
Difenoconazole Degradation by Novel Microbial Consortium TA01: Metabolic Pathway and Microbial Community Analysis

 ,
,
Abstract

1. Introduction
2. Results and Discussion
2.1. Determination of the Ability of Microbial Consortium to Degrade DIF
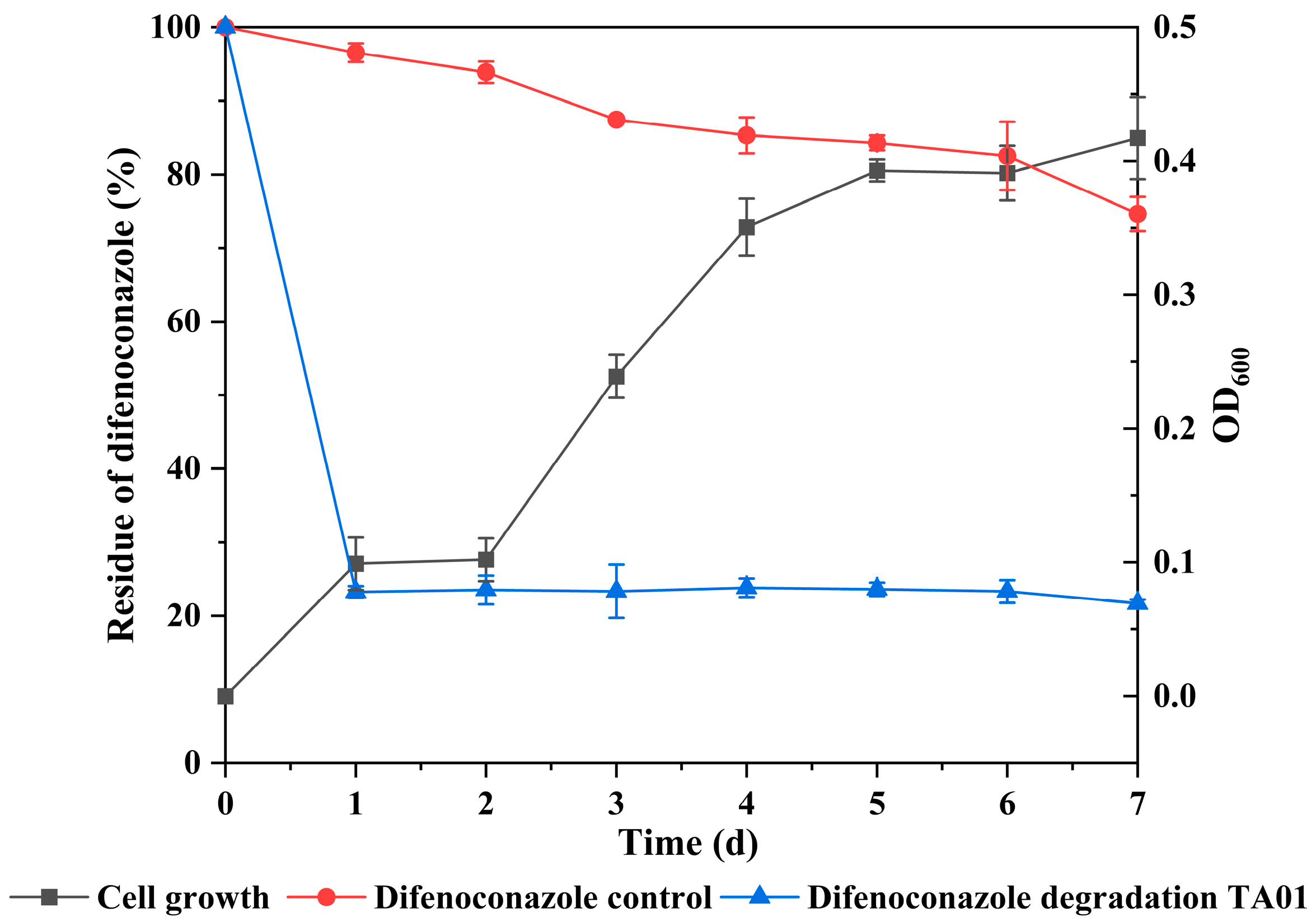
2.2. Curves of Growth and Degradation Relationship of Microbial Consortium TA01
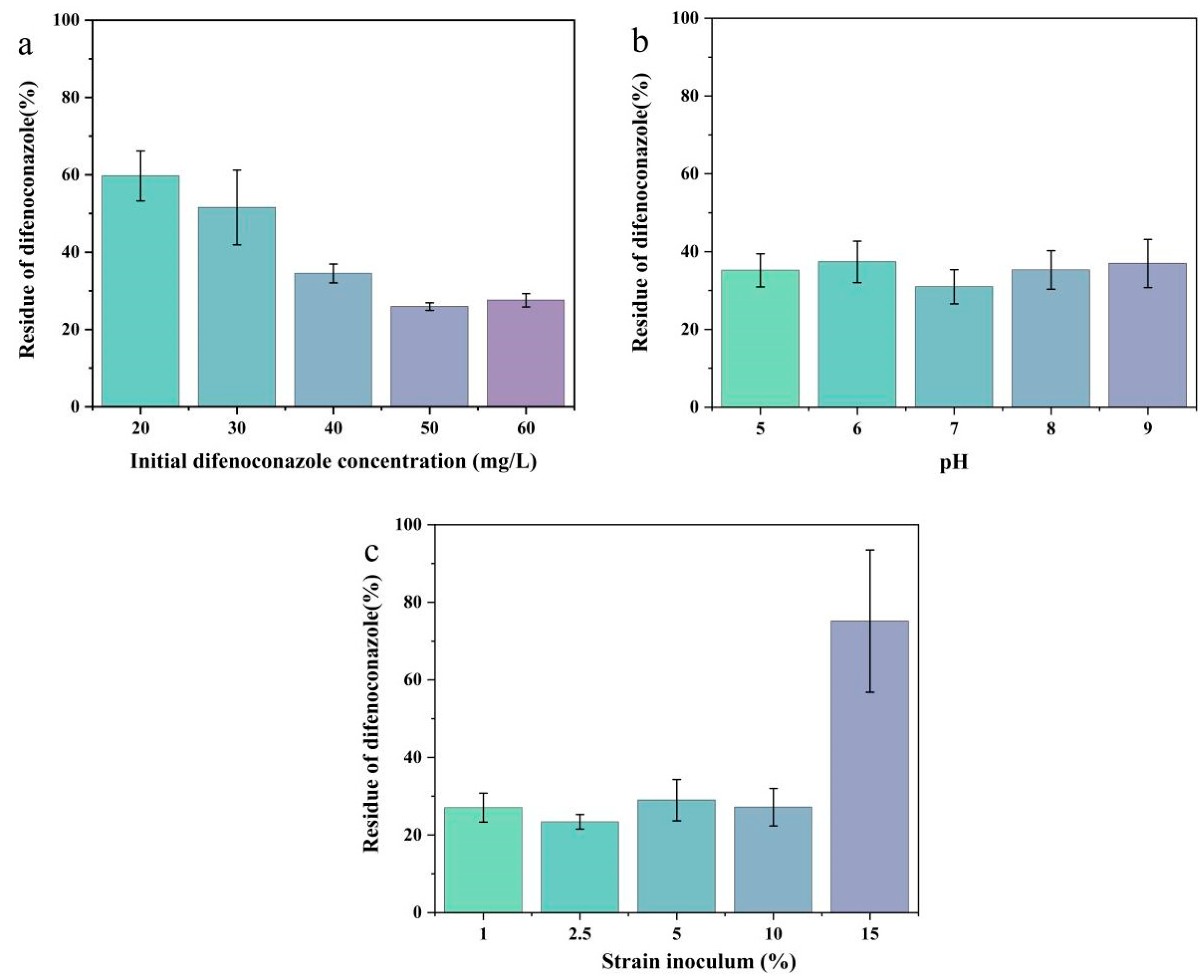
2.3. Effect of Culture Conditions on the Degradation of DIF by Microbial Consortium TA01
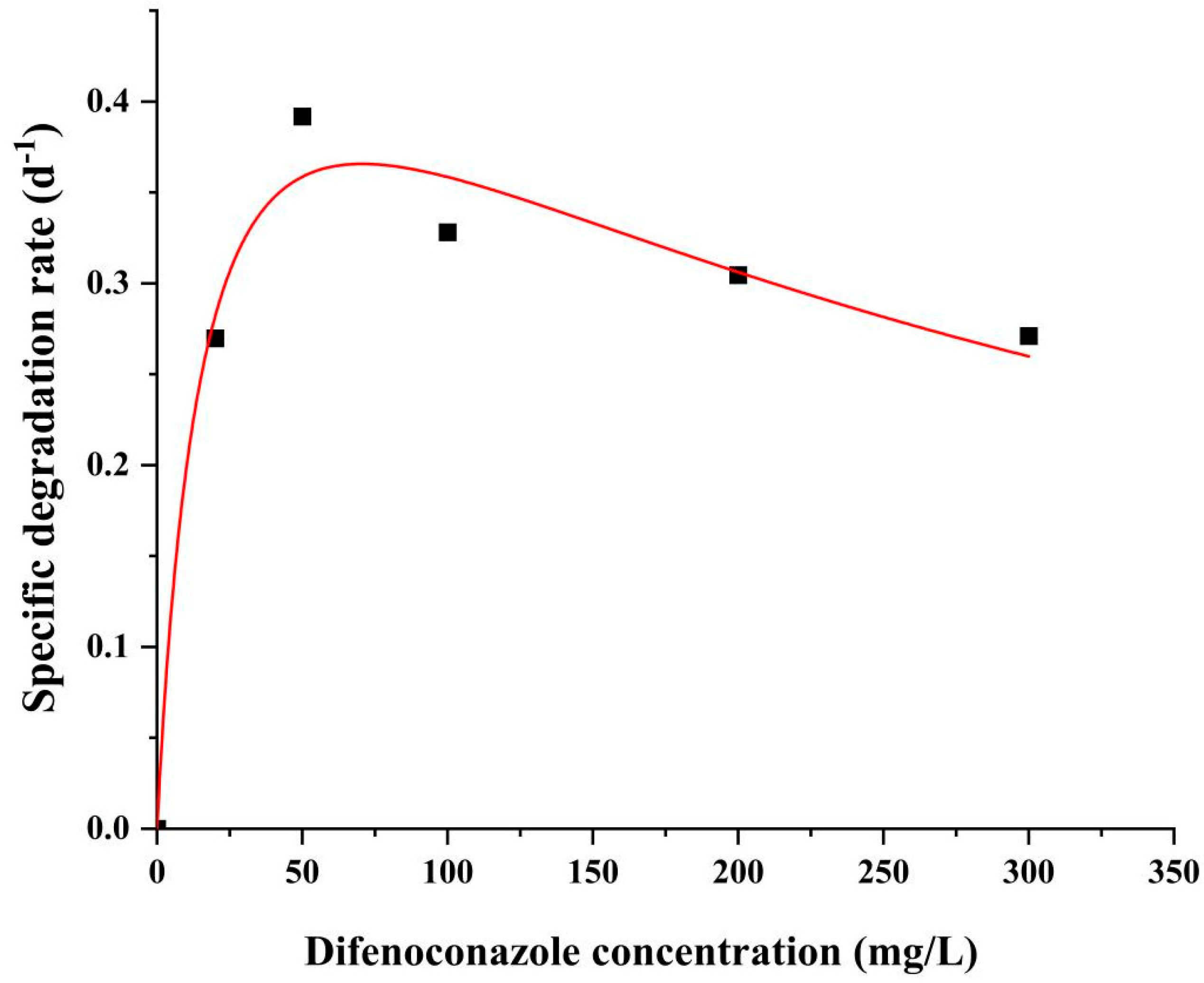
2.4. DIF Degradation Kinetics Analysis
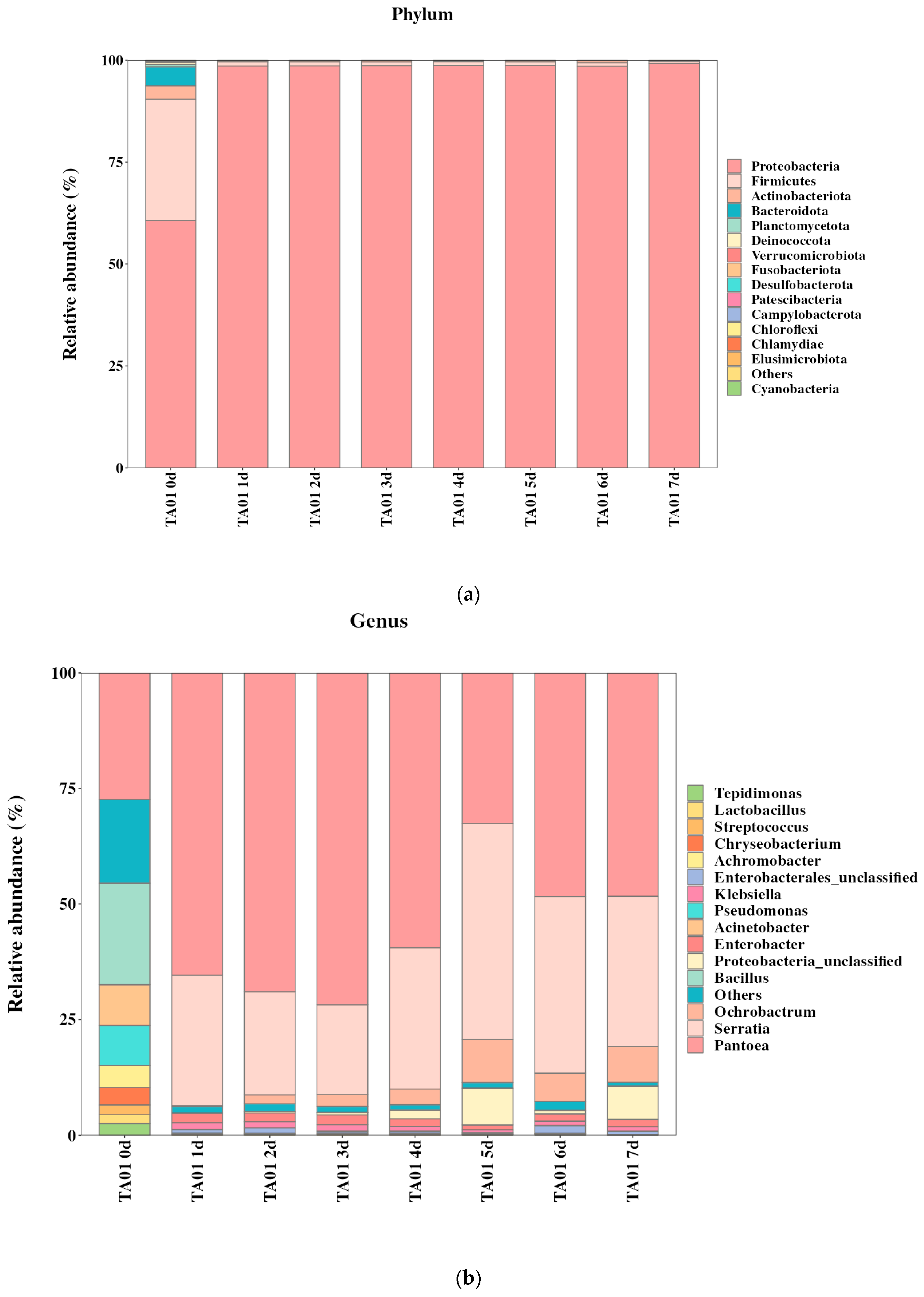
2.5. Community Structure of Degradative Flora and Analysis of Key Degradative Bacteria
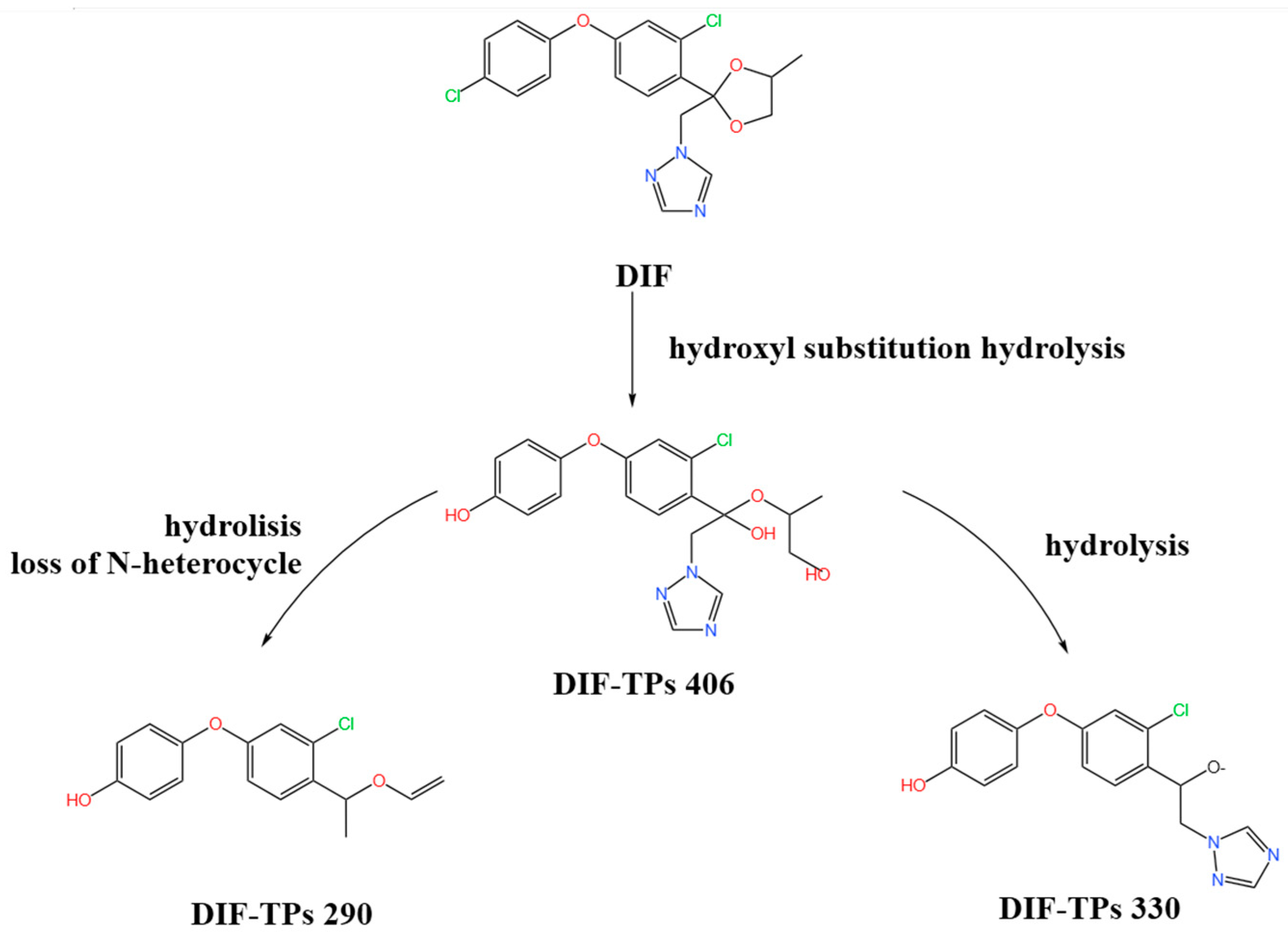
2.6. Characterization of DIF Degradation Products
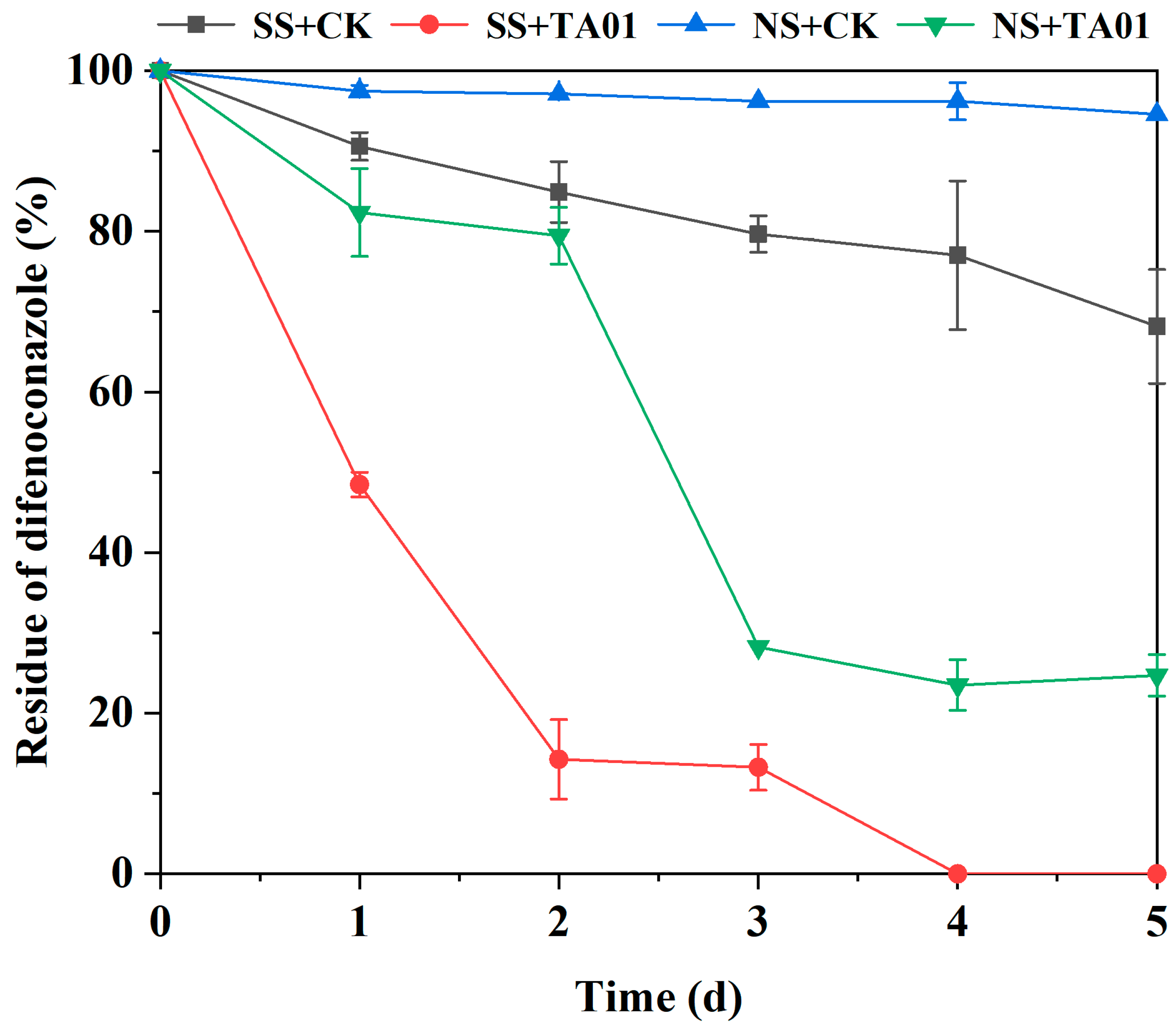
2.7. Biodegradation of DIF in Water–Sediment Simulated Pollution Systems
3. Materials and Methods
3.1. Enrichment and Screening of DIF-Degrading Microbial Consortia
3.2. Determination of DIF Degradation and Growth for Microbial Consortium TA01
3.3. Effect of Incubation Conditions on the Degradation of DIF by TA01
3.4. Kinetics of DIF Degradation
3.5. Analysis of the Community Structure and Key Degrading Bacteria of Microbial Consortium TA01
3.6. Identification of DIF Degradation Products
3.7. Bioremediation Potential of Microbial Consortium TA01
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Peng, K.; Pan, Y.; Tan, T.; Zeng, X.; Lin, M.; Jiang, S.; Zhao, Z.; Tian, F.; Zhao, X. Characterization and fungicide sensitivity of Colletotrichum godetiae causing sweet cherry fruit anthracnose in Guizhou, China. Front. Microbiol. 2022, 13, 923181. [Google Scholar]
- Zhou, J.; Wang, T.L.; Miao, Y.H.; Liu, D.H. Identification, biological characteristics, and fungicide screening of pathogen of black spot in Aconitum carmichaelii. Zhongguo Zhong Yao Za Zhi 2021, 47, 1215–1221. (In Chinese) [Google Scholar]
- Yin, H.; Zhou, J.; Lv, H.; Qin, N.; Chang, F.J.; Zhao, X. Identification, pathogenicity, and fungicide sensitivity of Ascochyta caulina (Teleomorph: Neocamarosporium calvescens) associated with black stem on quinoa in China. Plant Dis. 2020, 104, 2585–2597. [Google Scholar] [PubMed]
- Acosta-González, U.; Leyva-Mir, S.G.; Silva-Rojas, H.V.; Rebollar-Alviter, A. Preventive and Curative Effects of Treatments to Manage Strawberry Root and Crown Rot Caused by Neopestalotiopsis rosae. Plant Dis. 2024, 108, 1278–1288. [Google Scholar] [CrossRef]
- Maria, N.I.; Khan, R.; Alam, S.S.; Iqbal, O.; Akram, S.; Rajput, N.A.; Younas, M.U.; Qasim, M.; Ali, I.; Elsalahy, H.H.; et al. Unleashing the synergistic effect of promising fungicides: A breakthrough solution for combating powdery mildew in pea plants. Front. Microbiol. 2024, 15, 1448033. [Google Scholar]
- Khodadadi, F.; Martin, P.L.; Donahue, D.J.; Peter, K.A.; Aćimović, S.G. Characterizations of an emerging disease: Apple blotch caused by Diplocarpon coronariae (syn. Marssonina coronaria) in the mid-atlantic United States. Plant Dis. 2022, 106, 1803–1817. [Google Scholar]
- Park, J.; Kim, S.; Jo, M.; An, S.; Kim, Y.; Yoon, J.; Jeong, M.H.; Kim, E.Y.; Choi, J.; Kim, Y.; et al. Isolation and identification of Alternaria alternata from potato plants affected by leaf spot disease in Korea: Selection of Effective Fungicides. J. Fungi 2024, 10, 53. [Google Scholar] [CrossRef]
- Ao, X.; Shi, T.; Yang, W.; Ouyang, H.; Fan, R.; Siddiqui, J.A.; Wu, C.; Lv, Z.; Deng, S.; Chen, X. Biological characterization and in vitro fungicide screening of a new causal agent of walnut leaf spot in Guizhou Province, China. Front. Microbiol. 2024, 15, 1439487. [Google Scholar]
- Song, Y.J.; Zhao, N.L.; Dai, D.R.; Bao, R. Prospects of Pseudomonas in microbial fuel, bioremediation, and sustainability. ChemSusChem 2025, 18, e202401324. [Google Scholar] [CrossRef]
- Wang, C.; Hao, D.; Jiao, W.; Li, J.; Yuan, J.; Ma, Y.; Wang, X.; Xu, A.; Wang, M.; Wang, Y. Identification and fungicide sensitivity of Fusarium spp. associated with root rot of scutellaria baicalensis in Shanxi Province, China. Phytopathology 2024, 114, 1533–1541. [Google Scholar]
- Degani, O.; Dor, S.; Chen, A.; Orlov-Levin, V.; Stolov-Yosef, A.; Regev, D.; Rabinovitz, O. Molecular tracking and remote sensing to ealuate new chemical treatments against the maize late wilt disease causal agent, magnaporthiopsis maydis. J. Fungi 2020, 6, 54. [Google Scholar] [CrossRef]
- Khodadadi, F.; González, J.B.; Martin, P.L.; Giroux, E.; Bilodeau, G.J.; Peter, K.A.; Doyle, V.P.; Aćimović, S.G. Identification and characterization of Colletotrichum species causing apple bitter rot in New York and description of C. noveboracense sp. nov. Sci. Rep. 2020, 10, 11043. [Google Scholar]
- Yang, Q.; Deng, P.; Xing, D.; Liu, H.; Shi, F.; Hu, L.; Zou, X.; Nie, H.; Zuo, J.; Zhuang, Z.; et al. Developmental neurotoxicity of difenoconazole in zebrafish embryos. Toxics 2023, 11, 353. [Google Scholar] [CrossRef]
- Wu, X.; Han, H.; Xie, K.; He, N.; Yang, Z.; Jin, X.; Ma, S.; Dong, J. Difenoconazole disrupts carp intestinal physical barrier and causes inflammatory response via triggering oxidative stress and apoptosis. Pestic. Biochem. Physiol. 2023, 194, 105507. [Google Scholar] [CrossRef] [PubMed]
- Nataraj, B.; Hemalatha, D.; Malafaia, G.; Maharajan, K.; Ramesh, M. “Fishcide” effect of the fungicide difenoconazole in freshwater fish (Labeo rohita): A multi-endpoint approach. Sci. Total Environ. 2022, 857 Pt 2, 159425. [Google Scholar] [CrossRef]
- Zhai, Y.; Wang, S.; Zhang, B.; Tang, Y.; Wang, H.; Li, J.; Hu, Z.; Wang, Y.; Li, H.; Ge, R.S. The analysis of pesticides and fungicides in the inhibition of human and rat placental 3β-hydroxysteroid dehydrogenase activity: Mode of inhibition and mechanism. Toxicol. Lett. 2023, 379, 76–86. [Google Scholar] [CrossRef] [PubMed]
- Na, M.J.; Lee, W.Y.; Park, H.J. Difenoconazole induced damage of bovine mammary epithelial cells via ER stress and inflammatory response. Cells 2024, 13, 1715. [Google Scholar] [CrossRef]
- Qin, G.; Zhang, Q.; Zhang, Z.; Chen, Y.; Zhu, J.; Yang, Y.; Peijnenburg, W.J.G.M.; Qian, H. Understanding the ecological effects of the fungicide difenoconazole on soil and Enchytraeus crypticus gut microbiome. Environ. Pollut. 2023, 326, 121518. [Google Scholar] [CrossRef]
- Han, W.; Ye, Z.; Gu, Y.; Zhong, Y.; Gao, J.; Zhao, S.; Wang, S. Gut microbiota composition and gene expression changes induced in the Apis cerana exposed to acetamiprid and difenoconazole at environmentally realistic concentrations alone or combined. Front. Physiol. 2023, 14, 1174236. [Google Scholar] [CrossRef]
- Ganaie, M.I.; Jan, I.; Mayer, A.N.; Dar, A.A.; Mayer, I.A.; Ahmed, P.; Sofi, J.A. Health risk assessment of pesticide residues in drinking water of upper jhelum region in Kashmir Valley-India by GC-MS/MS. Int. J. Anal. Chem. 2023, 2023, 6802782. [Google Scholar] [CrossRef]
- Zhang, S.; Wang, F.; Wang, R.; Cai, M. Spatial assessment of triazole organic compounds in surface water from the coastal estuaries to the East China sea. Environ. Pollut. 2023, 320, 121024. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Tian, Z.; Han, A.; Li, J.; Luo, A.; Liu, R.; Zhang, Z. Integrative physiological, critical plant endogenous hormones, and transcriptomic analyses reveal the difenoconazole stress response mechanism in wheat (Triticum aestivum L.). Pestic. Biochem. Physiol. 2023, 197, 105688. [Google Scholar] [CrossRef]
- Liu, Y.; Liu, R.; Deng, Y.; Zheng, M.; Yu, S.; Nie, Y.; Li, J.Q.; Pan, C.; Zhou, Z.; Diao, J. Insights into the mechanism of flavor loss in strawberries induced by two fungicides integrating transcriptome and metabolome Analysis. J. Agric. Food Chem. 2023, 71, 3906–3919. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Feng, X.; Liu, X.; Zhang, L.; Mao, L.; Zhu, L.; Zheng, Y. Bioavailability assessment of difenoconazole to earthworms (Eisenia fetida) in soil by oleic acid-embedded cellulose acetate membrane. Sci. Total Environ. 2023, 905, 167276. [Google Scholar] [CrossRef]
- Altun, Ş.; Kadak, A.E.; Küçükgülmez, A.; Gülnaz, O.; Çelik, M. Explanation of difenoconazole removal by chitosan with Langmuir adsorption isotherm and kinetic modeling. Toxicol. Res. 2022, 39, 127–133. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Wang, L.; Sui, J.; Chen, F.; Wang, T.; Yang, J.; Chen, S.H.; Cui, X.; Yang, Y.; Zhang, W. Pathway elucidation and key enzymatic processes in the biodegradation of difenoconazole by Pseudomonas putida A-3. J. Agric. Food Chem. 2025, 73, 4770–4786. [Google Scholar] [CrossRef]
- Luong, T.T.; Nguyen, T.H.T.; Nguyen, T.D.; Le, V.T.; Pham, T.H.T.; Ho, T.T.; Nguyen, N.L. Degradation of triazole fungicides by plant growth-promoting bacteria from contaminated agricultural soil. J. Microbiol. Biotechnol. 2024, 34, 56–64. [Google Scholar] [CrossRef]
- Chen, X.; Peng, S.; Liu, M.; Wang, L.; Pang, K.; Zhang, L.; Cui, Z.; Liu, A. Highly efficient in-situ cleaner degradation of difenoconazole by two novel dominant strains: Microflora diversity, monoclonal isolation, growth factor optimization, intermediates, and pathways. Chemosphere 2023, 310, 136863. [Google Scholar] [CrossRef]
- Tan, Z.; Yang, X.; Chen, L.; Liu, Y.; Xu, H.J.; Li, Y.; Gong, B. Biodegradation Mechanism of Chloramphenicol by Aeromonas Media SZW3 and Genome Analysis. Bioresour. Technol. 2022, 344 Pt B, 126280. [Google Scholar] [CrossRef]
- Huang, Y.; Chen, W.J.; Li, J.; Ghorab, M.A.; Alansary, N.; El-Hefny, D.E.; El-Sayyad, G.S.; Mishra, S.; Zhang, X.; Bhatt, P.; et al. Novel mechanism and degradation kinetics of allethrin using Bacillus megaterium strain HLJ7 in contaminated soil/water environments. Environ. Res. 2022, 214 Pt 3, 113940. [Google Scholar] [CrossRef]
- Guo, Y.; Huang, Y.; Pang, S.; Zhou, T.; Lin, Z.; Yu, H.; Zhang, G.; Bhatt, P.; Chen, S. Novel mechanism and kinetics of tetramethrin degradation using an indigenous Gordonia Cholesterolivorans A16. Int. J. Mol. Sci. 2021, 22, 9242. [Google Scholar] [PubMed]
- Zhang, W.; Li, J.; Zhang, Y.; Wu, X.; Zhou, Z.; Huang, Y.; Zhao, Y.; Mishra, S.; Bhatt, P.; Chen, S. Characterization of a novel glyphosate-degrading bacterial species, Chryseobacterium sp. Y16C, and evaluation of its effects on microbial communities in glyphosate-contaminated soil. J. Hazard. Mater. 2022, 432, 128689. [Google Scholar] [PubMed]
- Sivasamy, S.; Rajangam, S.; Kanagasabai, T.; Bisht, D.; Prabhakaran, R.; Dhandayuthapani, S. Biocatalytic Potential of Pseudomonas Species in the Degradation of Polycyclic Aromatic Hydrocarbons. J. Basic Microbiol. 2025, 65, e2400448. [Google Scholar]
- Man, Y.; Stenrød, M.; Wu, C.; Almvik, M.; Holten, R.; Clarke, J.L.; Yuan, S.; Wu, X.; Xu, J.; Dong, F.; et al. Degradation of difenoconazole in water and soil: Kinetics, degradation pathways, transformation products identification and ecotoxicity assessment. J. Hazard. Mater. 2021, 418, 126303. [Google Scholar]
- Bhatt, P.; Rene, E.R.; Kumar, A.J.; Zhang, W.; Chen, S. Binding interaction of allethrin with esterase: Bioremediation potential and mechanism. Bioresour. Technol. 2020, 315, 123845. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample (d) | Sequences | Shannon | Simpson | Chao1 |
|---|---|---|---|---|
| 0 | 87,522 | 4.51 | 0.89 | 257.05 |
| 1 | 85,255 | 2.23 | 0.64 | 87.0 |
| 2 | 85,486 | 2.31 | 0.62 | 87.0 |
| 3 | 87,062 | 2.17 | 0.60 | 94.0 |
| 4 | 82,368 | 2.42 | 0.68 | 86.0 |
| 5 | 84,866 | 2.77 | 0.79 | 77.12 |
| 6 | 83,468 | 2.84 | 0.75 | 111.00 |
| 7 | 80,451 | 2.78 | 0.76 | 97.00 |
| Compounds | Chemical Structures | Molecular Formula | m/z [M+H] |
|---|---|---|---|
| DIF-TPs 406 | 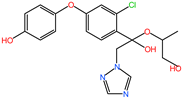 | C19H20ClN3O5 | 406.00 |
| DIF-TPs 330 | 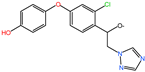 | C16H13ClN3O3 | 330.25 |
| DIF-TPs 290 | 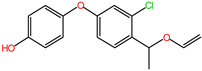 | C16H15ClO3 | 290.25 |
| Treatment | Regression Equation | k (d−1) | t1/2 (d) | R2 |
|---|---|---|---|---|
| SS + CK | Ct = 98.77213e−0.07067t | 0.07067 | 9.8 | 0.97974 |
| SS + TA01 | Ct = 100.84707e−0.81563t | 0.81563 | 0.9 | 0.98891 |
| NS + CK | Ct = 99.21675e−0.03094t | 0.03094 | 22.4 | 0.88518 |
| NS + TA01 | Ct = 106.36416e−0.30261t | 0.30261 | 2.3 | 0.86704 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, T.; Sui, J.; Zhou, Y.; Wang, L.; Yang, J.; Chen, F.; Cui, X.; Yang, Y.; Zhang, W. Difenoconazole Degradation by Novel Microbial Consortium TA01: Metabolic Pathway and Microbial Community Analysis. Int. J. Mol. Sci. 2025, 26, 3142. https://doi.org/10.3390/ijms26073142
Wang T, Sui J, Zhou Y, Wang L, Yang J, Chen F, Cui X, Yang Y, Zhang W. Difenoconazole Degradation by Novel Microbial Consortium TA01: Metabolic Pathway and Microbial Community Analysis. International Journal of Molecular Sciences. 2025; 26(7):3142. https://doi.org/10.3390/ijms26073142
Chicago/Turabian StyleWang, Tianyue, Jingyi Sui, Yi Zhou, Liping Wang, Jia Yang, Feiyu Chen, Xiuming Cui, Ye Yang, and Wenping Zhang. 2025. "Difenoconazole Degradation by Novel Microbial Consortium TA01: Metabolic Pathway and Microbial Community Analysis" International Journal of Molecular Sciences 26, no. 7: 3142. https://doi.org/10.3390/ijms26073142
APA StyleWang, T., Sui, J., Zhou, Y., Wang, L., Yang, J., Chen, F., Cui, X., Yang, Y., & Zhang, W. (2025). Difenoconazole Degradation by Novel Microbial Consortium TA01: Metabolic Pathway and Microbial Community Analysis. International Journal of Molecular Sciences, 26(7), 3142. https://doi.org/10.3390/ijms26073142