
Role of Endothelin-1 and Nitric Oxide in Acute Ischemic Stroke Leptomeningeal Collateral Activation

 , ,
, ,  , ,
, ,
Abstract
1. Introduction
2. Results
2.1. Patients’ Characteristics and Clinical Outcome
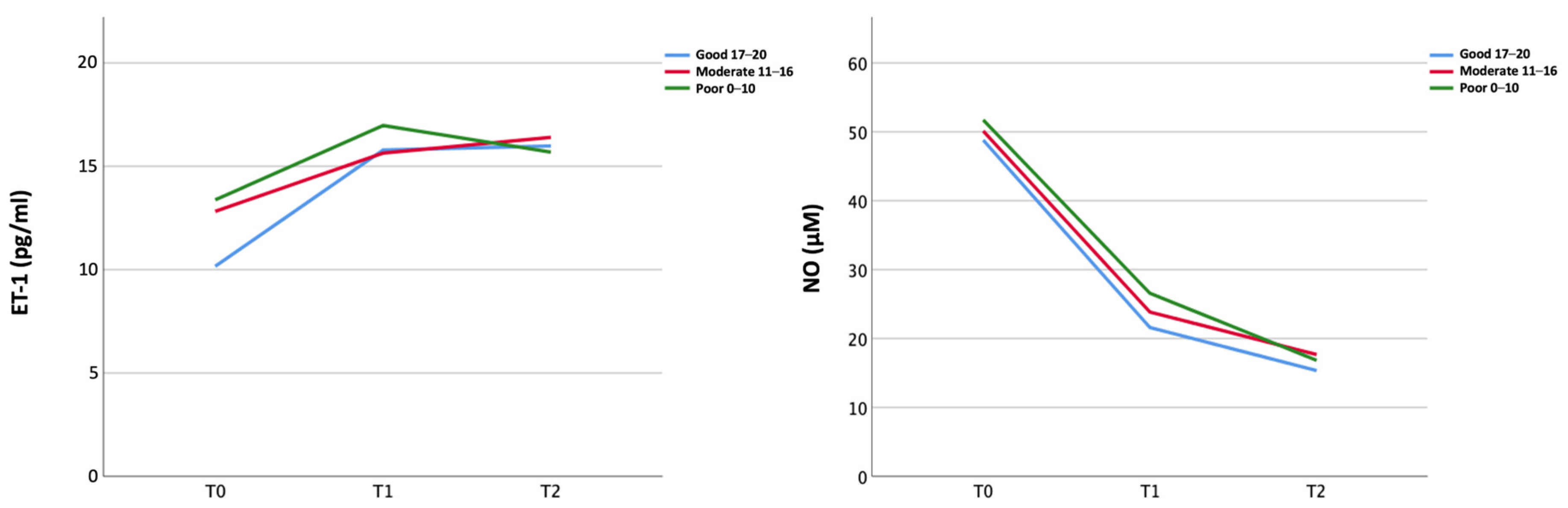
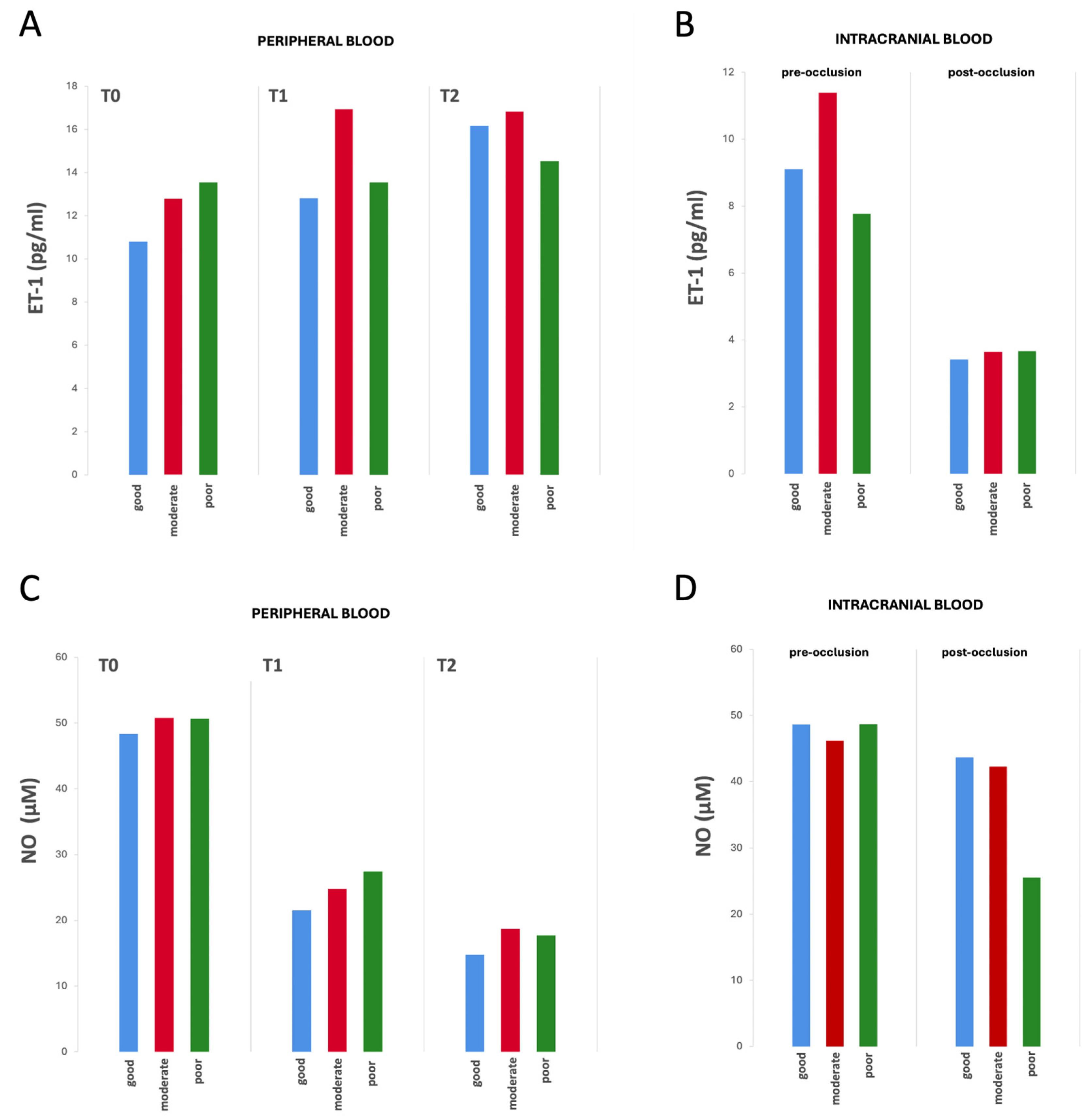
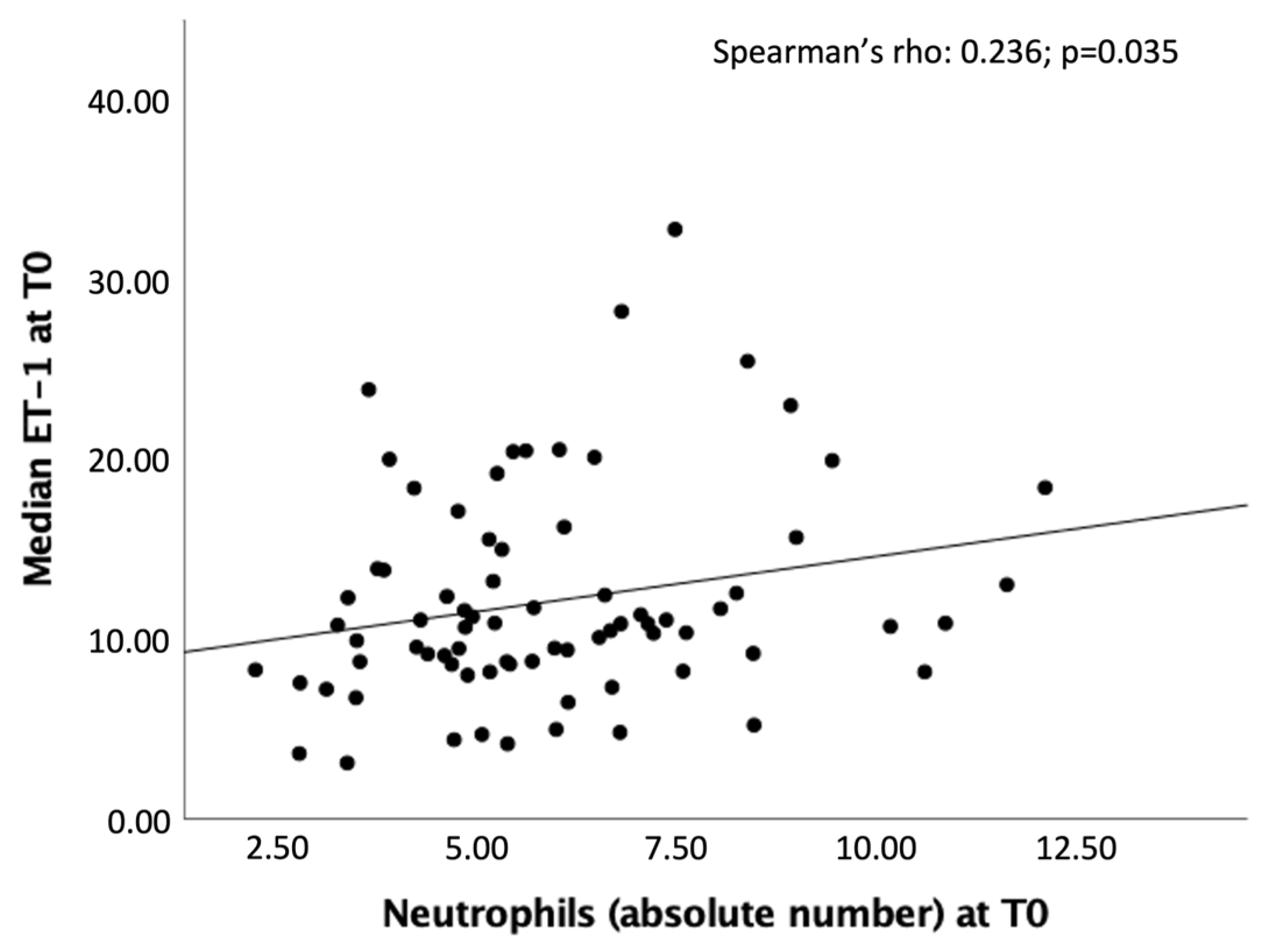
2.2. Biomarkers
3. Discussion
4. Materials and Methods
4.1. Patients
4.2. Laboratory Data
4.2.1. ET-1
4.2.2. NO
4.3. Radiological Data
4.4. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Vagal, A.; Aviv, R.; Sucharew, H.; Reddy, M.; Hou, Q.; Michel, P.; Jovin, T.; Tomsick, T.; Wintermark, M.; Khatri, P. Collateral Clock Is More Important Than Time Clock for Tissue Fate. Stroke 2018, 49, 2102–2107. [Google Scholar] [CrossRef]
- De Michele, M.; Lorenzano, S.; Bertuccini, L.; Iosi, F.; Toni, D. “Time lost is clot resolution lost”: The neglected perspective of the therapeutic time window for ischemic stroke. Front. Neurol. 2023, 14, 1177609. [Google Scholar] [CrossRef]
- Nogueira, R.G.; Jadhav, A.P.; Haussen, D.C.; Bonafe, A.; Budzik, R.F.; Bhuva, P.; Yavagal, D.R.; Ribo, M.; Cognard, C.; Hanel, R.A.; et al. Thrombectomy 6 to 24 Hours after Stroke with a Mismatch between Deficit and Infarct. N. Engl. J. Med. 2018, 378, 11–21. [Google Scholar] [CrossRef] [PubMed]
- Albers, G.W.; Marks, M.P.; Kemp, S.; Christensen, S.; Tsai, J.P.; Ortega-Gutierrez, S.; McTaggart, R.A.; Torbey, M.T.; Kim-Tenser, M.; Leslie-Mazwi, T.; et al. Thrombectomy for Stroke at 6 to 16 Hours with Selection by Perfusion Imaging. N. Engl. J. Med. 2018, 378, 708–718. [Google Scholar] [CrossRef]
- Rocha, M.; Jovin, T.G. Fast versus slow progressors of infarct growth in large vessel occlusion stroke: Clinical and research implications. Stroke 2017, 48, 2621–2627. [Google Scholar] [CrossRef] [PubMed]
- Jia, M.; Jin, F.; Li, S.; Ren, C.; Ruchi, M.; Ding, Y.; Zhao, W.; Ji, X. No-reflow after stroke reperfusion therapy: An emerging phenomenon to be explored. CNS Neurosci. Ther. 2024, 30, e14631. [Google Scholar] [CrossRef]
- Liebeskind, D.S. Collateral circulation. Stroke 2003, 34, 2279–2284. [Google Scholar] [CrossRef]
- Kofler, N.; Corti, F.; Rivera-Molina, F.; Deng, Y.; Toomre, D.; Simons, M. The Rab-effector protein RABEP2 regulates endosomal trafficking to mediate vascular endothelial growth factor receptor-2 (VEGFR2)-dependent signaling. J. Biol. Chem. 2018, 293, 4805–4817. [Google Scholar] [CrossRef] [PubMed]
- Logothetis, N.K.; Pauls, J.; Augath, M.; Trinath, T.; Oeltermann, A. Neurophysiological investigation of the basis of the fMRI signal. Nature 2001, 412, 150–157. [Google Scholar] [CrossRef]
- Morita, Y.; Fukuuchi, Y.; Koto, A.; Suzuki, N.; Isozumi, K.; Gotoh, J.; Shimizu, T.; Takao, M.; Aoyama, M. Rapid Changes in Pial Arterial Diameter and Cerebral Blood Flow Caused by Ipsilateral Carotid Artery Occlusion in Rats. Keio J. Med. 1997, 46, 120–127. [Google Scholar] [CrossRef]
- Maguida, G.; Shuaib, A. Collateral Circulation in Ischemic Stroke: An Updated Review. J. Stroke 2023, 25, 179–198. [Google Scholar] [CrossRef]
- Lundberg, J.O.; Weitzberg, E. Nitric oxide signaling in health and disease. Cell 2022, 185, 2853–2878. [Google Scholar] [CrossRef]
- Wang, Z.; Chen, G.; Chen, Z.-Q.; Mou, R.-T.; Feng, D.-X. The role of nitric oxide in stroke. Med. Gas. Res. 2017, 7, 194–203. [Google Scholar] [CrossRef]
- Iadecola, C.; Pelligrino, D.A.; Moskowitz, M.A.; Lassen, N.A. Nitric oxide synthase inhibition and cerebrovascular regulation. J. Cereb. Blood Flow Metab. 1994, 14, 175–192. [Google Scholar] [CrossRef]
- Atochin, D.N.; Yuzawa, I.; Li, Q.; Rauwerdink, K.M.; Malhotra, R.; Chang, J.; Brouckaert, P.; Ayata, C.; Moskowitz, M.A.; Bloch, K.D.; et al. Soluble Guanylate Cyclase α1β1 Limits Stroke Size and Attenuates Neurological Injury. Stroke 2010, 41, 1815–1819. [Google Scholar] [CrossRef]
- Ohkita, M.; Takaoka, M.; Shiota, Y.; Nojiri, R.; Matsumura, Y. Nitric oxide inhibits endothelin-1 production through the suppression of nuclear factor kB. Clin. Sci. 2002, 103, 68–71. [Google Scholar] [CrossRef]
- Kuruppu, S.; Rajapakse, N.W.; Dunstan, R.A.; Smith, A.I. Nitric oxide inhibits the production of soluble endothelin con-verting enzyme-1. Mol. Cell. Biochem. 2014, 396, 49–54. [Google Scholar] [CrossRef]
- Wiley, K.E.; Davenport, A.P. Novel nitric oxide donors reverse endothelin-1-mediated constriction in human blood vessels. J. Cardiovasc. Pharmacol. 2000, 36, S151–S152. [Google Scholar] [CrossRef]
- Wiley, K.E.; Davenport, A.P. Physiological antagonism of endothelin-1 in human conductance and resistance coronary artery. Br. J. Pharmacol. 2001, 133, 568–574. [Google Scholar] [CrossRef]
- Iadecola, C. The Neurovascular Unit Coming of Age: A Journey through Neurovascular Coupling in Health and Disease. Neuron 2017, 96, 17–42. [Google Scholar] [CrossRef]
- Džoljić, E.; Grbatinić, I.; Kostić, V. Why is nitric oxide important for our brain? Funct. Neurol. 2015, 30, 159–163. [Google Scholar]
- Shah, R. Endothelins in health and disease. Eur. J. Intern. Med. 2007, 18, 272–282. [Google Scholar] [CrossRef]
- Franceschini, R.; Gandolfo, C.; Cataldi, A.; Del Sette, M.; Rolandi, A.; Corsini, G.; Rolandi, E.; Barreca, T. Twenty-four-hour endo-thelin-1 secretory pattern in stroke patients. Biomed. Pharmacother. 2001, 55, 272–276. [Google Scholar]
- Farré, A.L.; Riesco, A.; Espinosa, G.; Digiuni, E.; Cernadas, M.R.; Alvarez, V.; Montón, M.; Rivas, F.; Gallego, M.J.; Egido, J. Effect of endothelin-1 on neutrophil adhesion to endothelial cells and perfused heart. Circulation 1993, 88, 1166–1171. [Google Scholar] [CrossRef]
- Hofman, F.M.; Chen, P.; Jeyaseelan, R.; Incardona, F.; Fisher, M.; Zidovetzki, R. Endothelin-1 induces production of the neutrophil chemotactic factor interleukin-8 by human brain-derived endothelial cells. Blood 1998, 92, 3064–3072. [Google Scholar] [CrossRef]
- Koehl, B.; Nivoit, P.; El Nemer, W.; Lenoir, O.; Hermand, P.; Pereira, C.; Brousse, V.; Guyonnet, L.; Ghinatti, G.; Benkerrou, M.; et al. The endothelin B receptor plays a crucial role in the adhesion of neutrophils to the endothelium in sickle cell disease. Haematologica 2017, 102, 1161–1172. [Google Scholar] [CrossRef]
- Ziv, I.; Fleminger, G.; Djaldetti, R.; Achiron, A.; Melamed, E.; Sokolovsky, M. Increased plasma endothelin-1 in acute ischemic stroke. Stroke 1992, 23, 1014–1016. [Google Scholar] [CrossRef]
- Estrada, V.; Téllez, M.J.; Moya, J.; Fernández-Durango, R.; Egido, J.; Cruz, A.F. High Plasma Levels of Endothelin-1 and Atrial Natriuretic Peptide in Patients with Acute Ischemic Stroke. Am. J. Hypertens. 1994, 7, 1085–1089. [Google Scholar] [CrossRef]
- Suzuki, H.; Sato, S.; Suzuki, Y.; Takekoshi, K.; Ishihara, N.; Shimoda, S. Increased endothelin concentration in CSF from patients with subarachnoid hemorrhage. Acta Neurol. Scand. 1990, 81, 553–554. [Google Scholar] [CrossRef]
- Lampl, Y.; Fleminger, G.; Gilad, R.; Galron, R.; Sarova-Pinhas, I.; Sokolovsky, M. Endothelin in cerebrospinal fluid and plasma of patients in the early stage of ischemic stroke. Stroke 1997, 28, 1951–1955. [Google Scholar] [CrossRef]
- Hamann, G.; Isenberg, E.; Strittmatter, M.; Moili, R.; Schimrigk, K. Big-endothelin in acute ischemic stroke. J. Stroke Cerebrovasc. Dis. 1993, 3, 256–260. [Google Scholar] [CrossRef]
- Haapaniemi, E.; Tatlisumak, T.; Hamel, K.; Soinne, L.; Lanni, C.; Opgenorth, T.J.; Kaste, M. Plasma Endothelin-1 Levels Neither Increase nor Correlate with Neurological Scores, Stroke Risk Factors, or Outcome in Patients with Ischemic Stroke. Stroke 2000, 31, 720–725. [Google Scholar] [CrossRef]
- Adams, H.P., Jr.; Bendixen, B.H.; Kappelle, L.J.; Biller, J.; Love, B.B.; Gordon, D.L.; Marsh, E.E., 3rd. Classification of Subtype of Acute Ischemic Stroke. Definitions for Use in a Multicenter Clinical Trial. TOAST. Trial of Org 10172 in Acute Stroke Treatment. Stroke 1993, 24, 35–41. [Google Scholar] [CrossRef]
- Lauer, T.; Preik, M.; Rassaf, T.; Strauer, B.E.; Deussen, A.; Feelisch, M.; Kelm, M. Plasma nitrite rather than nitrate reflects regional endothelial nitric oxide synthase activity but lacks intrinsic vasodilator action. Proc. Natl. Acad. Sci. USA 2001, 98, 12814–12819. [Google Scholar] [CrossRef]
- Menon, B.; Smith, E.; Modi, J.; Patel, S.; Bhatia, R.; Watson, T.; Hill, M.D.; Demchuk, A.; Goyal, M. Regional Leptomeningeal Score on CT Angiography Predicts Clinical and Imaging Outcomes in Patients with Acute Anterior Circulation Occlusions. Am. J. Neuroradiol. 2011, 32, 1640–1645. [Google Scholar] [CrossRef]
- Aviv, R.; Mandelcorn, J.; Chakraborty, S.; Gladstone, D.; Malham, S.; Tomlinson, G.; Fox, A.; Symons, S. Alberta Stroke Program Early CT Scoring of CT Perfusion in Early Stroke Visualization and Assessment. Am. J. Neuroradiol. 2007, 28, 1975–1980. [Google Scholar] [CrossRef]
- Zaidat, O.O.; Yoo, A.J.; Khatri, P.; Tomsick, T.A.; von Kummer, R.; Saver, J.L.; Marks, M.P.; Prabhakaran, S.; Kallmes, D.F.; Fitzsimmons, B.F.; et al. Recommendations on angiographic revascularization grading standards for acute ischemic stroke: A consensus statement. Stroke 2013, 44, 2650–2663. [Google Scholar] [CrossRef]
- Sims, J.R.; Gharai, L.R.; Schaefer, P.W.; Vangel, M.; Rosenthal, E.S.; Lev, M.H.; Schwamm, L. ABC/2 for rapid clinical estimate of infarct, perfusion, and mismatch volumes. Neurology 2009, 72, 2104–2110. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| All Patients n = 105 | Good n = 44 | Moderate n = 36 | Poor n = 22 | p-Value | |
|---|---|---|---|---|---|
| Demographics and Clinical Characteristics | |||||
| Age (years), mean (SD) | 76.0 (12.8) | 75.0 (11.1) | 77.1 (14.9) | 77.8 (10.1) | 0.604 |
| Sex (females) (%) | 66 (62.9) | 32 (72.7) | 20 (55.6) | 13 (59.1) | 0.248 |
| Pre-stroke mRS (%) | |||||
| 77 (73.3) | 38 (86.4) | 23 (63.9) | 14 (63.6) | 0.132 |
| 8 (7.6) | 2 (4.5) | 3 (8.3) | 3 (13.6) | |
| 6 (5.7) | 3 (6.8) | 3 (8.3) | 0 | |
| 11 (10.5) | 1 (2.3) | 6 (16.7) | 3 (13.6) | |
| 2 (1.9) | 0 | 1 (2.8) | 1 (4.5) | |
| 1 (1.0) | 0 | 0 | 1 (4.5) | |
| Pre-stroke mRS 0–1 (%) | 85 (81) | 40 (90.9) | 26 (72.2) | 17 (77.3) | 0.087 |
| Obesity (%) | 16/100 (16.0) | 8/42 (19.0) | 3/34 (8.8) | 5/21 (23.8) | 0.291 |
| Smoking (%) | 16/99 (16.2) | 8/43 (18.6) | 3/33 (9.1) | 5/20 (25.0) | 0.289 |
| Alcohol consumption (%) | 6/104 (5.8) | 1 (2.3) | 4/35 (11.4) | 1 (4.5) | 0.221 |
| Drug abuse (%) | 3/104 (2.9) | 0 | 2/35 (5.7) | 1 (4.5) | 0.294 |
| Hypertension (%) | 84/103 (81.6) | 33/43 (76.7) | 30/35 (85.7) | 19 (86.4) | 0.493 |
| Hyperlipidemia (%) | 56/100 (56.0) | 29/43 (67.4) | 17/34 (50.0) | 8/20 (40.0) | 0.089 |
| Atrial fibrillation (%) | 63/102 (61.8) | 25/43 (58.1) | 20/35 (57.1) | 18/21 (85.7) | 0.060 |
| Ischemic cardiopathy (%) | 32/104 (30.8) | 10 (22.7) | 11/35 (31.4) | 9 (40.9) | 0.301 |
| Diabetes mellitus (%) | 25/104 (24.0) | 8 (18.2) | 11/35 (31.4) | 6 (27.3) | 0.381 |
| Previous stroke (%) | 18/102 (97.1) | 5 (11.4) | 8/34 (23.5) | 4/21 (19.0) | 0.357 |
| Previous TIA (%) | 8/103 (7.8) | 2 (4.5) | 5/35 (14.3) | 1/21 (4.8) | 0.236 |
| Previous CEA (%) | 4/104 (3.8) | 1 (2.3) | 3/35 (8.6) | 0 | 0.203 |
| Carotid artery stenosis < 50% | 35/103 (34.0) | 15/43 (34.9) | 13/35 (37.1) | 7 (31.8) | 0.919 |
| Carotid artery stenosis (50–70%) | 7/103 (6.8) | 4/43 (9.3) | 3/35 (8.6) | 0 | 0.343 |
| Carotid artery stenosis > 70% | 15/104 (14.4) | 4/43 (9.3) | 5/35 (14.3) | 6 (27.3) | 0.146 |
| Stroke Characteristics | |||||
| Stroke onset on | 30/104 (28.8) | 14 (31.8) | 12/35 (34.3) | 3 (13.6) | 0.204 |
| |||||
| 22/105 (21.0) | 10/44 (22.7) | 9 (25.0) | 2 (9.1) | 0.312 |
| NIHSS, median (IQR) | |||||
| 15 (10–20) | 14 (10–20) | 15 (9–17) | 18 (14–22.50) | 0.035 |
| 10 (4–17) | 8 (2–14) | 10.50 (5.50–20) | 14 (10–17.25) | 0.090 |
| 9.50 (3–17) | 6 (2–16) | 10.50 (4–18) | 13.9 (7.8) | 0.095 |
| 5 (2–9) | 4 (1–9) | 5 (2–11) | 5 (4.50–14.25) | 0.281 |
| ASPECTS | |||||
| 8.22 (1.32) | 8.48 (1.19) | 8.24 (1.23) | 7.86 (1.21) | 0.155 |
| 8 (7–9) | 9 (8–9) | 8 (7.75–9) | 8 (7–8.25) | |
| Vessel occlusion site (%) | |||||
| 57/97 (58.8) | 24/41 (58.5) | 17/34 (50.0) | 13/19 (68.4) | |
| 29/97 (29.9) | 12/41 (29.3) | 14/34 (41.2) | 3/19 (15.8) | |
| 2/97 (2.1) | 0 | 1/34 (2.9) | 1/19 (5.3) | |
| 9/97 (9.3) | 5/41 (12.2) | 2/34 (5.9) | 2/19 (19.5) | 0.424 |
| Collateral status | <0.001 | ||||
| 14.65 (4.18) | 18.45 (1.0) | 13.72 (1.73) | 8.50 (2.33) | |
| 15.50 (11–18) | 18 (18–19) | 14 (12–15) | 8.50 (6.75–10) | |
| Recanalization treatment (%) | |||||
| 53 (50.5) | 20 (45.5) | 20 (55.6) | 12 (54.5) | 0.622 |
| 90/104 (85.3) | 38 (86.4) | 32/35 (91.4) | 18 (81.8) | 0.562 |
| 42 (41.2) | 15 (34.1) | 18 (50.0) | 9 (40.9) | 0.355 |
| MT technique | |||||
| 41/80 (51.2) | 17/36 (47.2) | 14/28 (50.0) | 10/15 (66.7) | 0.202 |
| 11/80 (13.8) | 3/36 (8.3) | 7/28 (25.0) | 1/15 (6.7) | |
| 23/80 (28.7) | 12/36 (33.3) | 7/28 (25.0) | 3/15 (20.0) | |
| |||||
| 5/80 (6.3) | 4736 (11.1) | 0 | 1/15 (6.7) | |
| Onset to IVT time (min), | 150.0 | 150.0 | 151.0 | 135.0 | |
| Median (IQR) | (120–190) | (131.25–205.0) | (110–190) | (100.0–177.0) | 0.635 |
| Onset to MT time (min), | 265.0 | 265.0 | 286.0 | 208.50 | 0.300 |
| Median (IQR) | (197.50–350) | (213–347.50) | (194.50–438.50) | (177.0–332.0) | |
| TICI (%) | |||||
| 7/83 (8.4) | 1/36 (2.8) | 3/30 (10.0) | 3/16 (18.8) | 0.090 |
| 5/83 (6.0) | 0 | 4/30 (13.3) | 0 | |
| 11/83 (13.3) | 5/36 (13.9) | 5/30 (16.7) | 1/16 (6.3) | |
| 17/83 (20.5) | 10/36 (27.8) | 5/30 (16.7) | 2/16 (12.5) | |
| 43/83 (51.8) | 20/36 (55.6) | 13/30 (43.3) | 10/16 (62.5) | |
| TICI 2b–3 (%) | 60/83 (72.3) | 30/36 (83.3) | 18/30 (60.0) | 12/16 (75.0) | 0.102 |
| Imaging for V measurement: | |||||
| 85/103 (82.5) | 36 (81.8) | 31/35 (88.6) | 17/21 (81.0) | |
| 18/103 (17.5) | 8 (18.2) | 4/35 (11.4) | 4/21 (19.0) | 0.655 |
| Infarct volume, median | 9.70 | 5.90 | 13.35 | 14.60 | 0.482 |
| (IQR) | (3.60–20.50) | (2.80–12.25) | (5.08–20.50) | (8.85–41.80) | |
| Stroke etiopathogenesis (%) | |||||
| 10/99 (10.1) | 5/41 (12.2) | 1/34 (2.9) | 4/21 (19.0) | 0.134 |
| 68/99 (68.7) | 25/41 (61.0) | 25/34 (73.5) | 17/21 (81.0) | |
| 7/99 (14.1) | 4/41 (9.8) | 3/34 (8.8) | 0 | |
| 14/99 (14.1) | 7/41 (17.1) | 5/34 (14.7) | 0 | |
| mRS 0–2 (n = 39) | mRS 3–6 (n = 61) | p-Value | |
|---|---|---|---|
| ET-1 at T1 (pg/mL), median (IQR) | 12.44 (10.61–16.58) | 16.95 (11.81–23.44) | 0.017 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Iacobucci, M.; Risitano, A.; Amisano, P.; Berto, I.; Carnevale, R.; Cammisotto, V.; Biraschi, F.; Cirelli, C.; Di Mascio, M.T.; Toni, D.; et al. Role of Endothelin-1 and Nitric Oxide in Acute Ischemic Stroke Leptomeningeal Collateral Activation. Int. J. Mol. Sci. 2025, 26, 3205. https://doi.org/10.3390/ijms26073205
Iacobucci M, Risitano A, Amisano P, Berto I, Carnevale R, Cammisotto V, Biraschi F, Cirelli C, Di Mascio MT, Toni D, et al. Role of Endothelin-1 and Nitric Oxide in Acute Ischemic Stroke Leptomeningeal Collateral Activation. International Journal of Molecular Sciences. 2025; 26(7):3205. https://doi.org/10.3390/ijms26073205
Chicago/Turabian StyleIacobucci, Marta, Angela Risitano, Paolo Amisano, Irene Berto, Roberto Carnevale, Vittoria Cammisotto, Francesco Biraschi, Carlo Cirelli, Maria Teresa Di Mascio, Danilo Toni, and et al. 2025. "Role of Endothelin-1 and Nitric Oxide in Acute Ischemic Stroke Leptomeningeal Collateral Activation" International Journal of Molecular Sciences 26, no. 7: 3205. https://doi.org/10.3390/ijms26073205
APA StyleIacobucci, M., Risitano, A., Amisano, P., Berto, I., Carnevale, R., Cammisotto, V., Biraschi, F., Cirelli, C., Di Mascio, M. T., Toni, D., Lorenzano, S., & De Michele, M. (2025). Role of Endothelin-1 and Nitric Oxide in Acute Ischemic Stroke Leptomeningeal Collateral Activation. International Journal of Molecular Sciences, 26(7), 3205. https://doi.org/10.3390/ijms26073205