

Novel 5-Oxopyrrolidine-3-carbohydrazides as Potent Protein Kinase Inhibitors: Synthesis, Anticancer Evaluation, and Molecular Modeling

 , , , , and
, , , , and 
Abstract

1. Introduction
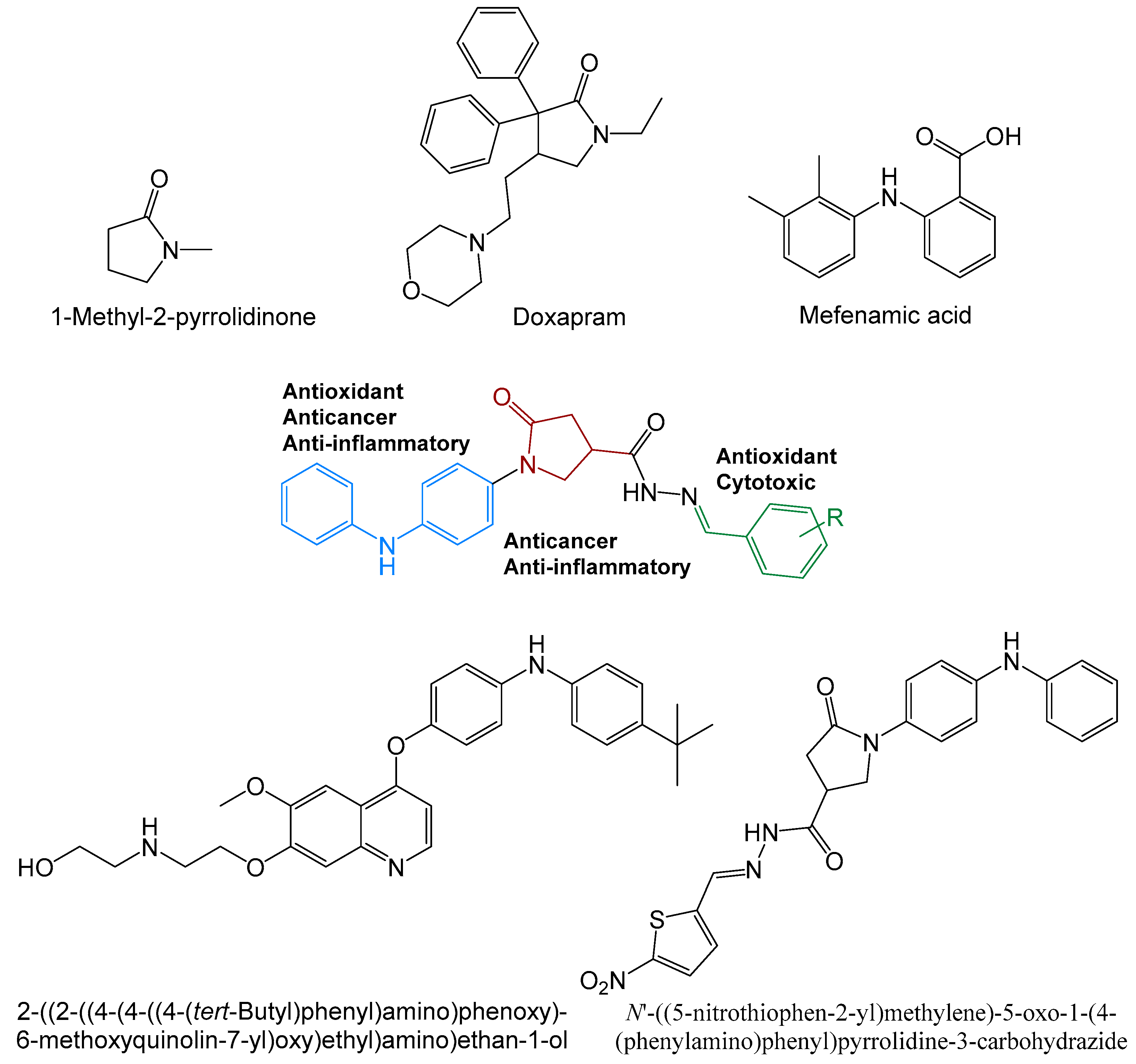
2. Results and Discussion
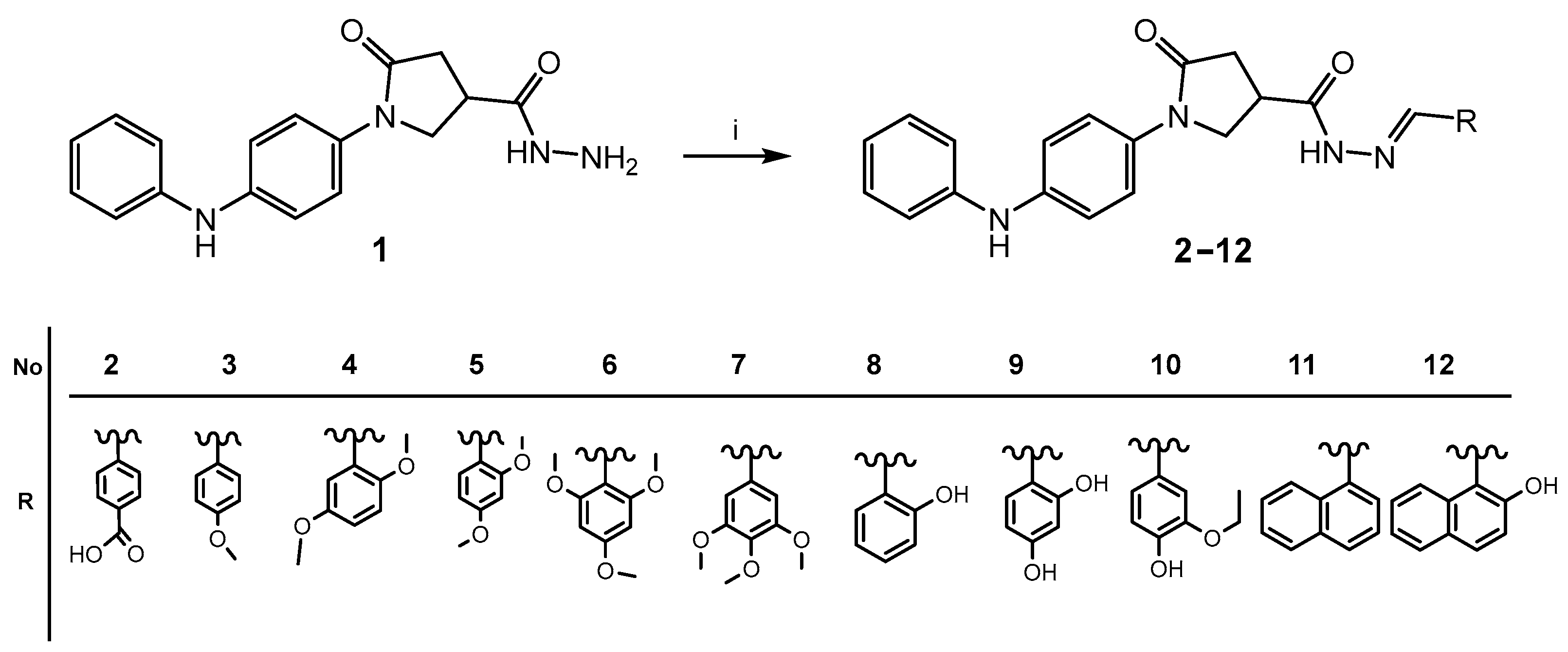
2.1. Chemistry
2.2. Pharmacology
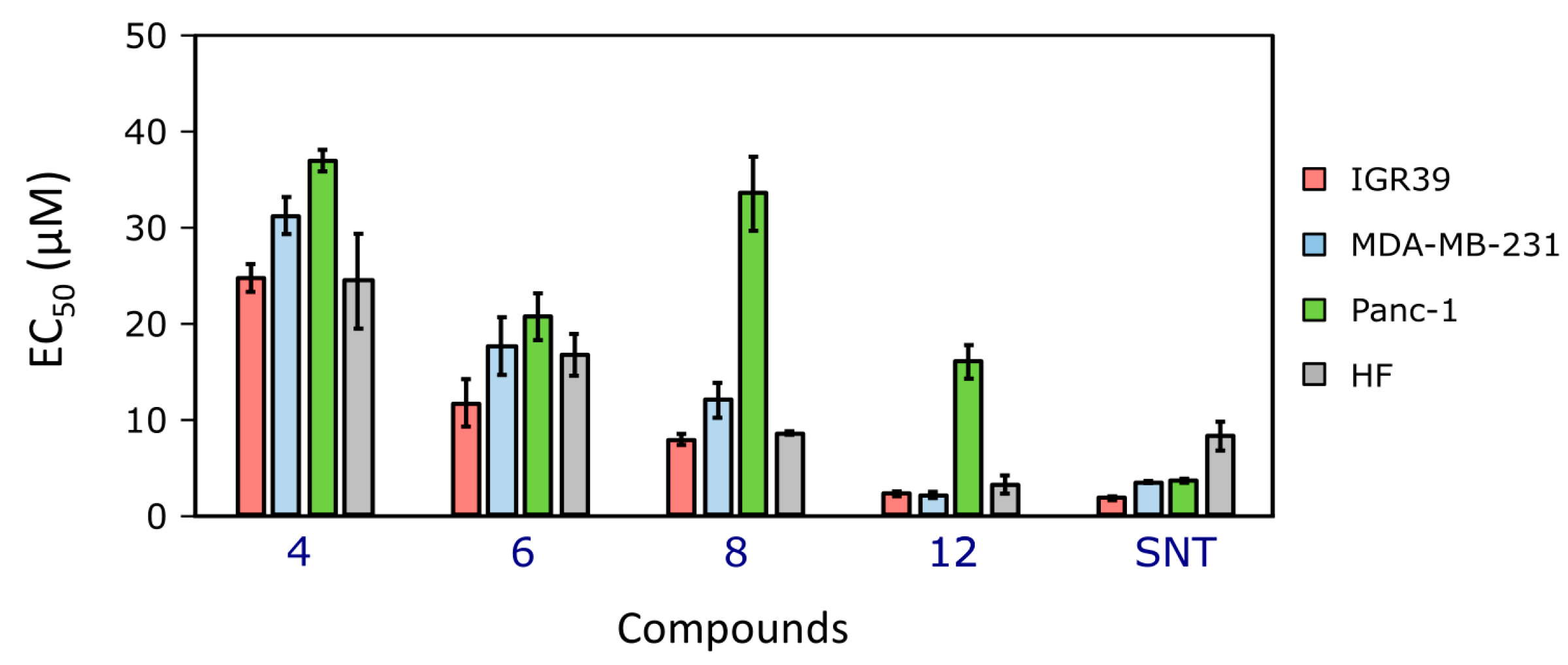
2.2.1. The Compounds’ Effects on Cancer Cell Viability
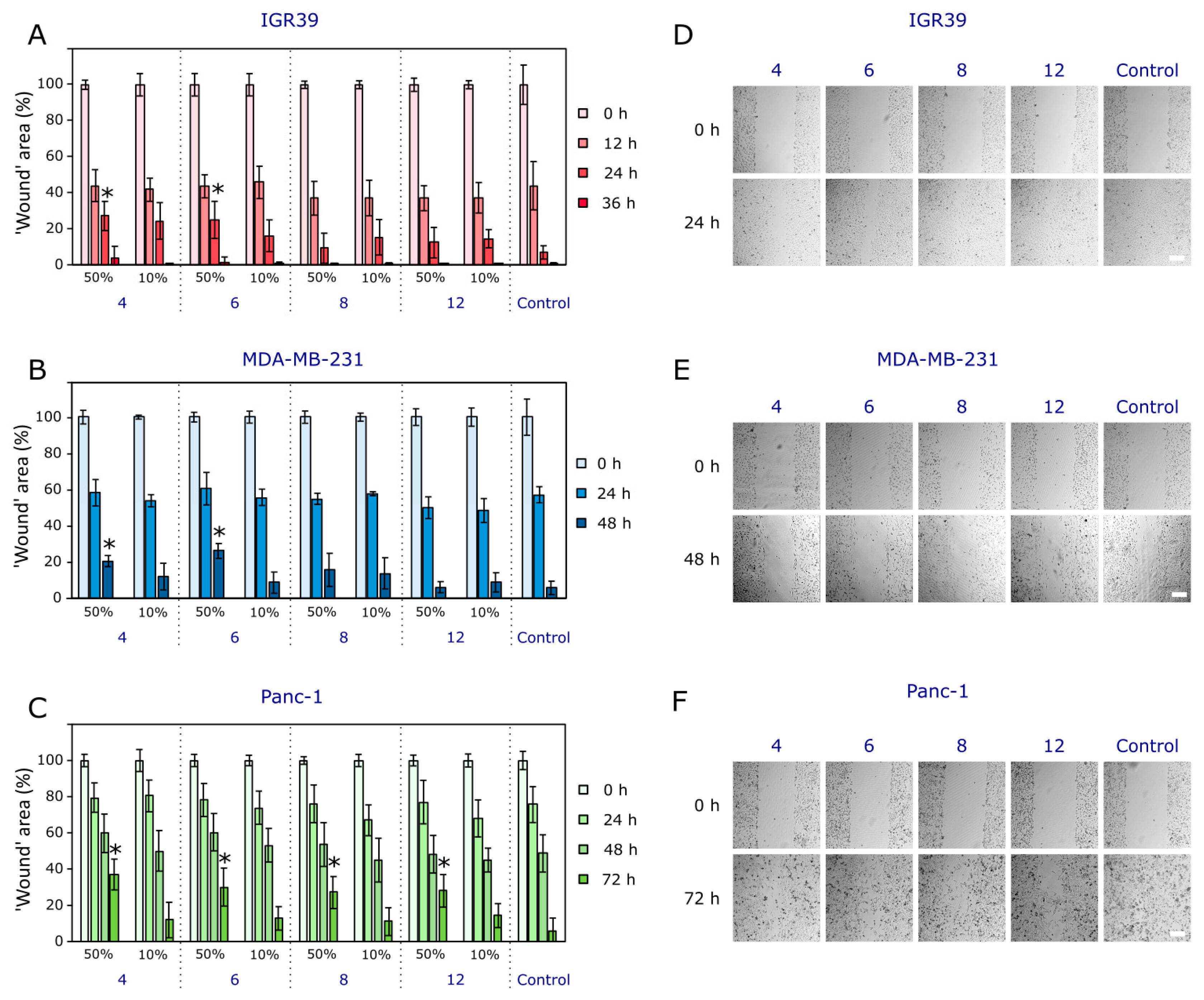
2.2.2. The Compounds’ Effects on Cancer Cell Migration
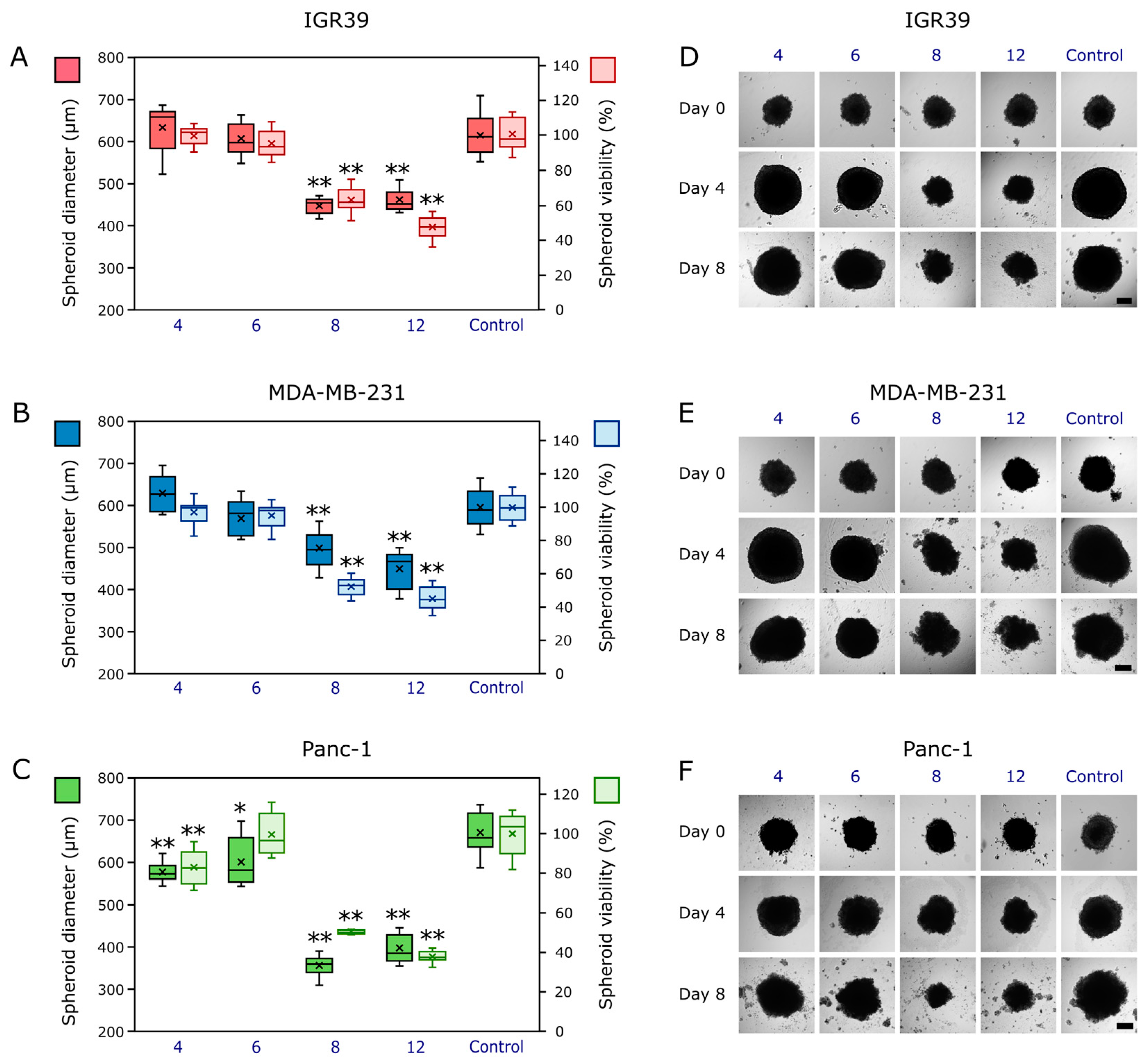
2.2.3. The Compounds’ Effects on Cancer Cell 3D Models (Cancer Spheroids)
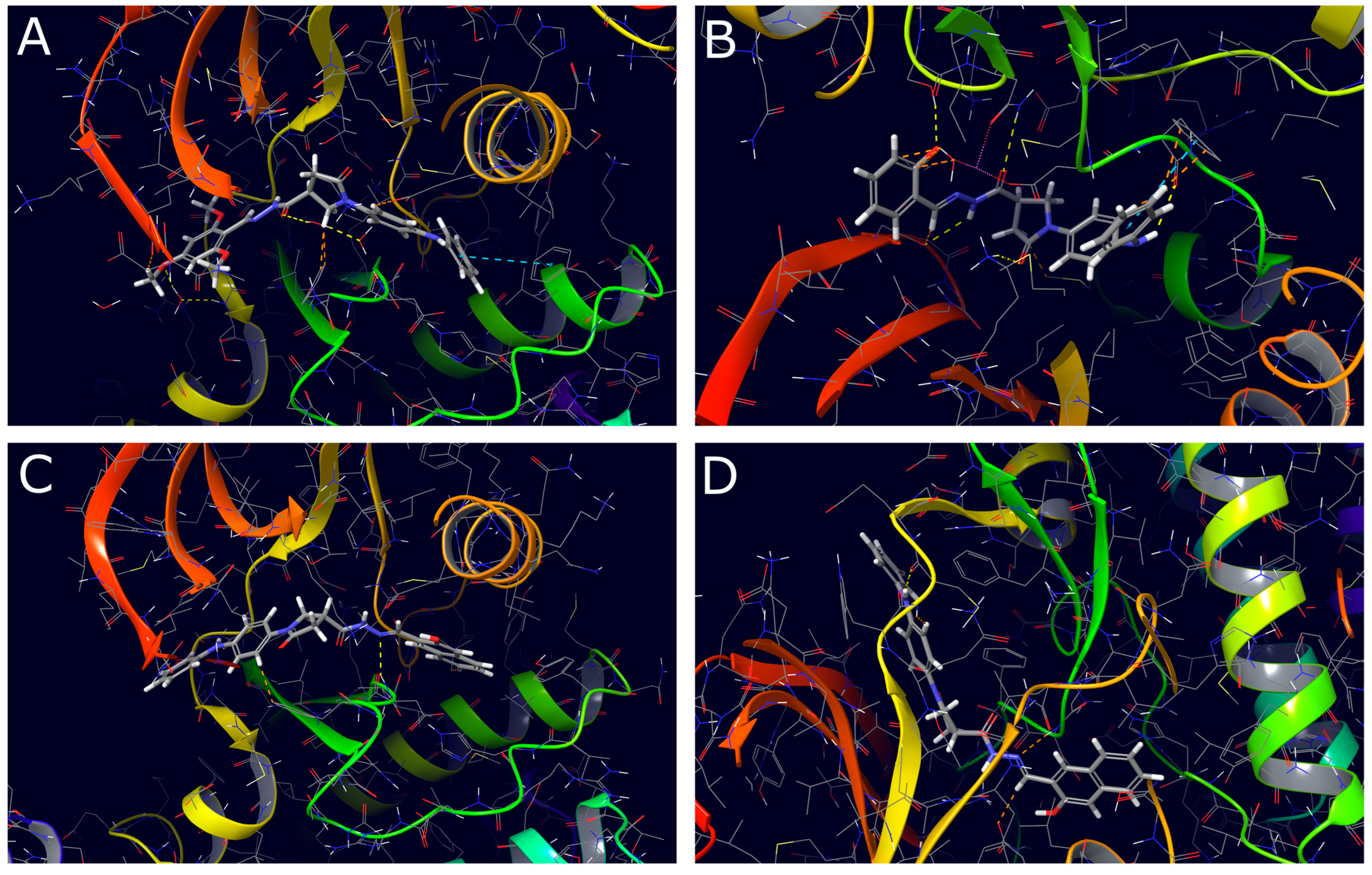
2.3. Molecular Docking Study
3. Materials and Methods
3.1. Chemistry
General Procedure for the Synthesis of Compounds 2–12
3.2. Biological Activity
3.2.1. Cell Culture
3.2.2. Cytotoxicity Assay
3.2.3. ‘Wound Healing’ Assay
3.2.4. Compound Activity Against Spheroids
3.2.5. Statistical Analysis
3.3. Molecular Docking
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Wagle, N.S.; Jemal, A. Cancer Statistics, 2023. CA A Cancer J. Clin. 2023, 73, 17–48. [Google Scholar] [CrossRef]
- Cooper, G.M. The Cell: A Molecular Approach. In The Development and Causes of Cancer, 2nd ed.; Sinauer Associates: Sunderland, MA, USA, 2000. Available online: https://www.ncbi.nlm.nih.gov/books/nbk9963/ (accessed on 8 June 2024).
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Theivendren, P.; Kunjiappan, S.; Mariappa Hegde, Y.; Vellaichamy, S.; Gopal, M.; Rajan Dhramalingam, S.; Kumar, S. Importance of Protein Kinase and Its Inhibitor: A Review. In Biochemistry; Kumar Singh, R., Ed.; IntechOpen: Rijeka, Croatia, 2021; Volume 24, ISBN 978-1-83880-906-5. [Google Scholar]
- Bhullar, K.S.; Lagarón, N.O.; McGowan, E.M.; Parmar, I.; Jha, A.; Hubbard, B.P.; Rupasinghe, H.P.V. Kinase-Targeted Cancer Therapies: Progress, Challenges and Future Directions. Mol. Cancer 2018, 17, 48. [Google Scholar] [CrossRef]
- Caksa, S.; Baqai, U.; Aplin, A.E. The Future of Targeted Kinase Inhibitors in Melanoma. Pharmacol. Ther. 2022, 239, 108200. [Google Scholar] [CrossRef] [PubMed]
- Bansal, I.; Pandey, A.K.; Ruwali, M. Small-Molecule Inhibitors of Kinases in Breast Cancer Therapy: Recent Advances, Opportunities, and Challenges. Front. Pharmacol. 2023, 14, 1244597. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.N.; Patil, K.; Ma, J.; Dufek, G.A.; Pai, S.B. Multifaceted Effects of Kinase Inhibitors on Pancreatic Cancer Cells Reveals Pivotal Entities with Therapeutic Implications. Biomedicines 2023, 11, 1716. [Google Scholar] [CrossRef]
- Gagic, Z.; Ruzic, D.; Djokovic, N.; Djikic, T.; Nikolic, K. In Silico Methods for Design of Kinase Inhibitors as Anticancer Drugs. Front. Chem. 2020, 7, 873. [Google Scholar] [CrossRef]
- Easty, D.J.; Gray, S.G.; O’Byrne, K.J.; O’Donnell, D.; Bennett, D.C. Receptor Tyrosine Kinases and Their Activation in Melanoma. Pigment. Cell Melanoma Res. 2011, 24, 446–461. [Google Scholar] [CrossRef]
- Livingstone, E.; Zimmer, L.; Vaubel, J.; Schadendorf, D. BRAF, MEK and KIT Inhibitors for Melanoma: Adverse Events and Their Management. Chin. Clin. Oncol. 2014, 3, 29. [Google Scholar]
- Iancu, G.; Serban, D.; Badiu, C.; Tanasescu, C.; Tudosie, M.; Tudor, C.; Costea, D.; Zgura, A.; Iancu, R.; Vasile, D. Tyrosine Kinase Inhibitors in Breast Cancer (Review). Exp. Ther. Med. 2021, 23, 114. [Google Scholar] [CrossRef]
- DiPippo, A.J.; Patel, N.K.; Barnett, C.M. Cyclin-Dependent Kinase Inhibitors for the Treatment of Breast Cancer: Past, Present, and Future. Pharmacotherapy 2016, 36, 652–667. [Google Scholar] [CrossRef] [PubMed]
- Grobbelaar, C.; Steenkamp, V.; Mabeta, P. Vascular Endothelial Growth Factor Receptors in the Vascularization of Pancreatic Tumors: Implications for Prognosis and Therapy. Curr. Issues Mol. Biol. 2025, 47, 179. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhang, T.; Tu, S.; Zhang, Z.; Meng, F. Identification of Novel Src Inhibitors: Pharmacophore-Based Virtual Screening, Molecular Docking and Molecular Dynamics Simulations. Molecules 2020, 25, 4094. [Google Scholar] [CrossRef] [PubMed]
- Zhu, C.; Wei, Y.; Wei, X. AXL Receptor Tyrosine Kinase as a Promising Anti-Cancer Approach: Functions, Molecular Mechanisms and Clinical Applications. Mol. Cancer 2019, 18, 153. [Google Scholar] [CrossRef]
- Mahajan, K.; Mahajan, N.P. ACK1 Tyrosine Kinase: Targeted Inhibition to Block Cancer Cell Proliferation. Cancer Lett. 2013, 338, 185–192. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Wang, X.; Li, L.; Han, B. Research Progress of the Functional Role of ACK1 in Breast Cancer. BioMed Res. Int. 2019, 2019, 1018034. [Google Scholar] [CrossRef]
- Lawrence, H.R.; Luo, Y.; Zhang, D.; Tindall, N.; Ozcan, S.; Huseyin, M.; Kazi, S.; Bandyopadhyay, S.; Mahajan, K.; Mahajan, N.P.; et al. Abstract 2511: New Inhibitors the Tyrosine Kinase ACK1/TNK2 Active in Prostate, Breast and Pancreatic Cancer. Cancer Res. 2014, 74, 2511. [Google Scholar] [CrossRef]
- Kumar, V.; Kumar, R.; Parate, S.; Yoon, S.; Lee, G.; Kim, D.; Lee, K.W. Identification of ACK1 Inhibitors as Anticancer Agents by Using Computer-Aided Drug Designing. J. Mol. Struct. 2021, 1235, 130200. [Google Scholar] [CrossRef]
- Shyam Sunder, S.; Sharma, U.C.; Pokharel, S. Adverse Effects of Tyrosine Kinase Inhibitors in Cancer Therapy: Pathophysiology, Mechanisms and Clinical Management. Signal Transduct. Target. Ther. 2023, 8, 262. [Google Scholar] [CrossRef]
- Yin, H.; Huang, T.; Shi, B.; Cao, W.; Yu, C.; Li, T.; Zhang, K.; Yao, C. Synthesis of 2-Pyrrolidinone Derivatives via N-Heterocyclic Carbene Catalyzed Radical Tandem Cyclization/Coupling Reactions. Org. Chem. Front. 2023, 10, 2695–2700. [Google Scholar] [CrossRef]
- Doxapram. DRUGBANK Online. Available online: https://go.drugbank.com/drugs/db00561 (accessed on 10 June 2024).
- Rolipram. DRUGBANK Online. Available online: https://go.drugbank.com/drugs/db01954 (accessed on 10 June 2024).
- 1-Methylpyrrolidinone. DRUGBANK Online. Available online: https://go.drugbank.com/drugs/db12521 (accessed on 10 June 2024).
- Albratty, M. Quantitative Structure–Activity Relationship Modeling and Docking of Some Synthesized Bioactive Oxopyrolidines against Staphylococcus Aureus. J. Saudi Chem. Soc. 2022, 26, 101509. [Google Scholar] [CrossRef]
- Zhu, X.-L.; Tian, X.-Q.; Xu, H.-H.; Wang, H.-M.; Chen, Q.-H.; Zeng, X.-H. Rhopaladins’ Analogue (E)-2-Aroyl-4-(4-Fluorobenzylidene)-5-Oxopyrrolidines Inhibit Proliferation, Promote Apoptosis and down-Regulation of E6/E7 mRNA in Cervical Cancer. Bioorg Med. Chem. Lett. 2020, 30, 127554. [Google Scholar] [CrossRef]
- Muralidharan, V.P.; Alagumuthu, M.; Iyer, S.K. Iodine Catalyzed Three Component Synthesis of 1-((2-Hydroxy Naphthalen-1-Yl)(Phenyl)(Methyl))Pyrrolidin-2-One Derivatives: Rationale as Potent PI3K Inhibitors and Anticancer Agents. Bioorg Med. Chem. Lett. 2017, 27, 2510–2514. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, Y.; Tokuhara, H.; Ohba, Y.; Okabe, A.; Nakayama, M.; Nakagawa, H.; Skene, R.; Hoffman, I.; Zou, H.; Yoshida, M. Efficient Synthesis of Tert-Butyl 3-Cyano-3-Cyclopropyl-2-Oxopyrrolidine-4-Carboxylates: Highly Functionalized 2-Pyrrolidinone Enabling Access to Novel Macrocyclic Tyk2 Inhibitors. Bioorg Med. Chem. Lett. 2020, 30, 126963. [Google Scholar] [CrossRef]
- Kumar, A.; Mishra, A.K. Pharmacological Applications of Diphenylamine and Its Derivative as Potent Bioactive Compound: A Review. Curr. Bioact. Comp. 2018, 14, 217–233. [Google Scholar] [CrossRef]
- Shimizu, T.; Fujiwara, Y.; Osawa, T.; Sakai, T.; Kubo, K.; Kubo, K.; Nishitoba, T.; Kimura, K.; Senga, T.; Murooka, H.; et al. Orally Active Anti-Proliferation Agents: Novel Diphenylamine Derivatives as FGF-R2 Autophosphorylation Inhibitors. Bioorg Med. Chem. Lett. 2004, 14, 875–879. [Google Scholar] [CrossRef] [PubMed]
- Abou-Seri, S.M. Synthesis and Biological Evaluation of Novel 2,4′-Bis Substituted Diphenylamines as Anticancer Agents and Potential Epidermal Growth Factor Receptor Tyrosine Kinase Inhibitors. Eur. J. Med. Chem. 2010, 45, 4113–4121. [Google Scholar] [CrossRef]
- Yan, X.-Y.; Leng, J.-F.; Chen, T.-T.; Zhao, Y.-J.; Kong, L.-Y.; Yin, Y. Design, Synthesis, and Biological Evaluation of Novel Diphenylamine Derivatives as Tubulin Polymerization Inhibitors Targeting the Colchicine Binding Site. Eur. J. Med. Chem. 2022, 237, 114372. [Google Scholar] [CrossRef]
- Zubrickė, I.; Jonuškienė, I.; Kantminienė, K.; Tumosienė, I.; Petrikaitė, V. Synthesis and In Vitro Evaluation as Potential Anticancer and Antioxidant Agents of Diphenylamine-Pyrrolidin-2-One-Hydrazone Derivatives. Int. J. Mol. Sci. 2023, 24, 16804. [Google Scholar] [CrossRef]
- Agu, P.C.; Afiukwa, C.A.; Orji, O.U.; Ezeh, E.M.; Ofoke, I.H.; Ogbu, C.O.; Ugwuja, E.I.; Aja, P.M. Molecular Docking as a Tool for the Discovery of Molecular Targets of Nutraceuticals in Diseases Management. Sci. Rep. 2023, 13, 13398. [Google Scholar] [CrossRef]
- Torres, P.H.M.; Sodero, A.C.R.; Jofily, P.; Silva-Jr, F.P. Key Topics in Molecular Docking for Drug Design. Int. J. Mol. Sci. 2019, 20, 4574. [Google Scholar] [CrossRef]
- Olszewski, M.; Stasevych, M.; Zvarych, V.; Maciejewska, N. 9,10-Dioxoanthracenyldithiocarbamates Effectively Inhibit the Proliferation of Non-Small Cell Lung Cancer by Targeting Multiple Protein Tyrosine Kinases. J. Enzym. Inhib. Med. Chem. 2024, 39, 2284113. [Google Scholar] [CrossRef] [PubMed]
- Maciejewska, N.; Olszewski, M.; Jurasz, J.; Baginski, M.; Stasevych, M.; Zvarych, V.; Folini, M.; Zaffaroni, N. Teloxantron Inhibits the Processivity of Telomerase with Preferential DNA Damage on Telomeres. Cell Death Dis. 2022, 13, 1005. [Google Scholar] [CrossRef] [PubMed]
- Tumosienė, I.; Kantminienė, K.; Klevinskas, A.; Petrikaitė, V.; Jonuškienė, I.; Mickevičius, V. Antioxidant and Anticancer Activity of Novel Derivatives of 3-[(4-Methoxyphenyl)Amino]Propanehydrazide. Molecules 2020, 25, 2980. [Google Scholar] [CrossRef]
- Tumosienė, I.; Jonuškienė, I.; Kantminienė, K.; Mickevičius, V.; Petrikaitė, V. Novel N-Substituted Amino Acid Hydrazone-Isatin Derivatives: Synthesis, Antioxidant Activity, and Anticancer Activity in 2D and 3D Models In Vitro. Int. J. Mol. Sci. 2021, 22, 7799. [Google Scholar] [CrossRef] [PubMed]
- Šermukšnytė, A.; Kantminienė, K.; Jonuškienė, I.; Tumosienė, I.; Petrikaitė, V. The Effect of 1,2,4-Triazole-3-Thiol Derivatives Bearing Hydrazone Moiety on Cancer Cell Migration and Growth of Melanoma, Breast, and Pancreatic Cancer Spheroids. Pharmaceuticals 2022, 15, 1026. [Google Scholar] [CrossRef]
- Kostova, I.; Saso, L. Advances in Research of Schiff-Base Metal Complexes as Potent Antioxidants. Curr. Med. Chem. 2013, 20, 4609–4632. [Google Scholar] [CrossRef]
- Rollas, S.; Küçükgüzel, S. Biological Activities of Hydrazone Derivatives. Molecules 2007, 12, 1910–1939. [Google Scholar] [CrossRef]
- Alam, M.; Verma, G.; Shaquiquzzaman, M.; Marella, A.; Akhtar, M.; Ali, M. A Review Exploring Biological Activities of Hydrazones. J. Pharm. Bioall Sci. 2014, 6, 69. [Google Scholar] [CrossRef]
- de Oliveira Carneiro Brum, J.; França, T.C.C.; LaPlante, S.R.; Villar, J.D.F. Synthesis and Biological Activity of Hydrazones and Derivatives: A Review. Mini Rev. Med. Chem. 2020, 20, 342–368. [Google Scholar] [CrossRef]
- Demurtas, M.; Baldisserotto, A.; Lampronti, I.; Moi, D.; Balboni, G.; Pacifico, S.; Vertuani, S.; Manfredini, S.; Onnis, V. Indole Derivatives as Multifunctional Drugs: Synthesis and Evaluation of Antioxidant, Photoprotective and Antiproliferative Activity of Indole Hydrazones. Bioorg Chem. 2019, 85, 568–576. [Google Scholar] [CrossRef] [PubMed]
- Han, M.İ.; Yeşil Baysal, Ö.D.; Başaran, G.Ş.; Sezer, G.; Telci, D.; Küçükgüzel, Ş.G. Design, Synthesis and Anticancer Activity Studies of Novel 4-Butylaminophenyl Hydrazide-Hydrazones as Apoptotic Inducers. Tetrahedron 2022, 115, 132797. [Google Scholar] [CrossRef]
- Manickam, M.; Boggu, P.R.; Cho, J.; Nam, Y.J.; Lee, S.J.; Jung, S.-H. Investigation of Chemical Reactivity of 2-Alkoxy-1,4-Naphthoquinones and Their Anticancer Activity. Bioorganic Med. Chem. Lett. 2018, 28, 2023–2028. [Google Scholar] [CrossRef]
- Marquina, S.; Maldonado-Santiago, M.; Sánchez-Carranza, J.N.; Antúnez-Mojica, M.; González-Maya, L.; Razo-Hernández, R.S.; Alvarez, L. Design, Synthesis and QSAR Study of 2′-Hydroxy-4′-Alkoxy Chalcone Derivatives That Exert Cytotoxic Activity by the Mitochondrial Apoptotic Pathway. Bioorganic Med. Chem. 2019, 27, 43–54. [Google Scholar] [CrossRef]
- Tisovský, P.; Csicsai, K.; Donovalová, J.; Šandrik, R.; Sokolík, R.; Gáplovský, A. Effect of a =X-NH-Fragment, (X = C, N), on Z/E Isomerization and ON/OFF Functionality of Isatin Arylhydrazones, ((Arylamino)Methylene)Indolin-2-Ones and Their Anions. Molecules 2020, 25, 3082. [Google Scholar] [CrossRef]
- Quiñonero, F.; Mesas, C.; Doello, K.; Cabeza, L.; Perazzoli, G.; Jimenez-Luna, C.; Rosa Rama, A.; Melguizo, C.; Prados, J. The Challenge of Drug Resistance in Pancreatic Ductal Adenocarcinoma: A Current Overview. Cancer Biol. Med. 2019, 16, 688–699. [Google Scholar] [CrossRef]
- Bedia, C.; Casas, J.; Andrieu-Abadie, N.; Fabriàs, G.; Levade, T. Acid Ceramidase Expression Modulates the Sensitivity of A375 Melanoma Cells to Dacarbazine. J. Biol. Chem. 2011, 286, 28200–28209. [Google Scholar] [CrossRef]
- Gentilcore, G.; Madonna, G.; Mozzillo, N.; Ribas, A.; Cossu, A.; Palmieri, G.; Ascierto, P.A. Effect of Dabrafenib on Melanoma Cell Lines Harbouring the BRAF V600D/R Mutations. BMC Cancer 2013, 13, 17. [Google Scholar] [CrossRef]
- Kapałczyńska, M.; Kolenda, T.; Przybyła, W.; Zajączkowska, M.; Teresiak, A.; Filas, V.; Ibbs, M.; Bliźniak, R.; Łuczewski, Ł.; Lamperska, K. 2D and 3D Cell Cultures—A Comparison of Different Types of Cancer Cell Cultures. Arch. Med. Sci. 2016, 14, 910–919. [Google Scholar] [CrossRef]
- Wong, C.C.; Cheng, K.-W.; Rigas, B. Preclinical Predictors of Anticancer Drug Efficacy: Critical Assessment with Emphasis on Whether Nanomolar Potency Should Be Required of Candidate Agents: TABLE 1. J. Pharmacol. Exp. Ther. 2012, 341, 572–578. [Google Scholar] [CrossRef]
- Zhang, L.; Luga, V.; Armitage, S.K.; Musiol, M.; Won, A.; Yip, C.M.; Plotnikov, S.V.; Wrana, J.L. A Lateral Signalling Pathway Coordinates Shape Volatility during Cell Migration. Nat. Commun. 2016, 7, 11714. [Google Scholar] [CrossRef] [PubMed]
- Riahi, R.; Yang, Y.; Zhang, D.D.; Wong, P.K. Advances in Wound-Healing Assays for Probing Collective Cell Migration. SLAS Technol. 2012, 17, 59–65. [Google Scholar] [CrossRef]
- Yakavets, I.; Francois, A.; Benoit, A.; Merlin, J.-L.; Bezdetnaya, L.; Vogin, G. Advanced Co-Culture 3D Breast Cancer Model for Investigation of Fibrosis Induced by External Stimuli: Optimization Study. Sci. Rep. 2020, 10, 21273. [Google Scholar] [CrossRef]
- Däster, S.; Amatruda, N.; Calabrese, D.; Ivanek, R.; Turrini, E.; Droeser, R.A.; Zajac, P.; Fimognari, C.; Spagnoli, G.C.; Iezzi, G.; et al. Induction of Hypoxia and Necrosis in Multicellular Tumor Spheroids Is Associated with Resistance to Chemotherapy Treatment. Oncotarget 2017, 8, 1725–1736. [Google Scholar] [CrossRef] [PubMed]
- Kairytė, K.; Vaickelionienė, R.; Grybaitė, B.; Anusevičius, K.; Mickevičius, V.; Petrikaitė, V. The Effect of 4-(Dimethylamino)Phenyl-5-Oxopyrrolidines on Breast and Pancreatic Cancer Cell Colony Formation, Migration, and Growth of Tumor Spheroids. Int. J. Mol. Sci. 2024, 25, 1834. [Google Scholar] [CrossRef]
- Tanda, E.T.; Vanni, I.; Boutros, A.; Andreotti, V.; Bruno, W.; Ghiorzo, P.; Spagnolo, F. Current State of Target Treatment in BRAF Mutated Melanoma. Front. Mol. Biosci. 2020, 7, 154. [Google Scholar] [CrossRef]
- Masuda, H.; Zhang, D.; Bartholomeusz, C.; Doihara, H.; Hortobagyi, G.N.; Ueno, N.T. Role of Epidermal Growth Factor Receptor in Breast Cancer. Breast Cancer Res. Treat. 2012, 136, 331–345. [Google Scholar] [CrossRef]
- Jeong, K.-Y.; Park, S.Y.; Park, M.H.; Kim, H.M. Suppressing Src-Mediated EGFR Signaling by Sustained Calcium Supply Targeting Triple-Negative Breast Cancer. Int. J. Mol. Sci. 2023, 24, 13291. [Google Scholar] [CrossRef]
- Lawrence, H.R.; Mahajan, K.; Luo, Y.; Zhang, D.; Tindall, N.; Huseyin, M.; Gevariya, H.; Kazi, S.; Ozcan, S.; Mahajan, N.P.; et al. Development of Novel ACK1/TNK2 Inhibitors Using a Fragment-Based Approach. J. Med. Chem. 2015, 58, 2746–2763. [Google Scholar] [CrossRef]
- Linderholm, B.K.; Hellborg, H.; Johansson, U.; Elmberger, G.; Skoog, L.; Lehtiö, J.; Lewensohn, R. Significantly Higher Levels of Vascular Endothelial Growth Factor (VEGF) and Shorter Survival Times for Patients with Primary Operable Triple-Negative Breast Cancer. Ann. Oncol. 2009, 20, 1639–1646. [Google Scholar] [CrossRef]
- Eggers, J.P.; Grandgenett, P.M.; Collisson, E.C.; Lewallen, M.E.; Tremayne, J.; Singh, P.K.; Swanson, B.J.; Andersen, J.M.; Caffrey, T.C.; High, R.R.; et al. Cyclin-Dependent Kinase 5 Is Amplified and Overexpressed in Pancreatic Cancer and Activated by Mutant K-Ras. Clin. Cancer Res. 2011, 17, 6140–6150. [Google Scholar] [CrossRef] [PubMed]
- Mahajan, K.; Mahajan, N.P. ACK1/TNK2 Tyrosine Kinase: Molecular Signaling and Evolving Role in Cancers. Oncogene 2015, 34, 4162–4167. [Google Scholar] [CrossRef] [PubMed]
- Murugappan, S.; Dastari, S.; Jungare, K.; Barve, N.M.; Shankaraiah, N. Hydrazide-Hydrazone/Hydrazone as Enabling Linkers in Anti-Cancer Drug Discovery: A Comprehensive Review. J. Mol. Struct. 2024, 1307, 138012. [Google Scholar] [CrossRef]
- Bytautaite, M.; Petrikaite, V. Comparative Study of Lipophilic Statin Activity in 2D and 3D in Vitro Models of Human Breast Cancer Cell Lines MDA-MB-231 and MCF-7. OncoTargets Ther. 2020, 13, 13201–13209. [Google Scholar] [CrossRef]
- Skaraitė, I.; Maccioni, E.; Petrikaitė, V. Anticancer Activity of Sunitinib Analogues in Human Pancreatic Cancer Cell Cultures under Normoxia and Hypoxia. Int. J. Mol. Sci. 2023, 24, 5422. [Google Scholar] [CrossRef]
- Schrödinger Release 2024-1: Maestro; Schrödinger LLC: New York, NY, USA, 2024.
- RCSB Protein Data Bank (RCSB PDB). Available online: https://www.rcsb.org (accessed on 10 October 2023).
- Schrödinger Release 2024-1: Protein Preparation Wizard Epik; Schrödinger, LLC: New York, NY, USA, 2024.
- Schrödinger Release 2024-1: LigPrep; Schrödinger, LLC: New York, NY, USA, 2024.
- Schrödinger Release 2024-1: Glide; Schrödinger, LLC: New York, NY, USA, 2024.



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Ligand | Non-Receptor Tyrosine Kinases | Serine Threonine Protein Kinases | ||||||
|---|---|---|---|---|---|---|---|---|
| ACK-1 (PDB: 5ZXB) | SCR (PDB: 3F3V) | BRAF (PDB: 1UWH) | MEK (PDB: 4U7Z) | |||||
| Docking Score kcal/mol | Glide Emodel kcal/mol | Docking Score kcal/mol | Glide Emodel kcal/mol | Docking Score kcal/mol | Glide Emodel kcal/mol | Docking Score kcal/mol | Glide Emodel kcal/mol | |
| 4 | −9.604 | −85.377 | −10.113 | −99.691 | −10.546 | −103.191 | −10.115 | −104.676 |
| 6 | −10.057 | −98.268 | −10.067 | −98.824 | −10.313 | −105.035 | −10.588 | −124.007 |
| 8 | −10.272 | −88.632 | −9.907 | −101.847 | −10.504 | −90.466 | −10.885 | −171.810 |
| 12 | −9.929 | −96.430 | −11.174 | −113.405 | −11.471 | −101.653 | −9.158 | −145.819 |
| Dasatinib | −9.412 | −108.418 | −9.758 | −115.364 | −10.428 | −101.336 | −8.052 | −104.318 |
| Trametinib | −7.171 | −67.301 | −5.636 | −37.185 | −10.256 | −62.904 | −9.545 | −111.156 |
| Vemurafenib | −8.322 | −86.139 | −7.201 | −70.350 | −11.313 | −96.824 | −7.275 | −89.902 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tumosienė, I.; Stasevych, M.; Zvarych, V.; Jonuškienė, I.; Kantminienė, K.; Petrikaitė, V. Novel 5-Oxopyrrolidine-3-carbohydrazides as Potent Protein Kinase Inhibitors: Synthesis, Anticancer Evaluation, and Molecular Modeling. Int. J. Mol. Sci. 2025, 26, 3162. https://doi.org/10.3390/ijms26073162
Tumosienė I, Stasevych M, Zvarych V, Jonuškienė I, Kantminienė K, Petrikaitė V. Novel 5-Oxopyrrolidine-3-carbohydrazides as Potent Protein Kinase Inhibitors: Synthesis, Anticancer Evaluation, and Molecular Modeling. International Journal of Molecular Sciences. 2025; 26(7):3162. https://doi.org/10.3390/ijms26073162
Chicago/Turabian StyleTumosienė, Ingrida, Maryna Stasevych, Viktor Zvarych, Ilona Jonuškienė, Kristina Kantminienė, and Vilma Petrikaitė. 2025. "Novel 5-Oxopyrrolidine-3-carbohydrazides as Potent Protein Kinase Inhibitors: Synthesis, Anticancer Evaluation, and Molecular Modeling" International Journal of Molecular Sciences 26, no. 7: 3162. https://doi.org/10.3390/ijms26073162
APA StyleTumosienė, I., Stasevych, M., Zvarych, V., Jonuškienė, I., Kantminienė, K., & Petrikaitė, V. (2025). Novel 5-Oxopyrrolidine-3-carbohydrazides as Potent Protein Kinase Inhibitors: Synthesis, Anticancer Evaluation, and Molecular Modeling. International Journal of Molecular Sciences, 26(7), 3162. https://doi.org/10.3390/ijms26073162