

Autophagy and Its Association with Macrophages in Clonal Hematopoiesis Leading to Atherosclerosis
Abstract
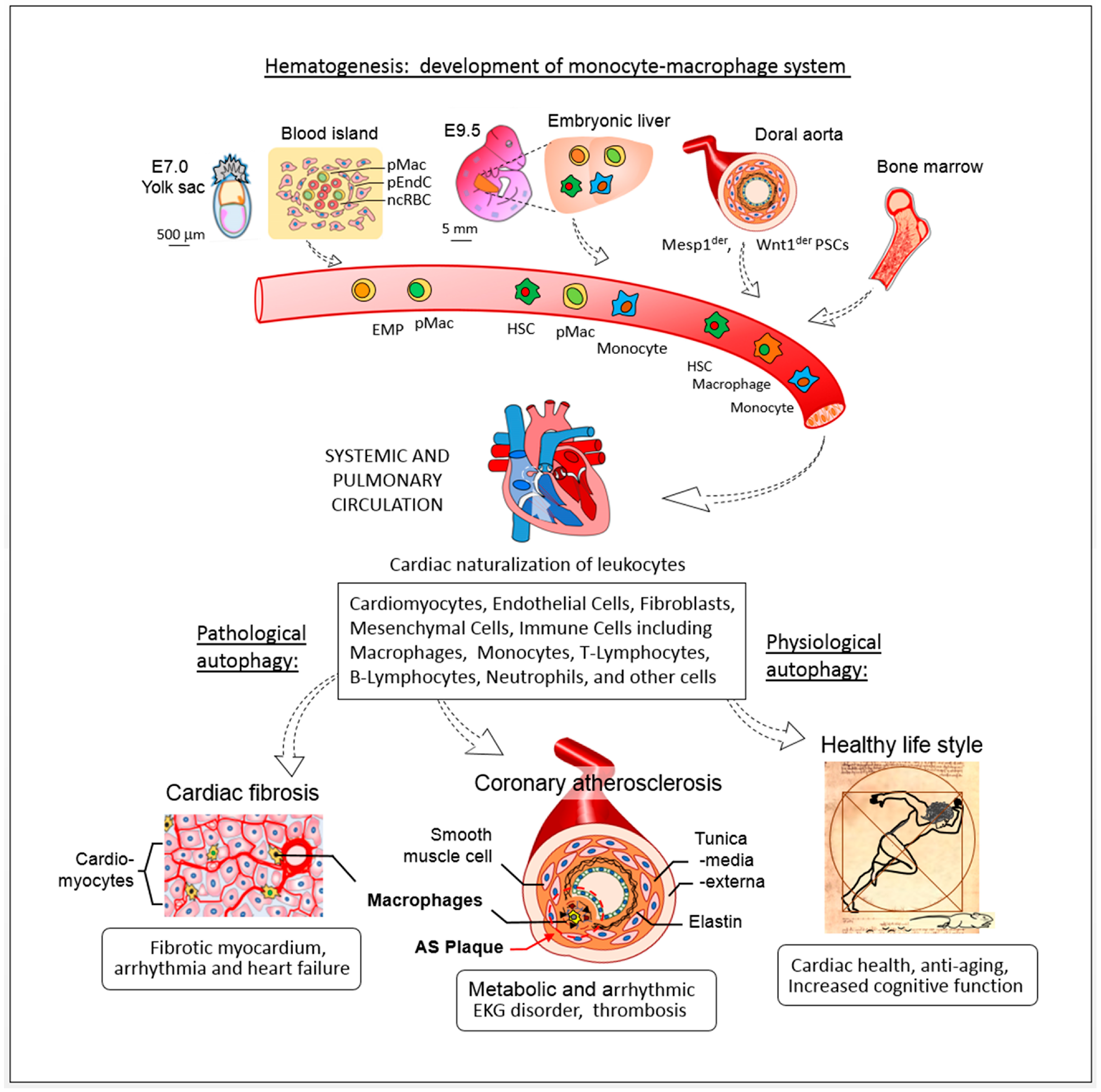
1. Introduction
2. Chaperone-Mediated Autophagy (CMA) Initiates in Inflammation and Atherosclerosis
2.1. Chaperone-Mediated Autophagy as a Specialized Form of Autophagy
2.2. The Protective Role of Chaperone-Mediated Autophagy in Atherosclerosis
3. Deficient Chaperone-Mediated Autophagy Promotes Lipid Accumulation in Macrophages
3.1. Macrophages and Their Fundemental Functions
3.2. Lipids Are Essential for Multiple Functions of Macrophages
3.3. CMA Plays an Important Role in Lipid Accumulation in Macrophages
3.4. PYCARD Modulates CMA-Mediated Lipid Regulation in Macrophages
3.5. p62-ATG5-Mediated Autophagy and CMA in Macrophages
4. Autophagy-Mediated Macrophage Pyroptosis in Atherosclerosis
4.1. Macrophage Pyroptosis
4.2. Pathological Studies and Mechanisms of Macrophage Pyroptosis
4.3. Mechanistic Insights and Therapeutic Targets of Autophagy-Mediated Macrophage Pyroptosis
4.4. Potential and Risk of Pharmacological Manipulation of Autophagic Pathways
5. Macrophage Autophagy and Clonal Hematopoiesis in Atherosclerosis
5.1. Clonal Hematopoiesis as a Risk Factor for Atherosclerosis
5.2. Mechanisms Linking Clonal Hematopoiesis to Atherosclerosis
5.3. Confirmed Function of Circulating Macrophages in the Cardiovascular System
5.4. TET2, JAK2, and DNMT3A-Associated Autophagy Could Lead Clonal Hematopoiesis to Atherosclerosis
5.4.1. JAK2 and Autophagic Regulation in Atherosclerosis
5.4.2. TET2 Mutations and Impaired Autophagy in Atherosclerosis
5.4.3. DNMT3A and Epigenetic Regulation of Autophagy in Atherosclerosis
5.5. The Role of the Monocyte-Macrophage System in Cardiovascular Health
6. Conclusions and Perspective
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Shao, B.Z.; Han, B.Z.; Zeng, Y.X.; Su, D.F.; Liu, C. The roles of macrophage autophagy in atherosclerosis. Acta Pharmacol. 2016, 37, 150–156. [Google Scholar] [CrossRef] [PubMed]
- Pan, W.; Zhang, J.; Zhang, L.; Zhang, Y.; Song, Y.; Han, L.; Tan, M.; Yin, Y.; Yang, T.; Jiang, T.; et al. Comprehensive view of macrophage autophagy and its application in cardiovascular diseases. Cell Prolif. 2024, 57, e13525. [Google Scholar] [CrossRef]
- Poznyak, A.V.; Nikiforov, N.G.; Wu, W.K.; Kirichenko, T.V.; Orekhov, A.N. Autophagy and Mitophagy as Essential Components of Atherosclerosis. Cells 2021, 10, 443. [Google Scholar] [CrossRef]
- Chen, S.; Saeed, A.F.U.H.; Liu, Q.; Jiang, Q.; Xu, H.; Xiao, G.G.; Rao, L.; Duo, Y. Macrophages in immunoregulation and therapeutics. Signal. Transduct. Target. Ther. 2023, 8, 207. [Google Scholar] [CrossRef]
- Hou, P.; Fang, J.; Liu, Z.; Shi, Y.; Agostini, M.; Bernassola, F.; Bove, P.; Candi, E.; Rovella, V.; Sica, G.; et al. Macrophage polarization and metabolism in atherosclerosis. Cell. Death Dis. 2023, 14, 691. [Google Scholar] [CrossRef] [PubMed]
- Yang, B.; Hang, S.; Xu, S.; Gao, Y.; Yu, W.; Zang, G.; Zhang, L.; Wang, Z. Macrophage polarisation and inflammatory mechanisms in atherosclerosis: Implications for prevention and treatment. Heliyon 2024, 10, e32073. [Google Scholar] [CrossRef]
- Wu, M.Y.; Lu, J.H. Autophagy and Macrophage Functions: Inflammatory Response and Phagocytosis. Cells 2019, 9, 70. [Google Scholar] [CrossRef] [PubMed]
- Moore, K.J.; Sheedy, F.J.; Fisher, E.A. Macrophages in atherosclerosis: A dynamic balance. Nat. Rev. Immunol. 2013, 13, 709–721. [Google Scholar] [CrossRef]
- Peled, M.; Fisher, E.A. Dynamic Aspects of Macrophage Polarization during Atherosclerosis Progression and Regression. Front. Immunol. 2014, 5, 579. [Google Scholar] [CrossRef]
- Ajoolabady, A.; Pratico, D.; Lin, L.; Mantzoros, C.S.; Bahijri, S.; Tuomilehto, J.; Ren, J. Inflammation in atherosclerosis: Pathophysiology and mechanisms. Cell Death Dis. 2024, 15, 817. [Google Scholar] [CrossRef]
- Nussenzweig, S.C.; Verma, S.; Finkel, T. The role of autophagy in vascular biology. Circ. Res. 2015, 116, 480–488. [Google Scholar] [CrossRef]
- Zhang, S.; Peng, X.; Yang, S.; Li, X.; Huang, M.; Wei, S.; Liu, J.; He, G.; Zheng, H.; Yang, L.; et al. The regulation, function, and role of lipophagy, a form of selective autophagy, in metabolic disorders. Cell Death Dis. 2022, 13, 132. [Google Scholar] [CrossRef] [PubMed]
- Narabayashi, K.; Ito, Y.; Eid, N.; Maemura, K.; Inoue, T.; Takeuchi, T.; Otsuki, Y.; Higuchi, K. Indomethacin suppresses LAMP-2 expression and induces lipophagy and lipoapoptosis in rat enterocytes via the ER stress pathway. J. Gastroenterol. 2015, 50, 541–554. [Google Scholar] [CrossRef]
- Wang, Z.; Gao, Z.; Zheng, Y.; Kou, J.; Song, D.; Yu, X.; Dong, B.; Chen, T.; Yang, Y.; Gao, X.; et al. Melatonin inhibits atherosclerosis progression via galectin-3 downregulation to enhance autophagy and inhibit inflammation. J. Pineal. Res. 2023, 74, e12855. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Li, C.; Lu, M.; Jiang, G.; Chen, S.; Li, H.; Lu, K. Pharmacological induction of autophagy reduces inflammation in macrophages by degrading immunoproteasome subunits. PLoS Biol. 2024, 22, e3002537. [Google Scholar] [CrossRef] [PubMed]
- Silvestre-Roig, C.; de Winther, M.P.; Weber, C.; Daemen, M.J.; Lutgens, E.; Soehnlein, O. Atherosclerotic plaque destabilization: Mechanisms, models, and therapeutic strategies. Circ. Res. 2014, 114, 214–226. [Google Scholar] [CrossRef]
- Xu, J.; Kitada, M.; Ogura, Y.; Koya, D. Relationship Between Autophagy and Metabolic Syndrome Characteristics in the Pathogenesis of Atherosclerosis. Front. Cell Dev. Biol. 2021, 9, 641852. [Google Scholar] [CrossRef]
- Zuriaga, M.A.; Fuster, J.J. Clonal hematopoiesis and atherosclerotic cardiovascular disease: A primer. Clin. Investig. Arterioscler. 2023, 35, 35–41. [Google Scholar] [CrossRef]
- Cobo, I.; Tanaka, T.; Glass, C.K.; Yeang, C. Clonal hematopoiesis driven by DNMT3A and TET2 mutations: Role in monocyte and macrophage biology and atherosclerotic cardiovascular disease. Curr. Opin. Hematol. 2022, 29, 1–7. [Google Scholar] [CrossRef]
- Liu, W.; Pircher, J.; Schuermans, A.; Ul Ain, Q.; Zhang, Z.; Honigberg, M.C.; Yalcinkaya, M.; Nakao, T.; Pournamadri, A.; Xiao, T.; et al. Jak2 V617F clonal hematopoiesis promotes arterial thrombosis via platelet activation and cross talk. Blood 2024, 143, 1539–1550. [Google Scholar] [CrossRef]
- Guo, H.; Wei, D.; Liu, R.; Zhang, C.; Jiang, S.; Wang, W.; Hu, H.; Shen, L.; Liang, X. A novel therapeutic strategy for atherosclerosis: Autophagy-dependent cholesterol efflux. J. Physiol. Biochem. 2022, 78, 557–572. [Google Scholar] [CrossRef]
- Amorós-Pérez, M.; Fuster, J.J. Clonal hematopoiesis driven by somatic mutations: A new player in atherosclerotic cardiovascular disease. Atherosclerosis 2020, 297, 120–126. [Google Scholar] [CrossRef]
- Tang, G.; Duan, F.; Li, W.; Wang, Y.; Zeng, C.; Hu, J.; Li, H.; Zhang, X.; Chen, Y.; Tan, H. Metformin inhibited Nod-like receptor protein 3 inflammasomes activation and suppressed diabetes-accelerated atherosclerosis in apoE−/− mice. Biomed Pharmacother. 2019, 119, 109410. [Google Scholar] [CrossRef]
- Gu, T.; Zhang, Z.; Liu, J.; Chen, L.; Tian, Y.; Xu, W.; Zeng, T.; Wu, W.; Lu, L. Chlorogenic Acid Alleviates LPS-Induced Inflammation and Oxidative Stress by Modulating CD36/AMPK/PGC-1α in RAW264.7 Macrophages. Int. J. Mol. Sci. 2023, 24, 13516. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; Qin, J.; Zhao, Q.; Yang, J.; Wei, X.; Huang, Y.; Xie, M.; Zhang, C.; Li, Y. Plaque-Targeted Rapamycin Spherical Nucleic Acids for Synergistic Atherosclerosis Treatment. Adv. Sci. 2022, 9, e2105875. [Google Scholar] [CrossRef]
- Grootaert, M.O.J.; Moulis, M.; Roth, L.; Martinet, W.; Vindis, C.; Bennett, M.R.; De Meyer, G.R.Y. Vascular smooth muscle cell death, autophagy and senescence in atherosclerosis. Cardiovasc. Res. 2018, 114, 622–634. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Zhu, R.; Jiang, H.; Yin, Q.; Gu, J.; Chen, J.; Ji, X.; Wu, X.; Fu, H.; Wang, H.; et al. Autophagy enhanced by curcumin ameliorates inflammation in atherogenesis via the TFEB-P300-BRD4 axis. Acta Pharm. Sin. B 2022, 12, 2280–2299. [Google Scholar] [CrossRef] [PubMed]
- Mizushima, N.; Komatsu, M. Autophagy: Renovation of cells and tissues. Cell 2011, 147, 728–741. [Google Scholar] [CrossRef]
- Qiao, L.; Ma, J.; Zhang, Z.; Sui, W.; Zhai, C.; Xu, D.; Wang, Z.; Lu, H.; Zhang, M.; Zhang, C.; et al. Deficient Chaperone-Mediated Autophagy Promotes Inflammation and Atherosclerosis. Circ. Res. 2021, 129, 1141–1157. [Google Scholar] [CrossRef]
- Yang, Q.; Wang, R.; Zhu, L. Chaperone-Mediated Autophagy. Adv. Exp. Med. Biol. 2019, 1206, 435–452. [Google Scholar] [CrossRef]
- Madrigal-Matute, J.; Cuervo, A.M.; Sluimer, J.C. Chaperone-mediated autophagy protects against atherosclerosis. Autophagy 2022, 18, 2505–2507. [Google Scholar] [CrossRef] [PubMed]
- Valdor, R.; Martinez-Vicente, M. The Role of Chaperone-Mediated Autophagy in Tissue Homeostasis and Disease Pathogenesis. Biomedicines 2024, 12, 257. [Google Scholar] [CrossRef] [PubMed]
- Kaushik, S.; Cuervo, A.M. The coming of age of chaperone-mediated autophagy. Nat. Rev. Mol. Cell Biol. 2018, 19, 365–381. [Google Scholar] [CrossRef]
- Qiao, L.; Hu, J.; Qiu, X.; Wang, C.; Peng, J.; Zhang, C.; Zhang, M.; Lu, H.; Chen, W. LAMP2A, LAMP2B and LAMP2C: Similar structures, divergent roles. Autophagy 2023, 19, 2837–2852. [Google Scholar] [CrossRef] [PubMed]
- Madrigal-Matute, J.; de Bruijn, J.; van Kuijk, K.; Riascos-Bernal, D.F.; Diaz, A.; Tasset, I.; Martín-Segura, A.; Gijbels, M.J.J.; Sander, B.; Kaushik, S.; et al. Protective role of chaperone-mediated autophagy against atherosclerosis. Proc. Natl. Acad. Sci. USA 2022, 119, e2121133119. [Google Scholar] [CrossRef]
- Grebe, A.; Hoss, F.; Latz, E. NLRP3 Inflammasome and the IL-1 Pathway in Atherosclerosis. Circ. Res. 2018, 122, 1722–1740. [Google Scholar] [CrossRef]
- Endicott, S.J.; Boynton, D.N., Jr.; Beckmann, L.J.; Miller, R.A. Long-lived mice with reduced growth hormone signaling have a constitutive upregulation of hepatic chaperone-mediated autophagy. Autophagy 2021, 17, 612–625. [Google Scholar] [CrossRef]
- Arias, E.; Cuervo, A.M. Pros and Cons of Chaperone-Mediated Autophagy in Cancer Biology. Trends. Endocrinol. Metab. 2020, 31, 53–66. [Google Scholar] [CrossRef]
- Madrigal-Matute, J.; Scorrano, L.; Sadoshima, J. Leducq Network: Modulating Autophagy to Treat Cardiovascular Disease. Circ. Res. 2018, 123, 323–325. [Google Scholar] [CrossRef]
- Dong, S.; Aguirre-Hernandez, C.; Scrivo, A.; Eliscovich, C.; Arias, E.; Bravo-Cordero, J.J.; Cuervo, A.M. Monitoring spatiotemporal changes in chaperone-mediated autophagy in vivo. Nat. Commun. 2020, 11, 645. [Google Scholar] [CrossRef]
- Schneider, J.L.; Villarroya, J.; Diaz-Carretero, A.; Patel, B.; Urbanska, A.M.; Thi, M.M.; Villarroya, F.; Santambrogio, L.; Cuervo, A.M. Loss of hepatic chaperone-mediated autophagy accelerates proteostasis failure in aging. Aging Cell. 2015, 14, 249–264. [Google Scholar] [CrossRef] [PubMed]
- Moore, K.J.; Tabas, I. Macrophages in the pathogenesis of atherosclerosis. Cell 2011, 145, 341–355. [Google Scholar] [CrossRef]
- Wen, J.H.; Li, D.Y.; Liang, S.; Yang, C.; Tang, J.X.; Liu, H.F. Macrophage autophagy in macrophage polarization, chronic inflammation and organ fibrosis. Front. Immunol. 2022, 13, 946832. [Google Scholar] [CrossRef]
- Li, X.; Zhu, X.; Wei, Y. Autophagy in Atherosclerotic Plaque Cells: Targeting NLRP3 Inflammasome for Self-Rescue. Biomolecules 2022, 13, 15. [Google Scholar] [CrossRef] [PubMed]
- Florance, I.; Ramasubbu, S. Current Understanding on the Role of Lipids in Macrophages and Associated Diseases. Int. J. Mol. Sci. 2022, 24, 589. [Google Scholar] [CrossRef]
- Rodriguez-Navarro, J.A.; Kaushik, S.; Koga, H.; Dall’Armi, C.; Shui, G.; Wenk, M.R.; Di Paolo, G.; Cuervo, A.M. Inhibitory effect of dietary lipids on chaperone-mediated autophagy. Proc. Natl. Acad. Sci. USA 2012, 109, E705–E714. [Google Scholar] [CrossRef]
- Ye, G.; Gao, H.; Wang, Z.; Lin, Y.; Liao, X.; Zhang, H.; Chi, Y.; Zhu, H.; Dong, S. PPARα and PPARγ activation attenuates total free fatty acid and triglyceride accumulation in macrophages via the inhibition of Fatp1 expression. Cell Death Dis. 2019, 10, 39. [Google Scholar] [CrossRef]
- Vogel, A.; Brunner, J.S.; Hajto, A.; Sharif, O.; Schabbauer, G. Lipid scavenging macrophages and inflammation. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2022, 1867, 159066. [Google Scholar] [CrossRef]
- Dib, L.; Koneva, L.A.; Edsfeldt, A.; Zurke, Y.X.; Sun, J.; Nitulescu, M.; Attar, M.; Lutgens, E.; Schmidt, S.; Lindholm, M.W.; et al. Lipid-associated macrophages transition to an inflammatory state in human atherosclerosis increasing the risk of cerebrovascular complications. Nat. Cardiovasc. Res. 2023, 2, 656–672. [Google Scholar] [CrossRef]
- Zhai, M.; Gong, S.; Luan, P.; Shi, Y.; Kou, W.; Zeng, Y.; Shi, J.; Yu, G.; Hou, J.; Yu, Q.; et al. Extracellular Traps from Activated Vascular Smooth Muscle Cells Drive the Progression of Atherosclerosis. Nat. Commun. 2022, 13, 7500. [Google Scholar] [CrossRef]
- Qiao, L.; Wang, H.F.; Xiang, L.; Ma, J.; Zhu, Q.; Xu, D.; Zheng, H.; Peng, J.Q.; Zhang, S.; Lu, H.X.; et al. Deficient Chaperone-Mediated Autophagy Promotes Lipid Accumulation in Macrophage. J. Cardiovasc. Transl. Res. 2021, 14, 661–669. [Google Scholar] [CrossRef]
- Sebastian, W.A.; Inoue, M.; Shimizu, N.; Sato, R.; Oguri, S.; Itonaga, T.; Kishimoto, S.; Shiraishi, H.; Hanada, T.; Ihara, K. Cardiac Manifestations of Human ACTA2 Variants Recapitulated in a Zebrafish Model. J. Hum. Genet. 2024, 69, 133–138. [Google Scholar] [CrossRef]
- Jackson, A.O.; Regine, M.A.; Subrata, C.; Long, S. Molecular Mechanisms and Genetic Regulation in Atherosclerosis. Int. J. Cardiol. Heart Vasc. 2018, 21, 36–44. [Google Scholar] [CrossRef]
- Madan, N.; Ghazi, A.R.; Kong, X.; Chen, E.S.; Shaw, C.A.; Edelstein, L.C. Functionalization of CD36 Cardiovascular Disease and Expression Associated Variants by Interdisciplinary High Throughput Analysis. PLoS Genet. 2019, 15, e1008287. [Google Scholar] [CrossRef]
- Wu, H.; Che, X.; Zheng, Q.; Wu, A.; Pan, K.; Shao, A.; Wu, Q.; Zhang, J.; Hong, Y. Caspases: A Molecular Switch Node in the Crosstalk between Autophagy and Apoptosis. Int. J. Biol. Sci. 2014, 10, 1072–1083. [Google Scholar] [CrossRef]
- Zhang, J.; Huang, J.; Gu, Y.; Xue, M.; Qian, F.; Wang, B.; Yang, W.; Yu, H.; Wang, Q.; Guo, X.; et al. Inflammation-Induced Inhibition of Chaperone-Mediated Autophagy Maintains the Immunosuppressive Function of Murine Mesenchymal Stromal Cells. Cell Mol. Immunol. 2021, 18, 1476–1488. [Google Scholar] [CrossRef] [PubMed]
- Tzeng, H.T.; Yang, J.L.; Tseng, Y.J.; Lee, C.H.; Chen, W.J.; Chyuan, I.T. Plasminogen Activator Inhibitor-1 Secretion by Autophagy Contributes to Melanoma Resistance to Chemotherapy through Tumor Microenvironment Modulation. Cancers 2021, 13, 1253. [Google Scholar] [CrossRef]
- Orenstein, S.J.; Cuervo, A.M. Chaperone-Mediated Autophagy: Molecular Mechanisms and Physiological Relevance. Semin. Cell Dev. Biol. 2010, 21, 719–726. [Google Scholar] [CrossRef] [PubMed]
- Sukhorukov, V.N.; Khotina, V.A.; Chegodaev, Y.S.; Ivanova, E.; Sobenin, I.A.; Orekhov, A.N. Lipid Metabolism in Macrophages: Focus on Atherosclerosis. Biomedicines 2020, 8, 262. [Google Scholar] [CrossRef]
- Hu, H.J.; Wang, X.H.; Zhang, T.Q.; Liu, Y.; Chen, Z.R.; Zhang, Z.Z.; Huang, H.; Tang, H.F.; Jiang, Z.S. PLK1 Promotes Cholesterol Efflux and Alleviates Atherosclerosis by Up-Regulating ABCA1 and ABCG1 Expression via the AMPK/PPARγ/LXRα Pathway. Biochim. Biophys. Acta Mol. Cell Biol. 2022, 1867, 159221. [Google Scholar] [CrossRef]
- Thevkar-Nagesh, P.; Habault, J.; Voisin, M.; Ruff, S.E.; Ha, S.; Ruoff, R.; Chen, X.; Rawal, S.; Zahr, T.; Szabo, G.; et al. Transcriptional Regulation of Acsl1 by CHREBP and NF-κB in Macrophages during Hyperglycemia and Inflammation. PLoS ONE 2022, 17, e0272986. [Google Scholar] [CrossRef]
- Zhou, Q.; Wang, Y.; Cheng, Y.; Zhou, J.; Liu, W.; Ma, X.; Tang, S.; Tang, S.; Tang, C. Ilexgenin A Inhibits Lipid Accumulation in Macrophages and Reduces the Progression of Atherosclerosis through PTPN2/ERK1/2/ABCA1 Signalling Pathway. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2024, 1869, 159533. [Google Scholar] [CrossRef]
- Cui, N.; Li, H.; Dun, Y.; Ripley-Gonzalez, J.W.; You, B.; Li, D.; Liu, Y.; Qiu, L.; Li, C.; Liu, S. Exercise Inhibits JNK Pathway Activation and Lipotoxicity via Macrophage Migration Inhibitory Factor in Nonalcoholic Fatty Liver Disease. Front. Endocrinol. 2022, 13, 961231. [Google Scholar] [CrossRef]
- Schlager, S.; Vujic, N.; Korbelius, M.; Duta-Mare, M.; Dorow, J.; Leopold, C.; Rainer, S.; Wegscheider, M.; Reicher, H.; Ceglarek, U.; et al. Lysosomal Lipid Hydrolysis Provides Substrates for Lipid Mediator Synthesis in Murine Macrophages. Oncotarget 2017, 8, 40037–40051. [Google Scholar] [CrossRef] [PubMed]
- Cho, K.Y.; Miyoshi, H.; Nakamura, A.; Greenberg, A.S.; Atsumi, T. Lipid Droplet Protein PLIN1 Regulates Inflammatory Polarity in Human Macrophages and Is Involved in Atherosclerotic Plaque Development by Promoting Stable Lipid Storage. J. Atheroscler. Thromb. 2023, 30, 170–181. [Google Scholar] [CrossRef] [PubMed]
- Tao, H.; Yancey, P.G.; Blakemore, J.L.; Zhang, Y.; Ding, L.; Jerome, W.G.; Brown, J.D.; Vickers, K.C.; Linton, M.F. Macrophage SR-BI Modulates Autophagy via VPS34 Complex and PPARα Transcription of Tfeb in Atherosclerosis. J. Clin. Investig. 2021, 131, e94229. [Google Scholar] [CrossRef]
- Li, M.; Wang, Z.; Wang, P.; Li, H.; Yang, L. TFEB: An Emerging Regulator in Lipid Homeostasis for Atherosclerosis. Front. Physiol. 2021, 12, 639920. [Google Scholar] [CrossRef]
- Pan, J.; Han, L.; Guo, J.; Wang, X.; Liu, D.; Tian, J.; Zhang, M.; An, F. AIM2 Accelerates the Atherosclerotic Plaque Progressions in ApoE-/- Mice. Biochem. Biophys. Res. Commun. 2018, 498, 487–494. [Google Scholar] [CrossRef]
- Wang, A.; Yue, K.; Zhong, W.; Zhang, G.; Zhang, X.; Wang, L. Targeted Delivery of Rapamycin and Inhibition of Platelet Adhesion with Multifunctional Peptide Nanoparticles for Atherosclerosis Treatment. J. Control Release 2024, 376, 753–765. [Google Scholar] [CrossRef]
- Hu, Y.; Liu, Y.; Zong, L.; Zhang, W.; Liu, R.; Xing, Q.; Liu, Z.; Yan, Q.; Li, W.; Lei, H.; et al. The Multifaceted Roles of GSDME-Mediated Pyroptosis in Cancer: Therapeutic Strategies and Persisting Obstacles. Cell Death Dis. 2023, 4, 836. [Google Scholar] [CrossRef]
- Wei, Y.; Lan, B.; Zheng, T.; Yang, L.; Zhang, X.; Cheng, L.; Tuerhongjiang, G.; Yuan, Z.; Wu, Y. GSDME-Mediated Pyroptosis Promotes the Progression and Associated Inflammation of Atherosclerosis. Nat. Commun. 2023, 14, 929. [Google Scholar] [CrossRef] [PubMed]
- Seimon, T.; Tabas, I. Mechanisms and Consequences of Macrophage Apoptosis in Atherosclerosis. J. Lipid Res. 2009, 50, S382–S387. [Google Scholar] [CrossRef]
- Kurdi, A.; De Meyer, G.R.; Martinet, W. Potential Therapeutic Effects of mTOR Inhibition in Atherosclerosis. Br. J. Clin. Pharmacol. 2016, 82, 1267–1279. [Google Scholar] [CrossRef] [PubMed]
- Xu, S.; Chen, H.; Ni, H.; Dai, Q. Targeting HDAC6 Attenuates Nicotine-Induced Macrophage Pyroptosis via NF-κB/NLRP3 Pathway. Atherosclerosis 2020, 317, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Fan, X.; Li, Q.; Wang, Y.; Zhang, D.M.; Zhou, J.; Chen, Q.; Sheng, L.; Passerini, A.G.; Sun, C. Non-Canonical NF-κB Contributes to Endothelial Pyroptosis and Atherogenesis Dependent on IRF-1. Transl. Res. 2023, 255, 1–13. [Google Scholar] [CrossRef]
- Xu, X.D.; Chen, J.X.; Zhu, L.; Xu, S.T.; Jiang, J.; Ren, K. The Emerging Role of Pyroptosis-Related Inflammasome Pathway in Atherosclerosis. Mol. Med. 2022, 28, 160. [Google Scholar] [CrossRef]
- Qian, Z.; Zhao, Y.; Wan, C.; Deng, Y.; Zhuang, Y.; Xu, Y.; Zhu, Y.; Lu, S.; Bao, Z. Pyroptosis in the Initiation and Progression of Atherosclerosis. Front. Pharmacol. 2021, 12, 652963. [Google Scholar] [CrossRef]
- Chen, Q.; Lv, J.; Yang, W.; Xu, B.; Wang, Z.; Yu, Z.; Wu, J.; Yang, Y.; Han, Y. Targeted Inhibition of STAT3 as a Potential Treatment Strategy for Atherosclerosis. Theranostics 2019, 9, 6424–6442. [Google Scholar] [CrossRef]
- Fan, X.; Han, J.; Zhong, L.; Zheng, W.; Shao, R.; Zhang, Y.; Shi, S.; Lin, S.; Huang, Z.; Huang, W.; et al. Macrophage-Derived GSDMD Plays an Essential Role in Atherosclerosis and Cross Talk Between Macrophages via the Mitochondria-STING-IRF3/NF-κB Axis. Arterioscler. Thromb. Vasc. Biol. 2024, 44, 1365–1378. [Google Scholar] [CrossRef]
- Sun, W.; Lu, H.; Dong, S.; Li, R.; Chu, Y.; Wang, N.; Zhao, Y.; Zhang, Y.; Wang, L.; Sun, L.; et al. Beclin1 Controls Caspase-4 Inflammasome Activation and Pyroptosis in Mouse Myocardial Reperfusion-Induced Microvascular Injury. Cell Commun. Signal. 2021, 19, 107. [Google Scholar] [CrossRef]
- Yang, G.; Zhang, Q.; Tan, J.; Xiong, Y.; Liang, Y.; Yan, J.; Gu, F.; Xu, Y. HMGB1 Induces Macrophage Pyroptosis in Chronic Endometritis. Int. Immunopharmacol. 2023, 123, 110706. [Google Scholar] [CrossRef]
- Peng, X.; Chen, H.; Li, Y.; Huang, D.; Huang, B.; Sun, D. Effects of NIX-Mediated Mitophagy on ox-LDL-Induced Macrophage Pyroptosis in Atherosclerosis. Cell Biol. Int. 2020, 44, 1481–1490. [Google Scholar] [CrossRef]
- Cao, Y.; Chen, X.; Pan, F.; Wang, M.; Zhuang, H.; Chen, J.; Lu, L.; Wang, L.; Wang, T. Xinmaikang-Mediated Mitophagy Attenuates Atherosclerosis via the PINK1/Parkin Signaling Pathway. Phytomedicine 2023, 119, 154955. [Google Scholar] [CrossRef]
- Joo, L.; Bradley, C.C.; Lin, S.H.; Scheet, P.A.; Nead, K.T. Causes of Clonal Hematopoiesis: A Review. Curr. Oncol. Rep. 2023, 25, 211–220. [Google Scholar] [CrossRef] [PubMed]
- Köhnke, T.; Majeti, R. Clonal Hematopoiesis: From Mechanisms to Clinical Intervention. Cancer Discov. 2021, 11, 2987–2997. [Google Scholar] [CrossRef] [PubMed]
- Páramo Fernández, J.A. Atherosclerosis and Clonal Hematopoiesis: A New Risk Factor. Clin. Investig. Arterioscler. 2018, 30, 133–136. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Yalcinkaya, M.; Maestre, I.F.; Olszewska, M.; Ampomah, P.B.; Heimlich, J.B.; Wang, R.; Vela, P.S.; Xiao, T.; Bick, A.G.; et al. Blockade of IL-6 Signaling Alleviates Atherosclerosis in Tet2-Deficient Clonal Hematopoiesis. Nat. Cardiovasc. Res. 2023, 2, 572–586. [Google Scholar] [CrossRef]
- Fidler, T.P.; Dunbar, A.; Kim, E.; Hardaway, B.; Pauli, J.; Xue, C.; Abramowicz, S.; Xiao, T.; O’Connor, K.; Sachs, N.; et al. Suppression of IL-1β Promotes Beneficial Accumulation of Fibroblast-Like Cells in Atherosclerotic Plaques in Clonal Hematopoiesis. Nat. Cardiovasc. Res. 2024, 3, 60–75. [Google Scholar] [CrossRef]
- Asada, S.; Kitamura, T. Clonal Hematopoiesis and Associated Diseases: A Review of Recent Findings. Cancer Sci. 2021, 112, 3962–3971. [Google Scholar] [CrossRef]
- Yalcinkaya, M.; Liu, W.; Thomas, L.A.; Olszewska, M.; Xiao, T.; Abramowicz, S.; Papapetrou, E.P.; Westerterp, M.; Wang, N.; Tabas, I.; et al. BRCC3-Mediated NLRP3 Deubiquitylation Promotes Inflammasome Activation and Atherosclerosis in Tet2 Clonal Hematopoiesis. Circulation 2023, 148, 1764–1777. [Google Scholar] [CrossRef]
- Abplanalp, W.T.; Mas-Peiro, S.; Cremer, S.; John, D.; Dimmeler, S.; Zeiher, A.M. Association of Clonal Hematopoiesis of Indeterminate Potential with Inflammatory Gene Expression in Patients with Severe Degenerative Aortic Valve Stenosis or Chronic Postischemic Heart Failure. JAMA Cardiol. 2020, 5, 1170–1175. [Google Scholar] [CrossRef] [PubMed]
- Polizio, A.H.; Park, E.; Walsh, K. Clonal Hematopoiesis: Connecting Aging and Inflammation in Atherosclerosis. Curr. Atheroscler. Rep. 2023, 25, 105–111. [Google Scholar] [CrossRef]
- Kiefer, K.C.; Cremer, S.; Pardali, E.; Assmus, B.; Abou-El-Ardat, K.; Kirschbaum, K.; Dorsheimer, L.; Rasper, T.; Berkowitsch, A.; Serve, H.; et al. Full Spectrum of Clonal Haematopoiesis-Driver Mutations in Chronic Heart Failure and Their Associations with Mortality. ESC. Heart. Fail. 2021, 8, 1873–1884. [Google Scholar] [CrossRef]
- Zekavat, S.M.; Viana-Huete, V.; Matesanz, N.; Jorshery, S.D.; Zuriaga, M.A.; Uddin, M.M.; Trinder, M.; Paruchuri, K.; Zorita, V.; Ferrer-Pérez, A.; et al. TP53-Mediated Clonal Hematopoiesis Confers Increased Risk for Incident Atherosclerotic Disease. Nat. Cardiovasc. Res. 2023, 2, 144–158. [Google Scholar] [CrossRef] [PubMed]
- Dutta, A.; Yang, Y.; Le, B.T.; Zhang, Y.; Abdel-Wahab, O.; Zang, C.; Mohi, G. U2af1 Is Required for Survival and Function of Hematopoietic Stem/Progenitor Cells. Leukemia 2021, 35, 2382–2398. [Google Scholar] [CrossRef]
- Díez-Díez, M.; Ramos-Neble, B.L.; de la Barrera, J.; Silla-Castro, J.C.; Quintas, A.; Vázquez, E.; Rey-Martín, M.A.; Izzi, B.; Sánchez-García, L.; García-Lunar, I.; et al. Unidirectional Association of Clonal Hematopoiesis with Atherosclerosis Development. Nat. Med. 2024, 30, 2857–2866. [Google Scholar] [CrossRef] [PubMed]
- Wynn, T.A.; Vannella, K.M. Macrophages in tissue repair, regeneration, and fibrosis. Immunity 2016, 44, 450–462. [Google Scholar] [CrossRef]
- Lendeckel, U.; Venz, S.; Wolke, C. Macrophages: Shapes and functions. ChemTexts 2022, 8, 12. [Google Scholar] [CrossRef]
- Wei, Q.; Deng, Y.; Yang, Q.; Zhan, A.; Wang, L. The markers to delineate different phenotypes of macrophages related to metabolic disorders. Front. Immunol. 2023, 14, 1084636. [Google Scholar] [CrossRef]
- Yunna, C.; Mengru, H.; Lei, W.; Weidong, C. Macrophage M1/M2 polarization. Eur. J. Pharmacol. 2020, 877, 173090. [Google Scholar] [CrossRef]
- Hussell, T.; Bell, T.J. Alveolar macrophages: Plasticity in a tissue-specific context. Nat. Rev. Immunol. 2014, 14, 81–93. [Google Scholar] [CrossRef] [PubMed]
- Misharin, A.V.; Morales-Nebreda, L.; Reyfman, P.A.; Cuda, C.M.; Walter, J.M.; McQuattie-Pimentel, A.C.; Chen, C.I.; Anekalla, K.R.; Joshi, N.; Williams, K.J.N.; et al. Monocyte-derived alveolar macrophages drive lung fibrosis and persist in the lung over the life span. J. Exp. Med. 2017, 214, 2387–2404. [Google Scholar] [CrossRef]
- Dong, T.; Hu, G.; Fan, Z.; Wang, H.; Gao, Y.; Wang, S.; Xu, H.; Yaffe, M.B.; Vander Heiden, M.G.; Lv, G.; et al. Activation of GPR3-β-arrestin2-PKM2 pathway in Kupffer cells stimulates glycolysis and inhibits obesity and liver pathogenesis. Nat. Commun. 2024, 15, 807. [Google Scholar] [CrossRef]
- Salter, M.W.; Stevens, B. Microglia emerge as central players in brain disease. Nat. Med. 2017, 23, 1018–1027. [Google Scholar] [CrossRef] [PubMed]
- Murray, P.J. Macrophage Polarization. Annu. Rev. Physiol. 2017, 79, 541–566. [Google Scholar] [CrossRef]
- Lapenna, A.; De Palma, M.; Lewis, C.E. Perivascular macrophages in health and disease. Nat. Rev. Immunol. 2018, 18, 689–702. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.; Zhang, H.; Tang, B.; Luo, Y.; Yang, Y.; Zhong, X.; Chen, S.; Xu, X.; Huang, S.; Liu, C. Macrophages in cardiovascular diseases: Molecular mechanisms and therapeutic targets. Signal Transduct. Target Ther. 2024, 9, 130. [Google Scholar] [CrossRef]
- Yu, B.; Jia, S.; Chen, Y.; Guan, R.; Chen, S.; Tang, W.; Bao, T.; Tian, Z. CXCL4 deficiency limits M4 macrophage infiltration and attenuates hyperoxia-induced lung injury. Mol. Med. 2024, 30, 253. [Google Scholar] [CrossRef]
- Naito, Y.; Takagi, T.; Higashimura, Y. Heme oxygenase-1 and anti-inflammatory M2 macrophages. Arch. Biochem. Biophys. 2014, 564, 83–88. [Google Scholar] [CrossRef]
- Lin, A.; Miano, J.M.; Fisher, E.A.; Misra, A. Chronic inflammation and vascular cell plasticity in atherosclerosis. Nat. Cardiovasc. Res. 2024, 3, 1408–1423. [Google Scholar] [CrossRef]
- Li, Y.; Sun, Z.; Zhang, L.; Yan, J.; Shao, C.; Jing, L.; Li, L.; Wang, Z. Role of Macrophages in the Progression and Regression of Vascular Calcification. Front. Pharmacol. 2020, 11, 661. [Google Scholar] [CrossRef] [PubMed]
- Hong, H.; Tian, X.Y. The Role of Macrophages in Vascular Repair and Regeneration after Ischemic Injury. Int. J. Mol. Sci. 2020, 21, 6328. [Google Scholar] [CrossRef]
- Ouimet, M.; Ediriweera, H.; Afonso, M.S.; Ramkhelawon, B.; Singaravelu, R.; Liao, X.; Bandler, R.C.; Rahman, K.; Fisher, E.A.; Rayner, K.J.; et al. microRNA-33 Regulates Macrophage Autophagy in Atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2017, 37, 1058–1067. [Google Scholar] [CrossRef]
- Wu, J.; Bai, X.; Yan, L.; Baimanov, D.; Cong, Y.; Quan, P.; Cai, R.; Guan, Y.; Bu, W.; Lin, B.; et al. Selective regulation of macrophage lipid metabolism via nanomaterials’ surface chemistry. Nat. Commun. 2024, 15, 8349. [Google Scholar] [CrossRef]
- Yan, J.; Horng, T. Lipid Metabolism in Regulation of Macrophage Functions. Trends. Cell. Biol. 2020, 30, 979–989. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Tang, X.; Yao, L.; Wang, Y.; Chen, Z.; Li, M.; Wu, N.; Wu, D.; Dai, X.; Jiang, H.; et al. Disruption of USP9X in macrophages promotes foam cell formation and atherosclerosis. J. Clin. Investig. 2022, 132, e154217. [Google Scholar] [CrossRef] [PubMed]
- Canfrán-Duque, A.; Rotllan, N.; Zhang, X.; Andrés-Blasco, I.; Thompson, B.M.; Sun, J.; Price, N.L.; Fernández-Fuertes, M.; Fowler, J.W.; Gómez-Coronado, D.; et al. Macrophage-Derived 25-Hydroxycholesterol Promotes Vascular Inflammation, Atherogenesis, and Lesion Remodeling. Circulation 2022, 147, 388–408. [Google Scholar] [CrossRef]
- Filali-Mouncef, Y.; Hunter, C.; Roccio, F.; Zagkou, S.; Dupont, N.; Primard, C.; Proikas-Cezanne, T.; Reggiori, F. The ménage à trois of autophagy, lipid droplets and liver disease. Autophagy 2022, 18, 50–72. [Google Scholar] [CrossRef]
- Emanuel, R.; Sergin, I.; Bhattacharya, S.; Turner, J.; Epelman, S.; Settembre, C.; Diwan, A.; Ballabio, A.; Razani, B. Induction of lysosomal biogenesis in atherosclerotic macrophages can rescue lipid-induced lysosomal dysfunction and downstream sequelae. Arterioscler. Thromb. Vasc. Biol. 2014, 34, 1942–1952. [Google Scholar] [CrossRef]
- Sergin, I.; Evans, T.D.; Zhang, X.; Bhattacharya, S.; Stokes, C.J.; Song, E.; Ali, S.; Dehestani, B.; Holloway, K.B.; Micevych, P.S.; et al. Exploiting macrophage autophagy-lysosomal biogenesis as a therapy for atherosclerosis. Nat. Commun. 2017, 8, 15750. [Google Scholar] [CrossRef]
- Kaushik, S.; Cuervo, A.M. Degradation of lipid droplet-associated proteins by chaperone-mediated autophagy facilitates lipolysis. Nat. Cell. Biol. 2015, 17, 759–770. [Google Scholar] [CrossRef]
- Schneider, J.L.; Suh, Y.; Cuervo, A.M. Deficient chaperone-mediated autophagy in liver leads to metabolic dysregulation. Cell. Metab. 2014, 20, 417–432. [Google Scholar] [CrossRef] [PubMed]
- Al-Rashed, F.; Haddad, D.; Al Madhoun, A.; Sindhu, S.; Jacob, T.; Kochumon, S.; Obeid, L.M.; Al-Mulla, F.; Hannun, Y.A.; Ahmad, R. ACSL1 is a key regulator of inflammatory and macrophage foaming induced by short-term palmitate exposure or acute high-fat feeding. iScience 2023, 26, 107145. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H. Lysosomal acid lipase and lipid metabolism: New mechanisms, new questions, and new therapies. Curr. Opin. Lipidol. 2018, 29, 218–223. [Google Scholar] [CrossRef]
- Vassiliou, E.; Farias-Pereira, R. Impact of Lipid Metabolism on Macrophage Polarization: Implications for Inflammation and Tumor Immunity. Int. J. Mol. Sci. 2023, 24, 12032. [Google Scholar] [CrossRef] [PubMed]
- Reiner, Ž.; Guardamagna, O.; Nair, D.; Soran, H.; Hovingh, K.; Bertolini, S.; Jones, S.; Ćorić, M.; Calandra, S.; Hamilton, J.; et al. Lysosomal acid lipase deficiency--an under-recognized cause of dyslipidaemia and liver dysfunction. Atherosclerosis 2014, 235, 21–30. [Google Scholar] [CrossRef]
- Li, J.; Yao, H.; Zhao, F.; An, J.; Wang, Q.; Mu, J.; Liu, Z.; Zou, M.H.; Xie, Z. Pycard deficiency inhibits microRNA maturation and prevents neointima formation by promoting chaperone-mediated autophagic degradation of AGO2/argonaute 2 in adipose tissue. Autophagy 2024, 20, 629–644. [Google Scholar] [CrossRef]
- Shen, W.; Wu, T.; Liu, Q.; Ke, B. Analysis of regulatory patterns of NLRP3 corpuscles and related genes and the role of macrophage polarization in atherosclerosis based on online database. Mol. Genet. Genom. 2024, 300, 7. [Google Scholar] [CrossRef]
- Elbarbary, R.A.; Maquat, L.E. Evaluating the susceptibility of AGO2-loaded microRNAs to degradation by nucleases in vitro. Methods 2019, 152, 18–22. [Google Scholar] [CrossRef]
- Malekmohammad, K.; Bezsonov, E.E.; Rafieian-Kopaei, M. Role of Lipid Accumulation and Inflammation in Atherosclerosis: Focus on Molecular and Cellular Mechanisms. Front. Cardiovasc. Med. 2021, 8, 707529. [Google Scholar] [CrossRef]
- Wang, D.W.; Peng, Z.J.; Ren, G.F.; Wang, G.X. The different roles of selective autophagic protein degradation in mammalian cells. Oncotarget 2015, 6, 37098–37116. [Google Scholar] [CrossRef] [PubMed]
- Hassanpour, M.; Rahbarghazi, R.; Nouri, M.; Aghamohammadzadeh, N.; Safaei, N.; Ahmadi, M. Role of autophagy in atherosclerosis: Foe or friend? J. Inflamm. 2019, 16, 8. [Google Scholar] [CrossRef]
- Sergin, I.; Razani, B. Self-eating in the plaque: What macrophage autophagy reveals about atherosclerosis. Trends. Endocrinol. Metab. 2014, 25, 225–234. [Google Scholar] [CrossRef] [PubMed]
- Wen, X.; Yang, Y.; Klionsky, D.J. Moments in autophagy and disease: Past and Present. Mol. Aspects. Med. 2021, 82, 100966. [Google Scholar] [CrossRef]
- Yao, R.; Shen, J. Chaperone-mediated autophagy: Molecular mechanisms, biological functions, and diseases. MedComm 2020, 4, e347. [Google Scholar] [CrossRef]
- Ni, D.; Mo, Z.; Yi, G. Recent insights into atherosclerotic plaque cell autophagy. Exp. Biol. Med. 2021, 246, 2553–2558. [Google Scholar] [CrossRef]
- Liu, W.J.; Ye, L.; Huang, W.F.; Guo, L.J.; Xu, Z.G.; Wu, H.L.; Yang, C.; Liu, H.F. p62 links the autophagy pathway and the ubiqutin-proteasome system upon ubiquitinated protein degradation. Cell. Mol. Biol. Lett. 2016, 21, 29. [Google Scholar] [CrossRef]
- Sergin, I.; Bhattacharya, S.; Emanuel, R.; Esen, E.; Stokes, C.J.; Evans, T.D.; Arif, B.; Curci, J.A.; Razani, B. Inclusion bodies enriched for p62 and polyubiquitinated proteins in macrophages protect against atherosclerosis. Sci. Signal. 2016, 9, ra2. [Google Scholar] [CrossRef]
- Kim, S.; Lee, W.; Cho, K. P62 Links the Autophagy Pathway and the Ubiquitin-Proteasome System in Endothelial Cells during Atherosclerosis. Int. J. Mol. Sci. 2021, 22, 7791. [Google Scholar] [CrossRef]
- Huang, X.; Yao, J.; Liu, L.; Chen, J.; Mei, L.; Huang, F.J.; Luo, D.; Wang, X.; Lin, C.; Chen, X.; et al. S-Acylation of p62 promotes p62 droplet recruitment into autophagosomes in mammalian autophagy. Mol. Cell. 2023, 83, 3485–3501. [Google Scholar] [CrossRef]
- Shi, Y.X.; Sun, Z.W.; Jia, D.L.; Wang, H.B. Autophagy deficiency promotes lung metastasis of prostate cancer via stabilization of TWIST1. Clin. Transl. Oncol. 2022, 24, 1403–1412. [Google Scholar] [CrossRef] [PubMed]
- Hennig, P.; Fenini, G.; Di Filippo, M.; Karakaya, T.; Beer, H.D. The Pathways Underlying the Multiple Roles of p62 in Inflammation and Cancer. Biomedicines 2021, 9, 707. [Google Scholar] [CrossRef]
- Wang, L.; Cano, M.; Handa, J.T. p62 provides dual cytoprotection against oxidative stress in the retinal pigment epithelium. Biochim. Biophys. Acta 2014, 1843, 1248–1258. [Google Scholar] [CrossRef] [PubMed]
- De Meyer, G.R.Y.; Zurek, M.; Puylaert, P.; Martinet, W. Programmed death of macrophages in atherosclerosis: Mechanisms and therapeutic targets. Nat. Rev. Cardiol. 2024, 21, 312–325. [Google Scholar] [CrossRef] [PubMed]
- Martinet, W.; Coornaert, I.; Puylaert, P.; De Meyer, G.R.Y. Macrophage Death as a Pharmacological Target in Atherosclerosis. Front. Pharmacol. 2019, 10, 306. [Google Scholar] [CrossRef]
- Yang, R.; Wang, D.; Ding, Y.; Liu, Q. Exploring biomarkers for autophagy-mediated macrophage pyroptosis in atherosclerosis. Cell. Biol. Int. 2023, 47, 1905–1925. [Google Scholar] [CrossRef]
- He, X.; Fan, X.; Bai, B.; Lu, N.; Zhang, S.; Zhang, L. Pyroptosis is a critical immune-inflammatory response involved in atherosclerosis. Pharmacol. Res. 2021, 165, 105447. [Google Scholar] [CrossRef]
- Lin, L.; Zhang, M.X.; Zhang, L.; Zhang, D.; Li, C.; Li, Y.L. Autophagy, Pyroptosis, and Ferroptosis: New Regulatory Mechanisms for Atherosclerosis. Front. Cell. Dev. Biol. 2022, 9, 809955. [Google Scholar] [CrossRef]
- Liu, Y.; Pan, R.; Ouyang, Y.; Gu, W.; Xiao, T.; Yang, H.; Tang, L.; Wang, H.; Xiang, B.; Chen, P. Pyroptosis in health and disease: Mechanisms, regulation and clinical perspective. Signal. Transduct. Target. Ther. 2024, 9, 245. [Google Scholar] [CrossRef]
- Zheng, Z.; Li, G. Mechanisms and Therapeutic Regulation of Pyroptosis in Inflammatory Diseases and Cancer. Int. J. Mol. Sci. 2020, 21, 1456. [Google Scholar] [CrossRef]
- Jiang, X.; Stockwell, B.R.; Conrad, M. Ferroptosis: Mechanisms, biology and role in disease. Nat. Rev. Mol. Cell. Biol. 2021, 22, 266–282. [Google Scholar] [CrossRef]
- Li, M.; Wang, Z.W.; Fang, L.J.; Cheng, S.Q.; Wang, X.; Liu, N.F. Programmed cell death in atherosclerosis and vascular calcification. Cell. Death. Dis. 2022, 13, 467. [Google Scholar] [CrossRef]
- Liu, C.; Jiang, Z.; Pan, Z.; Yang, L. The Function, Regulation and Mechanism of Programmed Cell Death of Macrophages in Atherosclerosis. Front Cell Dev. Biol. 2022, 9, 809516. [Google Scholar] [CrossRef] [PubMed]
- Gao, W.; Wang, X.; Zhou, Y.; Wang, X.; Yu, Y. Autophagy, ferroptosis, pyroptosis, and necroptosis in tumor immunotherapy. Signal. Transduct. Target. Ther. 2022, 7, 196. [Google Scholar] [CrossRef] [PubMed]
- Marchio, P.; Guerra-Ojeda, S.; Vila, J.M.; Aldasoro, M.; Victor, V.M.; Mauricio, M.D. Targeting Early Atherosclerosis: A Focus on Oxidative Stress and Inflammation. Oxid. Med. Cell. Longev. 2019, 2019, 8563845. [Google Scholar] [CrossRef] [PubMed]
- Jinnouchi, H.; Sato, Y.; Sakamoto, A.; Cornelissen, A.; Mori, M.; Kawakami, R.; Gadhoke, N.V.; Kolodgie, F.D.; Virmani, R.; Finn, A.V. Calcium deposition within coronary atherosclerotic lesion: Implications for plaque stability. Atherosclerosis 2020, 306, 85–95. [Google Scholar] [CrossRef]
- Mao, C.; Li, D.; Zhou, E.; Zhang, J.; Wang, C.; Xue, C. Nicotine exacerbates atherosclerosis through a macrophage-mediated endothelial injury pathway. Aging 2021, 13, 7627–7643. [Google Scholar] [CrossRef]
- Lin, J.; Shou, X.; Mao, X.; Dong, J.; Mohabeer, N.; Kushwaha, K.K.; Wang, L.; Su, Y.; Fang, H.; Li, D. Oxidized low density lipoprotein induced caspase-1 mediated pyroptotic cell death in macrophages: Implication in lesion instability? PLoS ONE 2013, 8, e62148. [Google Scholar] [CrossRef]
- Wang, K.; Sun, Q.; Zhong, X.; Zeng, M.; Zeng, H.; Shi, X.; Li, Z.; Wang, Y.; Zhao, Q.; Shao, F.; et al. Structural Mechanism for GSDMD Targeting by Autoprocessed Caspases in Pyroptosis. Cell 2020, 180, 941–955.e20. [Google Scholar] [CrossRef]
- Coll, R.C.; Robertson, A.A.; Chae, J.J.; Higgins, S.C.; Muñoz-Planillo, R.; Inserra, M.C.; Vetter, I.; Dungan, L.S.; Monks, B.G.; Stutz, A.; et al. A small-molecule inhibitor of the NLRP3 inflammasome for the treatment of inflammatory diseases. Nat. Med. 2015, 21, 248–255. [Google Scholar] [CrossRef]
- Sharma, A.; Choi, J.S.Y.; Stefanovic, N.; Al-Sharea, A.; Simpson, D.S.; Mukhamedova, N.; Jandeleit-Dahm, K.; Murphy, A.J.; Sviridov, D.; Vince, J.E.; et al. Specific NLRP3 Inhibition Protects Against Diabetes-Associated Atherosclerosis. Diabetes 2021, 70, 772–787. [Google Scholar] [CrossRef]
- Yao, F.; Jin, Z.; Zheng, Z.; Lv, X.; Ren, L.; Yang, J.; Chen, D.; Wang, B.; Yang, W.; Chen, L.; et al. HDAC11 promotes both NLRP3/Caspase-1/GSDMD and caspase-3/GSDME pathways causing pyroptosis via ERG in vascular endothelial cells. Cell Death Discov. 2022, 8, 112. [Google Scholar] [CrossRef]
- Jin, Y.; Liu, Y.; Xu, L.; Xu, J.; Xiong, Y.; Peng, Y.; Ding, K.; Zheng, S.; Yang, N.; Zhang, Z.; et al. Novel role for caspase 1 inhibitor VX765 in suppressing NLRP3 inflammasome assembly and atherosclerosis via promoting mitophagy and efferocytosis. Cell Death Dis. 2022, 13, 512. [Google Scholar] [CrossRef] [PubMed]
- Duewell, P.; Kono, H.; Rayner, K.J.; Sirois, C.M.; Vladimer, G.; Bauernfeind, F.G.; Abela, G.S.; Franchi, L.; Nuñez, G.; Schnurr, M.; et al. NLRP3 inflammasomes are required for atherogenesis and activated by cholesterol crystals. Nature 2010, 464, 1357–1361. [Google Scholar] [CrossRef] [PubMed]
- Luo, X.; Weng, X.; Bao, X.; Bai, X.; Lv, Y.; Zhang, S.; Chen, Y.; Zhao, C.; Zeng, M.; Huang, J.; et al. A novel anti-atherosclerotic mechanism of quercetin: Competitive binding to KEAP1 via Arg483 to inhibit macrophage pyroptosis. Redox. Biol. 2022, 57, 102511. [Google Scholar] [CrossRef]
- Zhang, S.; Lv, Y.; Luo, X.; Weng, X.; Qi, J.; Bai, X.; Zhao, C.; Zeng, M.; Bao, X.; Dai, X.; et al. Homocysteine promotes atherosclerosis through macrophage pyroptosis via endoplasmic reticulum stress and calcium disorder. Mol. Med. 2023, 29, 73. [Google Scholar] [CrossRef]
- Wang, R.; Wang, Y.; Mu, N.; Lou, X.; Li, W.; Chen, Y.; Fan, D.; Tan, H. Activation of NLRP3 inflammasomes contributes to hyperhomocysteinemia-aggravated inflammation and atherosclerosis in apoE-deficient mice. Lab. Investig. 2017, 97, 922–934. [Google Scholar] [CrossRef]
- Rocha, M.; Apostolova, N.; Diaz-Rua, R.; Muntane, J.; Victor, V.M. Mitochondria and T2D: Role of Autophagy, ER Stress, and Inflammasome. Trends Endocrinol. Metab. 2020, 31, 725–741. [Google Scholar] [CrossRef]
- Wang, Y.; Shi, P.; Chen, Q.; Huang, Z.; Zou, D.; Zhang, J.; Gao, X.; Lin, Z. Mitochondrial ROS promote macrophage pyroptosis by inducing GSDMD oxidation. J. Mol. Cell Biol. 2019, 11, 1069–1082. [Google Scholar] [CrossRef]
- Sun, S.; Gong, D.; Liu, R.; Wang, R.; Chen, D.; Yuan, T.; Wang, S.; Xing, C.; Lv, Y.; Du, G.; et al. Puerarin Inhibits NLRP3-Caspase-1-GSDMD-Mediated Pyroptosis via P2X7 Receptor in Cardiomyocytes and Macrophages. Int. J. Mol. Sci. 2023, 24, 13169. [Google Scholar] [CrossRef]
- Wu, P.; Chen, J.; Chen, J.; Tao, J.; Wu, S.; Xu, G.; Wang, Z.; Wei, D.; Yin, W. Trimethylamine N-oxide promotes apoE-/- mice atherosclerosis by inducing vascular endothelial cell pyroptosis via the SDHB/ROS pathway. J. Cell. Physiol. 2020, 235, 6582–6591. [Google Scholar] [CrossRef]
- Chen, W.; Xiao, W.; Liu, X.; Yuan, P.; Zhang, S.; Wang, Y.; Wu, W. Pharmacological manipulation of macrophage autophagy effectively rejuvenates the regenerative potential of biodegrading vascular graft in aging body. Bioact. Mater. 2021, 11, 283–299. [Google Scholar] [CrossRef]
- Utpal, B.K.; Mokhfi, F.Z.; Zehravi, M.; Sweilam, S.H.; Gupta, J.K.; Kareemulla, S.; Darwin, C.R.; Rao, A.A.; Kumar, V.V.; Krosuri, P.; et al. Resveratrol: A Natural Compound Targeting the PI3K/Akt/mTOR Pathway in Neurological Diseases. Mol. Neurobiol. 2024. [Google Scholar] [CrossRef]
- Nixon, R.A.; Rubinsztein, D.C. Mechanisms of autophagy-lysosome dysfunction in neurodegenerative diseases. Nat. Rev. Mol. Cell. Biol. 2024, 25, 926–946. [Google Scholar] [CrossRef] [PubMed]
- Sevriev, B.; Dimitrova, S.; Kehayova, G.; Dragomanova, S. Trehalose: Neuroprotective Effects and Mechanisms-An Updated Review. NeuroSci 2024, 5, 429–444. [Google Scholar] [CrossRef] [PubMed]
- Kocot, A.M.; Wróblewska, B. Nutritional strategies for autophagy activation and health consequences of autophagy impairment. Nutrition 2022, 103, 111686. [Google Scholar] [CrossRef]
- Debnath, J.; Gammoh, N.; Ryan, K.M. Autophagy and autophagy-related pathways in cancer. Nat. Rev. Mol. Cell. Biol. 2023, 24, 560–575. [Google Scholar] [CrossRef]
- Zhao, B.; Zhang, J.; Zhao, K.; Wang, B.; Liu, J.; Wang, C.; Zeng, L.; Zeng, X.; Luo, Y. Effect of rapamycin on hepatic metabolomics of non-alcoholic fatty liver rats based on non-targeted platform. J. Pharm. Biomed. Anal. 2025, 253, 116541. [Google Scholar] [CrossRef]
- Johnston, R.; Uthman, O.; Cummins, E. Canagliflozin, Dapagliflozin and Empagliflozin Monotherapy for Treating Type 2 Diabetes: Systematic Review and Economic Evaluation. Health Technol. Assess 2017, 21, 1–218. [Google Scholar] [CrossRef]
- Sun, R.Z.; Fan, Y.; Liang, X.; Gong, T.T.; Wang, Q.; Liu, H.; Shan, Z.Y.; Lei, L. Rapamycin and FTY720 Alleviate Atherosclerosis by Cross Talk of Macrophage Polarization and Autophagy. Biomed. Res. Int. 2018, 2018, 1010248. [Google Scholar] [CrossRef]
- Li, J.; Li, C.; Huang, Z.; Huang, C.; Liu, J.; Wu, T.; Xu, S.; Mai, P.; Geng, D.; Zhou, S.; et al. Empagliflozin Alleviates Atherosclerotic Calcification by Inhibiting Osteogenic Differentiation of Vascular Smooth Muscle Cells. Front. Pharmacol. 2023, 14, 1295463. [Google Scholar] [CrossRef]
- Suda, M.; Tchkonia, T.; Kirkland, J.L.; Minamino, T. Targeting Senescent Cells for the Treatment of Age-Associated Diseases. J. Biochem. 2024, 177, 177–187. [Google Scholar] [CrossRef]
- Gao, K.; Liu, Y.; Sun, C.; Wang, Y.; Bao, H.; Liu, G.; Ou, J.; Sun, P. TNF-α Induces Mitochondrial Dysfunction to Drive NLRP3/Caspase-1/GSDMD-Mediated Pyroptosis in MCF-7 Cells. Sci. Rep. 2024, 14, 25880. [Google Scholar] [CrossRef]
- Traughber, C.A.; Timinski, K.; Prince, A.; Bhandari, N.; Neupane, K.; Khan, M.R.; Opoku, E.; Opoku, E.; Brubaker, G.; Shin, J.; et al. Disulfiram Reduces Atherosclerosis and Enhances Efferocytosis, Autophagy, and Athereprotective Gut Microbiota in Hyperlipidemic Mice. J. Am. Heart Assoc. 2024, 13, e033881. [Google Scholar] [CrossRef] [PubMed]
- Xiang, L.; Wang, Y.; Liu, S.; Ying, L.; Zhang, K.; Liang, N.; Li, H.; Luo, G.; Xiao, L. Quercetin Attenuates KLF4-Mediated Phenotypic Switch of VSMCs to Macrophage-Like Cells in Atherosclerosis: A Critical Role for the JAK2/STAT3 Pathway. Int. J. Mol. Sci. 2024, 25, 7755. [Google Scholar] [CrossRef] [PubMed]
- Dotan, I.; Yang, J.; Ikeda, J. Macrophage Jak2 Deficiency Accelerates Atherosclerosis through Defects in Cholesterol Efflux. Commun. Biol. 2022, 5, 132. [Google Scholar] [CrossRef]
- Fidler, T.P.; Xue, C.; Yalcinkaya, M.; Hardaway, B.; Abramowicz, S.; Xiao, T.; Liu, W.; Thomas, D.G.; Hajebrahimi, M.A.; Pircher, J.; et al. The AIM2 Inflammasome Exacerbates Atherosclerosis in Clonal Haematopoiesis. Nature 2021, 592, 296–301. [Google Scholar] [CrossRef]
- Cheung, B.M.; Lauder, I.J.; Lau, C.P.; Kumana, C.R. Meta-Analysis of Large Randomized Controlled Trials to Evaluate the Impact of Statins on Cardiovascular Outcomes. Br. J. Clin. Pharmacol. 2004, 57, 640–651. [Google Scholar] [CrossRef]
- Yuan, J.; Liu, C.; Jiang, C.; Liu, N.; Yang, Z.; Xing, H. RSL3 Induces Ferroptosis by Activating the NF-κB Signalling Pathway to Enhance the Chemosensitivity of Triple-Negative Breast Cancer Cells to Paclitaxel. Sci. Rep. 2025, 15, 1654. [Google Scholar] [CrossRef]
- Ouyang, S.; You, J.; Zhi, C.; Li, P.; Lin, X.; Tan, X.; Ma, W.; Li, L.; Xie, W. Ferroptosis: The Potential Value Target in Atherosclerosis. Cell Death Dis. 2021, 12, 782. [Google Scholar] [CrossRef]
- Xu, X.; Zhou, R.; Duan, Q.; Miao, Y.; Zhang, T.; Wang, M.; Jones, O.D.; Xu, M.M. Circulating macrophages as the mechanistic link between mosaic loss of Y-chromosome and cardiac disease. Cell Biosci. 2023, 13, 135. [Google Scholar] [CrossRef]
- Sano, S.; Horitani, K.; Ogawa, H.; Halvardson, J.; Chavkin, N.W.; Wang, Y.; Sano, M.; Mattisson, J.; Hata, A.; Danielsson, M.; et al. Hematopoietic loss of Y chromosome leads to cardiac fibrosis and heart failure mortality. Science 2022, 377, 292–297. [Google Scholar] [CrossRef] [PubMed]
- Hulsmans, M.; Sam, F.; Nahrendorf, M. Monocyte and macrophage contributions to cardiac remodeling. J. Mol. Cell. Cardiol. 2016, 93, 149–155. [Google Scholar] [CrossRef]
- Zhou, X.; Li, Z.; Wang, Z.; Chen, E.; Wang, J.; Chen, F.; Jones, O.D.; Tan, T.; Chen, S.; Takeshima, H.; et al. Syncytium calcium signaling and macrophage function in the heart. Cell. Biosci. 2018, 8, 24. [Google Scholar] [CrossRef]
- Hulsmans, M.; Clauss, S.; Xiao, L.; Aguirre, A.D.; King, K.R.; Hanley, A.; Hucker, W.J.; Wülfers, E.M.; Seemann, G.; Courties, G.; et al. Macrophages facilitate electrical conduction in the heart. Cell 2017, 169, 510–522. [Google Scholar] [CrossRef] [PubMed]
- Oren, O.; Small, A.M.; Libby, P. Clonal hematopoiesis and atherosclerosis. J. Clin. Investig. 2024, 134, e180066. [Google Scholar] [CrossRef]
- Jaiswal, S.; Natarajan, P.; Silver, A.J.; Gibson, C.J.; Bick, A.G.; Shvartz, E.; McConkey, M.; Gupta, N.; Gabriel, S.; Ardissino, D.; et al. Clonal Hematopoiesis and Risk of Atherosclerotic Cardiovascular Disease. N. Engl. J. Med. 2017, 377, 111–121. [Google Scholar] [CrossRef]
- Marnell, C.S.; Bick, A.; Natarajan, P. Clonal hematopoiesis of indeterminate potential (CHIP): Linking somatic mutations, hematopoiesis, chronic inflammation and cardiovascular disease. J. Mol. Cell. Cardiol. 2021, 161, 98–105. [Google Scholar] [CrossRef]
- Fuster, J.J.; MacLauchlan, S.; Zuriaga, M.A.; Polackal, M.N.; Ostriker, A.C.; Chakraborty, R.; Wu, C.L.; Sano, S.; Muralidharan, S.; Rius, C.; et al. Clonal hematopoiesis associated with TET2 deficiency accelerates atherosclerosis development in mice. Science 2017, 355, 842–847. [Google Scholar] [CrossRef]
- Bick, A.G.; Weinstock, J.S.; Nandakumar, S.K.; Fulco, C.P.; Bao, E.L.; Zekavat, S.M.; Szeto, M.D.; Liao, X.; Leventhal, M.J.; Nasser, J.; et al. Inherited causes of clonal haematopoiesis in 97,691 whole genomes. Nature 2021, 586, 763–768. [Google Scholar] [CrossRef]
- Li, Z.; Duan, Q.; Cui, Y.; Jones, O.D.; Shao, D.; Zhang, J.; Gao, Y.; Cao, X.; Wang, S.; Li, J.; et al. Cardiac-Specific Expression of Cre Recombinase Leads to Age-Related Cardiac Dysfunction Associated with Tumor-like Growth of Atrial Cardiomyocyte and Ventricular Fibrosis and Ferroptosis. Int. J. Mol. Sci. 2023, 24, 3094. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Cui, Y.; Duan, Q.; Zhang, J.; Shao, D.; Cao, X.; Gao, Y.; Wang, S.; Li, J.; Jones, O.D.; et al. The Prognostic Significance of FKBP1A and Its Related Immune Infiltration in Liver Hepatocellular Carcinoma. Int. J. Mol. Sci. 2022, 23, 12797. [Google Scholar] [CrossRef] [PubMed]
- Uddin, M.M.; Saadatagah, S.; Niroula, A.; Yu, B.; Hornsby, W.E.; Ganesh, S.; Lannery, K.; Schuermans, A.; Honigberg, M.C.; Bick, A.G.; et al. Long-term longitudinal analysis of 4,187 participants reveals insights into determinants of clonal hematopoiesis. Nat. Commun. 2024, 15, 7858. [Google Scholar] [CrossRef]
- Walsh, K. The emergence of clonal hematopoiesis as a disease determinant. J. Clin. Investig. 2024, 134, e180063. [Google Scholar] [CrossRef] [PubMed]
- Izzi, B.; Fuster, J.J. Clonal Hematopoiesis and Cardiovascular Risk: Atherosclerosis, Thrombosis, and beyond. Hamostaseologie 2024, 44, 13–20. [Google Scholar] [CrossRef]
- Kinzhebay, A.; Salybekov, A.A. The Role of Somatic Mutations in Ischemic Stroke: CHIP’s Impact on Vascular Health. Neurol. Int. 2025, 17, 19. [Google Scholar] [CrossRef]
- Bird, L. Taking AIM2 at atherosclerotic plaques. Nat. Rev. Immunol. 2021, 21, 273. [Google Scholar] [CrossRef]
- Sano, S.; Walsh, K. Hematopoietic JAK2V617F-mediated clonal hematopoiesis: AIM2 understand mechanisms of atherogenesis. J. Cardiovasc. Aging 2021, 1, 5. [Google Scholar] [CrossRef]
- Du, L.; Wang, X.; Chen, S.; Guo, X. The AIM2 inflammasome: A novel biomarker and target in cardiovascular disease. Pharmacol. Res. 2022, 186, 106533. [Google Scholar] [CrossRef]
- Jaiswal, S.; Libby, P. Clonal haematopoiesis: Connecting ageing and inflammation in cardiovascular disease. Nat. Rev. Cardiol. 2020, 17, 137–144. [Google Scholar] [CrossRef]
- Sano, S.; Oshima, K.; Wang, Y.; MacLauchlan, S.; Katanasaka, Y.; Sano, M.; Zuriaga, M.A.; Yoshiyama, M.; Goukassian, D.; Cooper, M.A.; et al. Tet2-Mediated Clonal Hematopoiesis Accelerates Heart Failure Through a Mechanism Involving the IL-1β/NLRP3 Inflammasome. J. Am. Coll. Cardiol. 2017, 71, 875–886. [Google Scholar] [CrossRef]
- Tall, A.R.; Levine, R.L. Cardiovascular disease: Commonality with cancer. Nature 2017, 543, 45–47. [Google Scholar] [CrossRef]
- Zhu, Y.P.; Hedrick, C.C.; Gaddis, D.E. Hematopoietic stem cells gone rogue. Science 2017, 355, 798–799. [Google Scholar] [CrossRef] [PubMed]
- Lafuse, W.P.; Wozniak, D.J.; Rajaram, M.V.S. Role of Cardiac Macrophages on Cardiac Inflammation, Fibrosis and Tissue Repair. Cells 2020, 10, 51. [Google Scholar] [CrossRef]
- Frangogiannis, N.G. Cardiac fibrosis: Cell biological mechanisms, molecular pathways and therapeutic opportunities. Mol. Aspects. Med. 2019, 65, 70–99. [Google Scholar] [CrossRef]
- Zeiher, A.; Braun, T. Mosaic loss of Y chromosome during aging. Science 2022, 377, 266–267. [Google Scholar] [CrossRef] [PubMed]
- Polizio, A.H.; Marino, L.; Min, K.D.; Yura, Y.; Rolauer, L.; Cochran, J.D.; Evans, M.A.; Park, E.; Doviak, H.; Miura-Yura, E.; et al. Experimental TET2 Clonal Hematopoiesis Predisposes to Renal Hypertension Through an Inflammasome-Mediated Mechanism. Circ. Res. 2024, 135, 933–950. [Google Scholar] [CrossRef]
- Hu, F.; Song, D.; Yan, Y.; Huang, C.; Shen, C.; Lan, J.; Chen, Y.; Liu, A.; Wu, Q.; Sun, L.; et al. IL-6 regulates autophagy and chemotherapy resistance by promoting BECN1 phosphorylation. Nat. Commun. 2021, 12, 3651. [Google Scholar] [CrossRef] [PubMed]
- González-Rodríguez, P.; Cheray, M.; Füllgrabe, J.; Salli, M.; Engskog-Vlachos, P.; Keane, L.; Cunha, V.; Lupa, A.; Li, W.; Ma, Q.; et al. The DNA methyltransferase DNMT3A contributes to autophagy long-term memory. Autophagy 2021, 17, 1259–1277. [Google Scholar] [CrossRef]
- de la Calle-Fabregat, C.; Calafell-Segura, J.; Gardet, M.; Dunsmore, G.; Mulder, K.; Ciudad, L.; Silvin, A.; Moreno-Càceres, J.; Corbí, Á.L.; Muñoz-Pinedo, C.; et al. NF-κB and TET2 promote macrophage reprogramming in hypoxia that overrides the immunosuppressive effects of the tumor microenvironment. Sci. Adv. 2024, 10, 5226. [Google Scholar] [CrossRef]
- Epelman, S.; Lavine, K.J.; Beaudin, A.E.; Sojka, D.K.; Carrero, J.A.; Calderon, B.; Brija, T.; Gautier, E.L.; Ivanovm, S.; Satpathy, A.T.; et al. Embryonic and adult-derived resident cardiac macrophages are maintained through distinct mechanisms at steady state and during inflammation. Immunity 2014, 40, 91–104. [Google Scholar] [CrossRef] [PubMed]
- Epelman, S.; Lavine, K.J.; Randolph, G.J. Origin and functions of tissue macrophages. Immunity 2014, 41, 21–35. [Google Scholar] [CrossRef]
- Svedberg, F.R.; Guilliams, M. Cellular origin of human cardiac macrophage populations. Nat. Med. 2018, 24, 1091–1092. [Google Scholar] [CrossRef] [PubMed]
- Bajpai, G.; Schneider, C.; Wong, N.; Bredemeyer, A.; Hulsmans, M.; Nahrendorf, M.; Epelman, S.; Kreisel, D.; Liu, Y.; Itoh, A.; et al. The human heart contains distinct macrophage subsets with divergent origins and functions. Nat. Med. 2018, 24, 1234–1245. [Google Scholar] [CrossRef] [PubMed]
- Delgado, M.G.; Lennon-Duménil, A.M. How cell migration helps immune sentinels. Front. Cell. Dev. Biol. 2022, 10, 932472. [Google Scholar] [CrossRef]
- Peet, C.; Ivetic, A.; Bromage, D.I.; Shah, A.M. Cardiac monocytes and macrophages after myocardial infarction. Cardiovasc. Res. 2020, 116, 1101–1112. [Google Scholar] [CrossRef]
- Deniset, J.F.; Belke, D.; Lee, W.Y.; Jorch, S.K.; Deppermann, C.; Hassanabad, A.F.; Turnbull, J.D.; Teng, G.; Rozich, I.; Hudspeth, K.; et al. Gata6+ Pericardial Cavity Macrophages Relocate to the Injured Heart and Prevent Cardiac Fibrosis. Immunity 2019, 51, 131–140. [Google Scholar] [CrossRef]
- Chandrakanthan, V.; Rorimpandey, P.; Zanini, F.; Chacon, D.; Olivier, J.; Joshi, S.; Kang, Y.C.; Knezevic, K.; Huang, Y.; Qiao, Q.; et al. Mesoderm-derived PDGFRA+ cells regulate the emergence of hematopoietic stem cells in the dorsal aorta. Nat. Cell. Biol. 2022, 24, 1211–1225. [Google Scholar] [CrossRef]
- Samokhvalov, I.M.; Samokhvalova, N.I.; Nishikawa, S. Cell tracing shows the contribution of the yolk sac to adult haematopoiesis. Nature 2007, 446, 1056–1061. [Google Scholar] [CrossRef]
- Yokomizo, T.; Ideue, T.; Morino-Koga, S.; Tham, C.Y.; Sato, T.; Takeda, N.; Kubota, Y.; Kurokawa, M.; Komatsu, N.; Ogawa, M.; et al. Independent origins of fetal liver haematopoietic stem and progenitor cells. Nature 2022, 609, 779–784. [Google Scholar] [CrossRef]
- Wattrus, S.J.; Smith, M.L.; Rodrigues, C.P.; Hagedorn, E.J.; Kim, J.W.; Budnik, B.; Zon, L.I. Quality assurance of hematopoietic stem cells by macrophages determines stem cell clonality. Science 2022, 377, 1413–1419. [Google Scholar] [CrossRef]
- Forsberg, L.A.; Rasi, C.; Malmqvist, N.; Davies, H.; Pasupulati, S.; Pakalapati, G.; Sandgren, J.; Diaz de Ståhl, T.; Zaghlool, A.; Giedraitis, V.; et al. Mosaic loss of chromosome Y in peripheral blood is associated with shorter survival and higher risk of cancer. Nat. Genet. 2014, 46, 624–628. [Google Scholar] [CrossRef]
- Duan, Q.C.; Gao, Y.; Cao, X.; Wang, S.; Xu, M.M.; Jones, O.D.; Xu, X.H. Mosaic loss of chromosome Y in peripheral blood cells is associated with age-related macular degeneration in men. Cell. Biosci. 2022, 12, 73. [Google Scholar] [CrossRef]
- Zhang, Q.; Zhao, L.; Yang, Y.; Li, S.; Liu, Y.; Chen, C. Mosaic loss of chromosome Y promotes leukemogenesis and clonal hematopoiesis. JCI Insight. 2022, 7, e153768. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, H.; Zhang, S.; Mizushima, N. Autophagy genes in biology and disease. Nat. Rev. Genet. 2023, 24, 382–400. [Google Scholar] [CrossRef]
- Levine, B.; Kroemer, G. Biological Functions of Autophagy Genes: A Disease Perspective. Cell 2019, 176, 11–42. [Google Scholar] [CrossRef] [PubMed]
- Opoku, E.; Traughber, C.A.; Zhang, D.; Iacano, A.J.; Khan, M.; Han, J.; Smith, J.D.; Gulshan, K. Gasdermin D Mediates Inflammation-Induced Defects in Reverse Cholesterol Transport and Promotes Atherosclerosis. Front. Cell. Dev. Biol. 2021, 9, 715211. [Google Scholar] [CrossRef]
- Wang, Y.; Liu, X.; Shi, H.; Yu, Y.; Li, M.; Chen, R. NLRP3 inflammasome, an immune-inflammatory target in pathogenesis and treatment of cardiovascular diseases. Clin. Transl. Med. 2020, 10, 91–106. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
| Autophagy Related Cellular Processes | Involved Autophagy Genes for Functional Atherosclerosis | |||
|---|---|---|---|---|
| Signal Molecules Related to Promotion | Signal Molecules Related to Inhibition | |||
| Genes Involved | Ref. | Genes Involved | Ref. | |
| Chaperone-Mediated Autophagy | CD68, ERK, HSC70, LAMP-2A, LAL | [33,34,35,50] | ACSL1, ACTA2, COL1A1-3, COX2, CD36, caspase-1, HMGB1, iNOS, IL-1β, IL-18, NLRP3, P53, PAI-1, SR-A | [29,51,52,53,54,55,56,57,58] |
| Lipid Accumulation in Macrophage | Akt, MAPK, mTOR, NF-κB, SR-A | [56,59] | AMPK, ACSL1, ABCA1, FABP4, JNK, LAL, PPAR, PLIN/ADRP, SR-B, TFEB | [47,60,61,62,63,64,65,66,67] |
| Macrophage Pyroptosis | AIM2, caspase-1, DAMPs, GSDMD, GSDME, IL-1β, IL-18, mTOR, NLRP3, NF-κB, PAMPs, ROS, STAT3, SESN2, STING, TNF-α | [68,69,70,71,72,73,74,75,76,77,78,79] | beclin-1, HMGB1, LC3, PINK1/Parkin | [80,81,82,83] |
| Clonal Hematopoiesis | ASXL1, CBL, DNMT3A, cAMP, IDH2, IL-1β, IL-18, IL-6, JAK2, MCP-1, NLRP3, PPM1D, SF3B1, SRSF2, TET2, TP53, TNF-α, U2AF1 | [84,85,86,87,88,89,90,91,92,93,94,95] | IL-10, IL-13, TGF-β | [96] |
| Pathway | Drug Targeting Molecule | Agent Product | Biological Process/Mechanism of Action | Preclinical/Clinical Data | Class/Phase | References | |
|---|---|---|---|---|---|---|---|
| Autophagy associated drugs/molecules against atherosclerosis | Lipid Accumulation in Macrophage | SGLT2 | Enpagliflozin, Dagliliflozin | Activation of autophagy through the AMPK signaling pathway → clears intracellular lipid accumulation and damaged organelles, reducing inflammatory responses | Improved glycemic control in T2DM and reduces the risk of cardiovascular adverse cardiovascular events | Class II | [76,179] |
| Adiponectin receptor agonists | AdipoRon | Activate AMPK → enhance macrophage autophagy →promote cholesterol efflux | Cholesterol efflux increased by 40%, autophagy marker LC3-II/LC3-I ratio increased by 3 times | N/A | [62] | ||
| mTOR | Rapamycin | Inhibit mTOR→activation of autophagy→promote the burial effect | Rapamycin significantly reduced the atherosclerotic lesions in the aorta in a high-fat diet-induced atherosclerosis mouse model | Phase II | [69,180] | ||
| SGLT2 | Empagliflozin | Activation AMPK→inhibition of the mTORC1 activity→deregulate the inhibitory effect of mTOR on autophagy | Reduced mortality in patients with type 2 diabetes mellitus complicated with atherosclerotic cardiovascular disease | Phase III | [33,181] | ||
| TBK1/IKKε | BX795 | The TBK1/IKKε signaling pathway activates autophagy → clears damaged organelles and lipid accumulation within cells → reduces the production of inflammatory factors | TNF- α and IL-6 secretion were reduced by 70%, and autophagy-related genes (Atg 5) were upregulated | N/A | [182] | ||
| Macrophage Pyroptosis | Colchicine | Colchicine, low dose | Inhibition of NLRP3 inflammasome activation; it may interfere with autophagosome-lysosome fusion | Reduce the risk of cardiovascular events and improve coronary plaque stability | Class II | [74,183] | |
| NLRP3 | Tranilast | Inhibition of the NLRP3 protein →reduce in IL-1 β release; Increase the LC3-II/LC3-I ratio and reduce p62 accumulation | Significantly reduced the atherosclerotic plaque formation in the aorta | Phase II | [78] | ||
| Caspase-1 | VX-765 | Blocking caspase-1 downstream of the inflammasome → inhibition of the GSDMD-mediated pyroptosis | The plaque pyroptotic cells were decreased by 55%, and the plaque stability was enhanced | N/A | [67] | ||
| Gasdermin | Disulfiram | Blocking of the gasdermin D pore tract → inhibition of pyroptosis | Inhibition of pyroptosis reduced the plaque necrotic core | Phase I | [184] | ||
| GSDME | Gasdermin E inhibitor | Blocking of the GSDME shearing →inhibit pyrodead pore formation | Plaque area decreased by 30%, fibrous cap thickness increased by 50%, and 60% decreased macrophage pyroptosis markers (caspase-3 activity) | N/A | [80,185] | ||
| Clonal Hematopoiesis | JAK2 | Fedratinib | Inhibition of the expansion of the JAK 2 mutant clones → reduction of the proinflammatory macrophages | Reduce IL-6, and TNF- α secretion | N/A | [185,186,187] | |
| TET2 | N/A | Reduce the methylation level of the Beclin1 promoter region → promote the expression of Beclin1 | Increase the expression of autophagy marker LC3-II, while reducing the accumulation of autophagy disorder marker p62 | N/A | [81] | ||
| Drugs or molecules of other pathways against atherosclerosis | Statins | Atorvastatin, Rosuvastatin | Inhibits HMG-CoA reductase → lowers LDL-C | LDL-C decreased to <100 mg/dL | Class I | [188] | |
| PCSK9 | Alirocumab, Elosulfase | Blocking of the PCSK 9→increasing the LDL receptor expression →reduce LDL-C | Lower plasma LDL-C levels by 50–60% | Class I | [189] | ||
| IL-1β | Canakinumab | Neutralization of IL-1 β→inhibition of the NLRP3 inflammasome | Significant reduction in the hs-CRP levels | Class II | [29] | ||
| Antiplatelet drugs | Aspirin, Clopidogrel | Inhibition of platelet aggregation | Could significantly reduce the risk of stent thrombosis and cardiovascular events | Class I | [87,183] | ||
| GPX4 | RSL3 | Triggering of macrophage iron death by induction of lipid peroxidation →reduce intraplaque necrotic core | The plaque necrotic core was reduced by 40% | N/A | [67,189] | ||
| NLRP3/TRPV2 | Tranilast | Inhibition of the NLRP3 inflammasome blocks the TRPV 2 channels → synergy anti-inflammatory | Spot collagen content increased by 25% and macrophage apoptosis decreased by 40% | N/A | [78] | ||
| CCR2 | RS504393 | Blocking CCR2 → inhibition of monocyte recruitment to the plaques | The plaque macrophage number decreased by 45% and the overall plaque load decreased by 28% | N/A | [190] | ||
| PLK1 | BI-2536 | Inhibition of PLK1 → blocking of macrophage proliferation → reduce cell accumulation within the plaque | The proliferating macrophages (Ki67 +) were reduced by 60% and plaque stability was improved | N/A | [60] | ||
| Anti-aging drugs | Dasatinib, Quercetin | Reduced inflammatory factor release in senescent macrophages | Elimination of senescent clonal hematopoietic cells → lighten SASP | N/A | [31] | ||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, S.; Zhou, X.; Duan, Q.; Niu, S.; Li, P.; Feng, Y.; Zhang, Y.; Xu, X.; Gong, S.-P.; Cao, H. Autophagy and Its Association with Macrophages in Clonal Hematopoiesis Leading to Atherosclerosis. Int. J. Mol. Sci. 2025, 26, 3252. https://doi.org/10.3390/ijms26073252
Li S, Zhou X, Duan Q, Niu S, Li P, Feng Y, Zhang Y, Xu X, Gong S-P, Cao H. Autophagy and Its Association with Macrophages in Clonal Hematopoiesis Leading to Atherosclerosis. International Journal of Molecular Sciences. 2025; 26(7):3252. https://doi.org/10.3390/ijms26073252
Chicago/Turabian StyleLi, Shuanhu, Xin Zhou, Qinchun Duan, Shukun Niu, Pengquan Li, Yihan Feng, Ye Zhang, Xuehong Xu, Shou-Ping Gong, and Huiling Cao. 2025. "Autophagy and Its Association with Macrophages in Clonal Hematopoiesis Leading to Atherosclerosis" International Journal of Molecular Sciences 26, no. 7: 3252. https://doi.org/10.3390/ijms26073252
APA StyleLi, S., Zhou, X., Duan, Q., Niu, S., Li, P., Feng, Y., Zhang, Y., Xu, X., Gong, S.-P., & Cao, H. (2025). Autophagy and Its Association with Macrophages in Clonal Hematopoiesis Leading to Atherosclerosis. International Journal of Molecular Sciences, 26(7), 3252. https://doi.org/10.3390/ijms26073252