 50 ng/μL vector and 100 ng/μL TP mRNA were co-injected into one-cell stage embryos.
4.12. TUNEL Assay
TUNEL staining was conducted according to the manufacturer’s instructions of the One Step TUNEL Apoptosis Assay Kit (Beyotime, C1089, Shanghai, China). In brief, embryos were fixed in 4% PFA at RT for 30 min and permeated with 0.3% Triton X-100/PBS at RT for 5 min, then incubated with the TUNEL-detected buffer (10% TdT enzyme, and 90% Cy3-labeled buffer) in the dark at 37 °C for 60 min. Finally, the embryos were washed with PBS and were imaged under a fluorescent microscope (Zeiss, Axio Observer7) using 550 nm excitation and 570 nm emission.
4.13. EdU Staining Assay
Hair cell regeneration assessment was conducted utilizing EdU Apollo 567 In Vitro Kit (ACMEC AC10869, Shanghai, China). The experiments were conducted according to the protocol provided by the manufacturer. In brief, 6 dpf larvae were treated with 300 μM neomycin for 30 min and then incubated with 2.5μM EDU-A buffer for 6 h. Then, the larvae were anesthetized with 0.016% (w/v) anesthetic tricaine and fixed with 4% PFA at RT for 30 min, penetrated with 0.5% Triton X-100 (Coolaber, Shanghai, China) in PBS for 20 min at RT, and then incubated with the 1×straining buffer which contains buffer B-E in the dark at RT for 30 min. Finally, the larvae were washed with 0.5% Triton X-100 in PBS for 15 min and incubated with DAPI for 5 min in the dark. The signal was obtained with a Zeiss LSM780 confocal microscope.
4.14. Statistical Analysis
All the experiments were repeated at least three times. The data are presented as mean ± SD. An unpaired two-tailed Student’s t-test was used for statistical analysis by GraphPad Prism 8.0 Software. A value of p < 0.05 between groups was considered statistically significant.
5. Conclusions
In this study, we have discovered that tekt3 is specifically expressed in both utricular and neuromast HCs, but that a kinocilium defect, i.e., a curved or bubbled cilium tip, was only evident in neuromast HCs of a zebrafish tekt3 mutant. Moreover, neuromast functions such as the intake of FM1-43 or neomycin and the response to vibrational stimulation were reduced in the tekt3 mutant. These defects could be rescued by injecting the full-length of normal tekt3 mRNA into one-cell stage embryos. In addition, the tekt3 mutant exhibited a lower cell proliferation rate and lower apoptotic signal in neomycin-damaged neuromasts, consistent with a late start of neuromast HC regeneration after neomycin treatment. Our study has demonstrated a specific role of Tekt3 in maintaining the integrity and functions of neuromast HCs in zebrafish.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/ijms26073115/s1.
Author Contributions
Conceptualization, D.L. and D.S.; methodology, D.S., L.Z. and S.L.; software, D.L.; validation, D.L., D.S., S.L. and L.Z.; formal analysis, D.S.; investigation, D.S., S.L. and L.Z.; resources, D.L.; data curation, D.S.; writing—original draft preparation, D.S.; writing—review and editing, D.L.; visualization, S.L.; supervision, D.L.; project administration, D.L.; funding acquisition, D.L. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the National Natural Science Foundation of China, (grant number 2018YFA0801006), Natural Science Foundation of China (31771618), Guangdong Natural Science Foundation (2022A1515011878) and Shenzhen-Hong Kong Institute of Brain Science-Shenzhen Fundamental Research Institutions (2021SHIBS0002) to D.L.
Institutional Review Board Statement
All animal procedures were approved by the Animal Care and Use Committee of Southern University of Science and Technology (permission No. SUSTech-JY20240415; approval date: 15 April 2024) and were consistent with the National Institutes of Health Guide for the Care and Use of Laboratory Animals.
Informed Consent Statement
Not applicable.
Data Availability Statement
The data presented in this study are available in the article and Supplementary Materials.
Acknowledgments
We thank Shizheng Zhao of Hong Kong University of Science and Technology for technical support in tekt3 expression vector construction.
Conflicts of Interest
The authors declare no conflicts of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript; or in the decision to publish the results.
References
- Kikuchi, T.; Takasaka, T.; Tonosaki, A.; Watanabe, H. Fine structure of guinea pig vestibular kinocilium. Acta Otolaryngol. 1989, 108, 26–30. [Google Scholar] [CrossRef] [PubMed]
- Erickson, T.; Biggers, W.P., 3rd; Williams, K.; Butland, S.E.; Venuto, A. Regionalized Protein Localization Domains in the Zebrafish Hair Cell Kinocilium. J. Dev. Biol. 2023, 11, 28. [Google Scholar] [CrossRef] [PubMed]
- Sharkova, M.; Chow, E.; Erickson, T.; Hocking, J.C. The morphological and functional diversity of apical microvilli. J. Anat. 2023, 242, 327–353. [Google Scholar] [CrossRef] [PubMed]
- Bastin, B.R.; Meha, S.M.; Khindurangala, L.; Schneider, S.Q. Cooption of regulatory modules for tektin paralogs during ciliary band formation in a marine annelid larva. Dev. Biol. 2023, 503, 95–110. [Google Scholar] [CrossRef]
- Gu, Y.; Zhao, Y.; Ichikawa, M. Tektin makes a microtubule a “micropillar”. Cell 2023, 186, 2725–2727. [Google Scholar] [CrossRef]
- Gui, M.; Farley, H.; Anujan, P.; Anderson, J.R.; Maxwell, D.W.; Whitchurch, J.B.; Botsch, J.J.; Qiu, T.; Meleppattu, S.; Singh, S.K.; et al. De novo identification of mammalian ciliary motility proteins using cryo-EM. Cell 2021, 184, 5791–5806.e19. [Google Scholar] [CrossRef]
- Leung, M.R.; Zeng, J.; Wang, X.; Roelofs, M.C.; Huang, W.; Zenezini Chiozzi, R.; Hevler, J.F.; Heck, A.J.R.; Dutcher, S.K.; Brown, A.; et al. Structural specializations of the sperm tail. Cell 2023, 186, 2880–2896.e17. [Google Scholar] [CrossRef]
- Zhou, L.; Liu, H.; Liu, S.; Yang, X.; Dong, Y.; Pan, Y.; Xiao, Z.; Zheng, B.; Sun, Y.; Huang, P.; et al. Structures of sperm flagellar doublet microtubules expand the genetic spectrum of male infertility. Cell 2023, 186, 2897–2910.e19. [Google Scholar] [CrossRef]
- Erickson, T.; Nicolson, T. Identification of sensory hair-cell transcripts by thiouracil-tagging in zebrafish. BMC Genom. 2015, 16, 842. [Google Scholar] [CrossRef]
- Dong, Z.; Xu, J.; Pan, J. Identification of Regulators for Ciliary Disassembly by a Chemical Screen. ACS Chem. Biol. 2021, 16, 2665–2672. [Google Scholar] [CrossRef]
- Shi, T.; Beaulieu, M.O.; Saunders, L.M.; Fabian, P.; Trapnell, C.; Segil, N.; Crump, J.G.; Raible, D.W. Single-cell transcriptomic profiling of the zebrafish inner ear reveals molecularly distinct hair cell and supporting cell subtypes. eLife 2023, 12, e82978. [Google Scholar] [CrossRef] [PubMed]
- Shim, H.; Kim, J.H.; Kim, C.Y.; Hwang, S.; Kim, H.; Yang, S.; Lee, J.E.; Lee, I. Function-driven discovery of disease genes in zebrafish using an integrated genomics big data resource. Nucleic Acids Res. 2016, 44, 9611–9623. [Google Scholar] [CrossRef] [PubMed]
- Roy, A.; Yan, W.; Burns, K.H.; Matzuk, M.M. Tektin3 encodes an evolutionarily conserved putative testicular microtubules-related protein expressed preferentially in male germ cells. Mol. Reprod. Dev. 2004, 67, 295–302. [Google Scholar] [CrossRef]
- Liu, Y.; Li, Y.; Meng, L.; Li, K.; Gao, Y.; Lv, M.; Guo, R.; Xu, Y.; Zhou, P.; Wei, Z.; et al. Bi-allelic human TEKT3 mutations cause male infertility with oligoasthenoteratozoospermia owing to acrosomal hypoplasia and reduced progressive motility. Hum. Mol. Genet. 2023, 32, 1730–1740. [Google Scholar] [CrossRef]
- Gui, M.; Croft, J.T.; Zabeo, D.; Acharya, V.; Kollman, J.M.; Burgoyne, T.; Höög, J.L.; Brown, A. SPACA9 is a lumenal protein of human ciliary singlet and doublet microtubules. Proc. Natl. Acad. Sci. USA 2022, 119, e2207605119. [Google Scholar] [CrossRef] [PubMed]
- Takiguchi, H.; Murayama, E.; Kaneko, T.; Kurio, H.; Toshimori, K.; Iida, H. Characterization and subcellular localization of Tektin 3 in rat spermatozoa. Mol. Reprod. Dev. 2011, 78, 611–620. [Google Scholar] [CrossRef]
- Roy, A.; Lin, Y.N.; Agno, J.E.; DeMayo, F.J.; Matzuk, M.M. Tektin 3 is required for progressive sperm motility in mice. Mol. Reprod. Dev. 2009, 76, 453–459. [Google Scholar] [CrossRef]
- Iida, H.; Honda, Y.; Matsuyama, T.; Shibata, Y.; Inai, T. Tektin 4 is located on outer dense fibers, not associated with axonemal tubulins of flagella in rodent spermatozoa. Mol. Reprod. Dev. 2006, 73, 929–936. [Google Scholar] [CrossRef]
- Murayama, E.; Yamamoto, E.; Kaneko, T.; Shibata, Y.; Inai, T.; Iida, H. Tektin5, a new Tektin family member, is a component of the middle piece of flagella in rat spermatozoa. Mol. Reprod. Dev. 2008, 75, 650–658. [Google Scholar] [CrossRef]
- Shimasaki, S.; Yamamoto, E.; Murayama, E.; Kurio, H.; Kaneko, T.; Shibata, Y.; Inai, T.; Iida, H. Subcellular localization of Tektin2 in rat sperm flagellum. Zool. Sci. 2010, 27, 755–761. [Google Scholar] [CrossRef]
- Lush, M.E.; Diaz, D.C.; Koenecke, N.; Baek, S.; Boldt, H.; St Peter, M.K.; Gaitan-Escudero, T.; Romero-Carvajal, A.; Busch-Nentwich, E.M.; Perera, A.G.; et al. scRNA-Seq reveals distinct stem cell populations that drive hair cell regeneration after loss of Fgf and Notch signaling. eLife 2019, 8, e44431. [Google Scholar] [CrossRef] [PubMed]
- Peng, J. Gene redundancy and gene compensation: An updated view. J. Genet. Genom. 2019, 46, 329–333. [Google Scholar] [CrossRef]
- Lee, J.; Chung, Y.D. Ciliary subcompartments: How are they established and what are their functions? BMB Rep. 2015, 48, 380–387. [Google Scholar] [CrossRef] [PubMed]
- Soares, H.; Carmona, B.; Nolasco, S.; Viseu Melo, L.; Gonçalves, J. Cilia Distal Domain: Diversity in Evolutionarily Conserved Structures. Cells 2019, 8, 160. [Google Scholar] [CrossRef] [PubMed]
- Ylikoski, J. Surface structures of vestibular end organs in surgical specimens. ORL 1980, 42, 336–345. [Google Scholar] [CrossRef] [PubMed]
- Qian, F.; Wei, G.; Gao, Y.; Wang, X.; Gong, J.; Guo, C.; Wang, X.; Zhang, X.; Zhao, J.; Wang, C.; et al. Single-cell RNA-sequencing of zebrafish hair cells reveals novel genes potentially involved in hearing loss. Cell. Mol. Life Sci. 2022, 79, 385. [Google Scholar] [CrossRef] [PubMed]
- Jones, C.; Chen, P. Primary cilia in planar cell polarity regulation of the inner ear. Curr. Top. Dev. Biol. 2008, 85, 197–224. [Google Scholar]
- Moon, K.H.; Ma, J.H.; Min, H.; Koo, H.; Kim, H.; Ko, H.W.; Bok, J. Dysregulation of sonic hedgehog signaling causes hearing loss in ciliopathy mouse models. eLife 2020, 9, e56551. [Google Scholar] [CrossRef]
- Jędrychowska, J.; Vardanyan, V.; Wieczor, M.; Marciniak, A.; Czub, J.; Amini, R.; Jain, R.; Shen, H.; Choi, H.; Kuznicki, J.; et al. Mutant analysis of Kcng4b reveals how the different functional states of the voltage-gated potassium channel regulate ear development. Dev. Biol. 2024, 513, 50–62. [Google Scholar] [CrossRef]
- Tona, Y.; Wu, D.K. Live imaging of hair bundle polarity acquisition demonstrates a critical timeline for transcription factor Emx2. eLife 2020, 9, e59282. [Google Scholar] [CrossRef]
- Webb, S.W.; Grillet, N.; Andrade, L.R.; Xiong, W.; Swarthout, L.; Della Santina, C.C.; Kachar, B.; Müller, U. Regulation of PCDH15 function in mechanosensory hair cells by alternative splicing of the cytoplasmic domain. Development 2011, 138, 1607–1617. [Google Scholar] [CrossRef] [PubMed]
- Zhong, Y.; Zhang, N.; Zhao, F.; Chang, S.; Chen, W.; Cao, Q.; Sun, L.; Wang, Y.; Gong, Z.; Lu, L.; et al. RBFOX1 and Working Memory: From Genome to Transcriptome Revealed Posttranscriptional Mechanism Separate from Attention-Deficit/Hyperactivity Disorder. Biol. Psychiatry Glob. Open Sci. 2022, 3, 1042–1052. [Google Scholar] [CrossRef] [PubMed]
- Meyers, J.R.; MacDonald, R.B.; Duggan, A.; Lenzi, D.; Standaert, D.G.; Corwin, J.T.; Corey, D.P. Lighting up the senses: FM1-43 loading of sensory cells through nonselective ion channels. J. Neurosci. 2003, 23, 4054–4065. [Google Scholar] [CrossRef] [PubMed]
- Gale, J.E.; Marcotti, W.; Kennedy, H.J.; Kros, C.J.; Richardson, G.P. FM1-43 dye behaves as a permeant blocker of the hair-cell mechanotransducer channel. J. Neurosci. 2001, 21, 7013–7025. [Google Scholar] [CrossRef]
- Santos, F.; MacDonald, G.; Rubel, E.W.; Raible, D.W. Lateral line hair cell maturation is a determinant of aminoglycoside susceptibility in zebrafish (Danio rerio). Hear. Res. 2006, 213, 25–33. [Google Scholar] [CrossRef]
- Shen, Q.; Zhao, F.; Zhang, N.; Zheng, L.; Su, D.; Qian, Y.; Xin, L.; Mingxia, S.; Hongxu, Z.; Chen, F.; et al. Embryonic exposure of estrogen and BPA in zebrafish leads to ADHD-like and ASD-like phenotypes, respectively. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2025, 137, 111293. [Google Scholar] [CrossRef]
- Higgs, D.M.; Souza, M.J.; Wilkins, H.R.; Presson, J.C.; Popper, A.N. Age- and size-related changes in the inner ear and hearing ability of the adult zebrafish (Danio rerio). J. Assoc. Res. Otolaryngol. 2002, 3, 174–184. [Google Scholar] [CrossRef]
- Hardy, K.; Amariutei, A.E.; De Faveri, F.; Hendry, A.; Marcotti, W.; Ceriani, F. Functional development and regeneration of hair cells in the zebrafish lateral line. J. Physiol. 2021, 599, 3913–3936. [Google Scholar] [CrossRef]
- Levi, R.; Akanyeti, O.; Ballo, A.; Liao, J.C. Frequency response properties of primary afferent neurons in the posterior lateral line system of larval zebrafish. J. Neurophysiol. 2015, 113, 657–668. [Google Scholar] [CrossRef]
- Higgs, D.M.; Radford, C.A. The contribution of the lateral line to ‘hearing’ in fish. J. Exp. Biol. 2013, 216 Pt 8, 1484–1490. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).