
Mitochondrial Cardiolipin-Targeted Tetrapeptide, SS-31, Exerts Neuroprotective Effects Within In Vitro and In Vivo Models of Spinal Cord Injury
 , , and
, , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
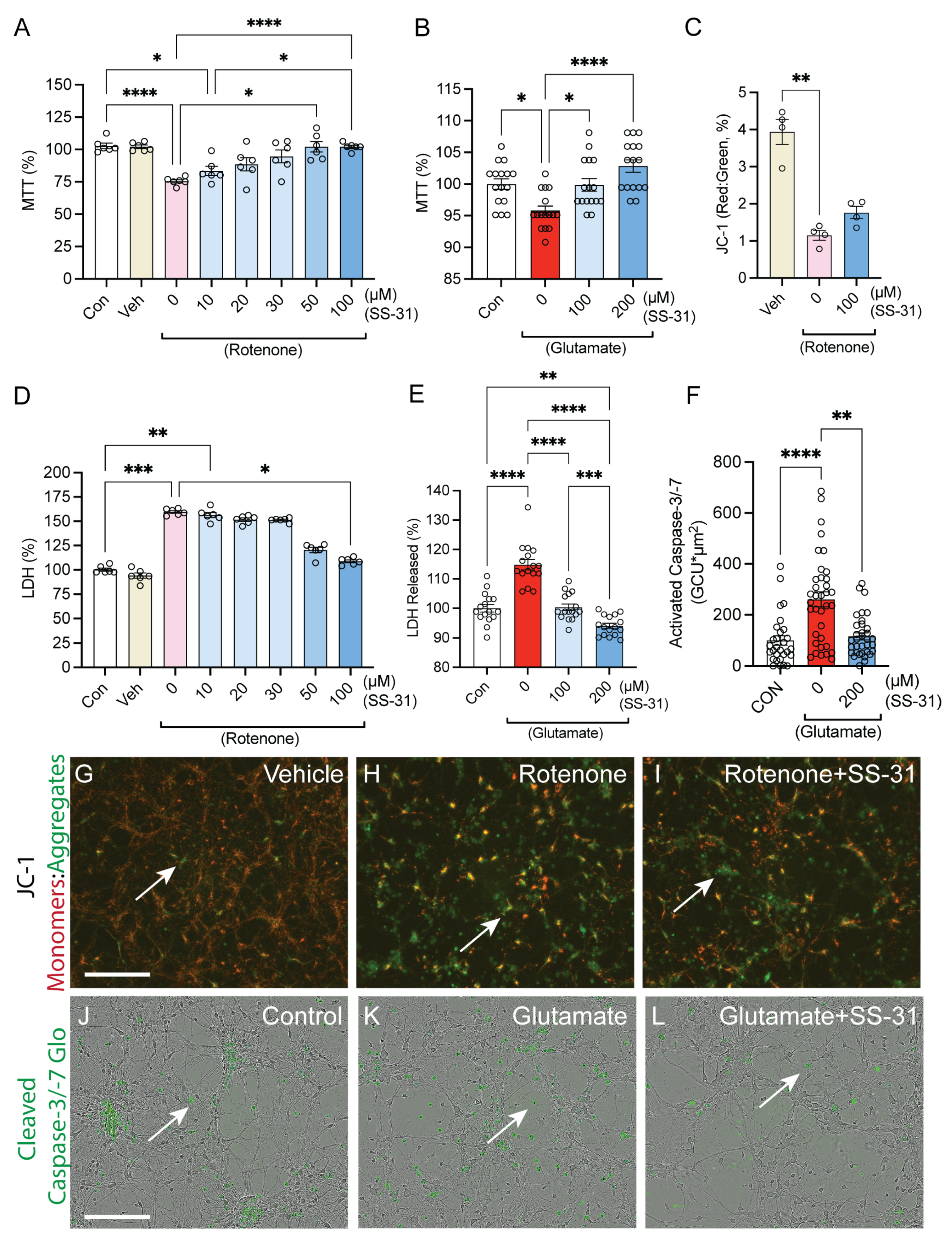
2.1. SS-31 Protected Spinal Cord Neurons from In Vitro Cell Death Induced by Rotenone or Glutamatergic Excitotoxicity
2.2. SS-31 Protected Spinal Cord Neurons from In Vitro Neurite Degeneration Induced by Glutamatergic Excitotoxicity
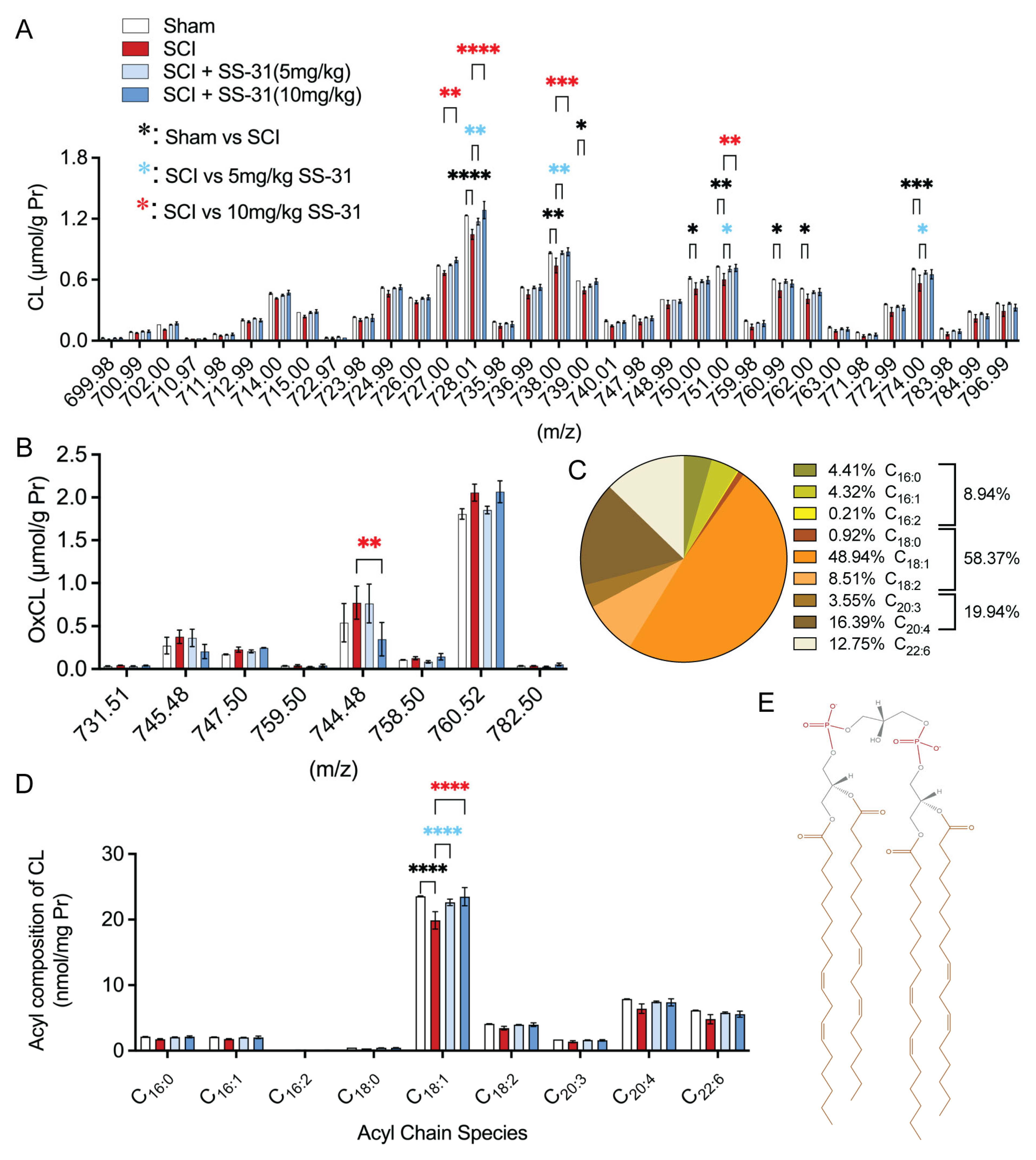
2.3. SS-31 Attenuated Cardiolipin Alterations in the Injured Spinal Cord After In Vivo SCI

2.4. SS-31 Improved Functional Locomotor Recovery After SCI
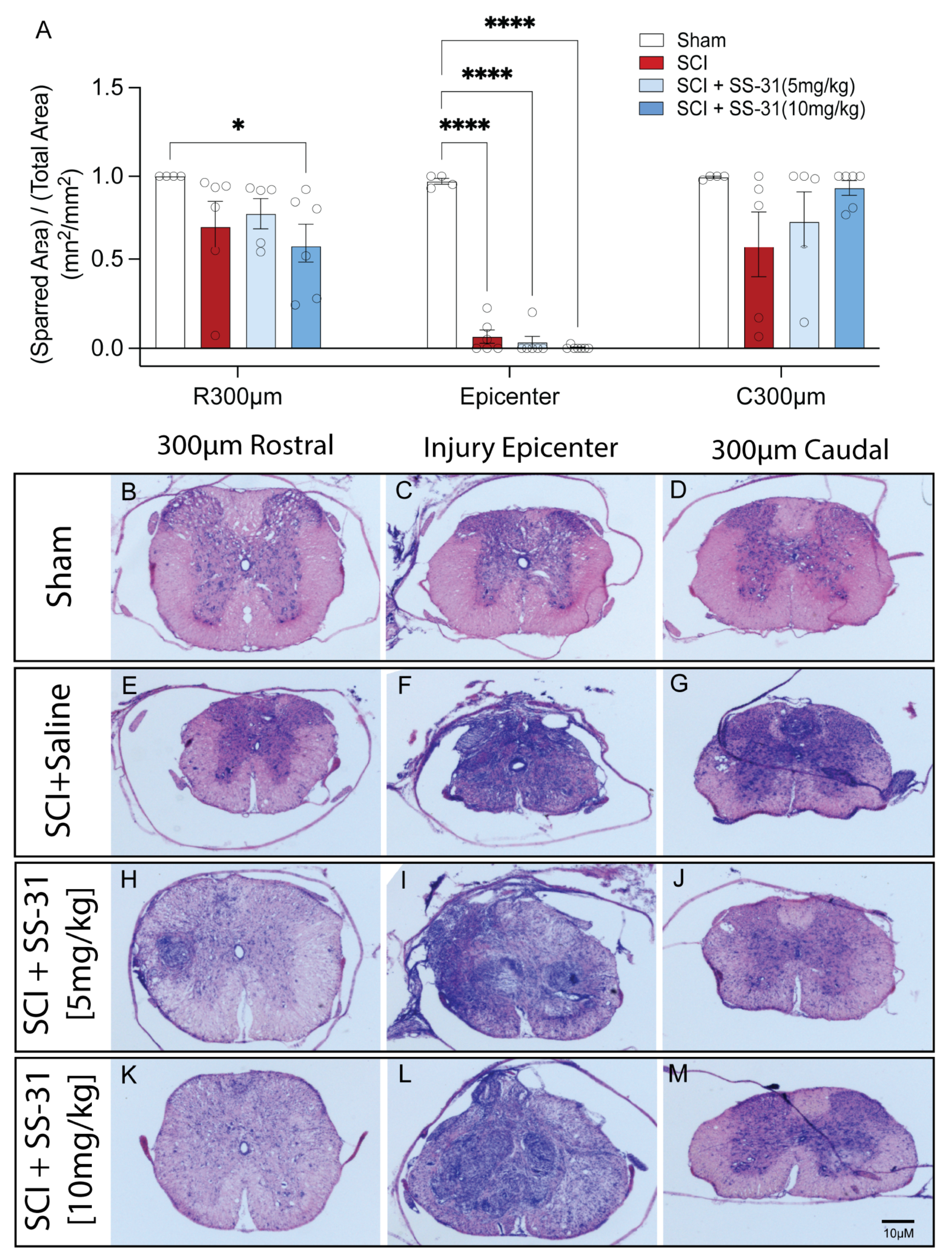
2.5. SS-31 Did Not Reduce Tissue Damage After SCI
3. Discussion
4. Materials and Methods
4.1. Reagents
4.2. Animals
4.3. Spinal Cord Neuronal Culture, Cell Treatment and Viability Assessment
4.4. Mitochondrial Membrane Potential (MMP, Δψm) Assay
4.5. Apoptotic Analysis
4.6. Spinal Cord Injury and Treatment
4.7. Lipidomics
4.8. Behavioral Assessments
4.9. Histological Assessments
4.10. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sinescu, C.; Popa, F.; Grigorean, V.; Onose, G.; Sandu, A.; Popescu, M.; Burnei, G.; Strambu, V.; Popa, C. Molecular basis of vascular events following spinal cord injury. J. Med. Life 2010, 3, 254–261. [Google Scholar] [PubMed]
- GBD 2016 Traumatic Brain Injury and Spinal Cord Injury Collaborators. Global, regional, and national burden of traumatic brain injury and spinal cord injury, 1990–2016: A systematic analysis for the Global Burden of Disease Study 2016. Lancet Neurol. 2019, 18, 56–87. [Google Scholar] [CrossRef] [PubMed]
- Fehlings, M.G.; Singh, A.; Tetreault, L.; Kalsi-Ryan, S.; Nouri, A. Global prevalence and incidence of traumatic spinal cord injury. Clin. Epidemiol. 2014, 6, 309–331. [Google Scholar] [CrossRef]
- Rabchevsky, A.G.; Patel, S.P.; Springer, J.E. Pharmacological interventions for spinal cord injury: Where do we stand? How might we step forward? Pharmacol. Ther. 2011, 132, 15–29. [Google Scholar] [CrossRef]
- Liu, N.K.; Xu, X.M. Neuroprotection and its molecular mechanism following spinal cord injury. Neural Reg. Res. 2012, 7, 2051–2062. [Google Scholar]
- Liu, N.-K.; Xu, X.-M. Phospholipase A2 and its Molecular Mechanism after Spinal Cord Injury. Mol. Neurobiol. 2010, 41, 197–205. [Google Scholar] [CrossRef]
- Farooqui, A.A.; Horrocks, L.A. Phospholipase A2-generated lipid mediators in the brain: The good, the bad, and the ugly. Neuroscientist 2006, 12, 245–260. [Google Scholar] [CrossRef]
- Sullivan, P.G.; Rabchevsky, A.G.; Keller, J.N.; Lovell, M.; Sodhi, A.; Hart, R.P.; Scheff, S.W. Intrinsic differences in brain and spinal cord mitochondria: Implication for therapeutic interventions. J. Comp. Neurol. 2004, 474, 524–534. [Google Scholar] [CrossRef]
- Liu, N.-K.; Zhang, Y.P.; Titsworth, W.L.; Jiang, X.; Han, S.; Lu, P.-H.; Shields, C.B.; Xu, X.-M. A novel role of phospholipase A2in mediating spinal cord secondary injury. Ann. Neurol. 2006, 59, 606–619. [Google Scholar] [CrossRef]
- Liu, N.; Deng, L.; Zhang, Y.P.; Lu, Q.; Wang, X.; Hu, J.; Oakes, E.; Bonventre, J.V.; Shields, C.B.; Xu, X. Cytosolic phospholipase A2 protein as a novel therapeutic target for spinal cord injury. Ann. Neurol. 2014, 75, 644–658. [Google Scholar] [CrossRef]
- Liu, N.-K.; Titsworth, W.; Xu, X.-M. Phospholipase A2 in CNS disorders: Implication on traumatic spinal cord and brain injuries. In Handbook of Neurochemistry and Molecular Neurobiology; Lajtha, A., Banik, N., Ray, S.K., Eds.; Springer: Boston, MA, USA, 2009; pp. 321–341. [Google Scholar]
- Farooqui, A.A.; Ong, W.-Y.; Horrocks, L.A. Biochemical Aspects of Neurodegeneration in Human Brain: Involvement of Neural Membrane Phospholipids and Phospholipases A2. Neurochem. Res. 2004, 29, 1961–1977. [Google Scholar] [CrossRef] [PubMed]
- Christie, W.W.; Han, X. (Eds.) Chapter 1—Lipids: Their structures and occurrence. In Lipid Analysis, 4th ed.; Woodhead Publishing: Sawston, UK, 2012; pp. 3–19. [Google Scholar]
- Hall, E.D.; Braughler, J.M. Role of Lipid Peroxidation in Post-Traumatic Spinal Cord Degeneration: A Review. Central Nerv. Syst. Trauma 1986, 3, 281–294. [Google Scholar] [CrossRef]
- Braughler, J.M.; Hall, E.D. Involvement of lipid peroxidation in CNS injury. J. Neurotrauma 1992, 9, S1–S7. [Google Scholar] [PubMed]
- Shamas-Din, A.; Bindner, S.; Chi, X.; Leber, B.; Andrews, D.W.; Fradin, C. Distinct lipid effects on tBid and Bim activation of membrane permeabilization by pro-apoptotic Bax. Biochem. J. 2015, 467, 495–505. [Google Scholar] [CrossRef] [PubMed]
- Paradies, G.; Paradies, V.; Ruggiero, F.M.; Petrosillo, G. Role of Cardiolipin in Mitochondrial Function and Dynamics in Health and Disease: Molecular and Pharmacological Aspects. Cells 2019, 8, 728. [Google Scholar] [CrossRef]
- Schlame, M.; Rua, D.; Greenberg, M.L. The biosynthesis and functional role of cardiolipin. Prog. Lipid Res. 2000, 39, 257–288. [Google Scholar] [CrossRef]
- Mohammadyani, D.; Yanamala, N.; Samhan-Arias, A.K.; Kapralov, A.A.; Stepanov, G.; Nuar, N.; Planas-Iglesias, J.; Sanghera, N.; Kagan, V.E.; Klein-Seetharaman, J. Structural characterization of cardiolipin-driven activation of cytochrome c into a peroxidase and membrane perturbation. Biochim. Biophys. Acta (BBA)—Biomembr. 2018, 1860, 1057–1068. [Google Scholar] [CrossRef]
- Liu, N.-K.; Deng, L.-X.; Wang, M.; Lu, Q.-B.; Wang, C.; Wu, X.; Wu, W.; Wang, Y.; Qu, W.; Han, Q.; et al. Restoring mitochondrial cardiolipin homeostasis reduces cell death and promotes recovery after spinal cord injury. Cell Death Dis. 2022, 13, 1058. [Google Scholar] [CrossRef]
- Zhu, Y.; Wang, H.; Fang, J.; Dai, W.; Zhou, J.; Wang, X.; Zhou, M. SS-31 Provides Neuroprotection by Reversing Mitochondrial Dysfunction after Traumatic Brain Injury. Oxidative Med. Cell. Longev. 2018, 2018, 4783602. [Google Scholar] [CrossRef]
- Zhu, L.-L.; Li, M.-Q.; He, F.; Zhou, S.-B.; Jiang, W. Mitochondria Targeted Peptide Attenuates Mitochondrial Dysfunction, Controls Inflammation and Protects Against Spinal Cord Injury-Induced Lung Injury. Cell. Physiol. Biochem. 2017, 44, 388–400. [Google Scholar] [CrossRef]
- Ji, J.; Kline, A.E.; Amoscato, A.; Samhan-Arias, A.K.; Sparvero, L.J.; Tyurin, V.A.; Tyurina, Y.Y.; Fink, B.; Manole, M.D.; Puccio, A.M.; et al. Lipidomics identifies cardiolipin oxidation as a mitochondrial target for redox therapy of brain injury. Nat. Neurosci. 2012, 15, 1407–1413. [Google Scholar] [CrossRef] [PubMed]
- Birk, A.V.; Chao, W.M.; Bracken, C.; Warren, J.D.; Szeto, H.H. Targeting mitochondrial cardiolipin and the cytochrome c/cardiolipin complex to promote electron transport and optimize mitochondrial ATP synthesis. Br. J. Pharmacol. 2014, 171, 2017–2028. [Google Scholar] [CrossRef] [PubMed]
- Szeto, H.H. First-in-class cardiolipin-protective compound as a therapeutic agent to restore mitochondrial bioenergetics. Br. J. Pharmacol. 2014, 171, 2029–2050. [Google Scholar] [CrossRef]
- Zhao, K.; Zhao, G.-M.; Wu, D.; Soong, Y.; Birk, A.V.; Schiller, P.W.; Szeto, H.H. Cell-permeable Peptide Antioxidants Targeted to Inner Mitochondrial Membrane inhibit Mitochondrial Swelling, Oxidative Cell Death, and Reperfusion Injury. J. Biol. Chem. 2004, 279, 34682–34690. [Google Scholar] [CrossRef] [PubMed]
- Birk, A.V.; Liu, S.; Soong, Y.; Mills, W.; Singh, P.; Warren, J.D.; Seshan, S.V.; Pardee, J.D.; Szeto, H.H. The Mitochondrial-Targeted Compound SS-31 Re-Energizes Ischemic Mitochondria by Interacting with Cardiolipin. J. Am. Soc. Nephrol. 2013, 24, 1250–1261. [Google Scholar] [CrossRef] [PubMed]
- Graham, Z.A.; DeBerry, J.J.; Cardozo, C.P.; Bamman, M.M. A 50 kdyne contusion spinal cord injury with or without the drug SS-31 was not associated with major changes in muscle mass or gene expression 14 d after injury in young male mice. Physiol. Rep. 2021, 9, e14751. [Google Scholar] [CrossRef] [PubMed]
- Graham, Z.A.; DeBerry, J.J.; Cardozo, C.P.; Bamman, M.M. SS-31 does not prevent or reduce muscle atrophy 7 days after a 65 kdyne contusion spinal cord injury in young male mice. Physiol. Rep. 2022, 10, e15266. [Google Scholar] [CrossRef]
- Zhang, H.; Chen, Y.; Li, F.; Wu, C.; Cai, W.; Ye, H.; Su, H.; He, M.; Yang, L.; Wang, X.; et al. Elamipretide alleviates pyroptosis in traumatically injured spinal cord by inhibiting cPLA2-induced lysosomal membrane permeabilization. J. Neuroinflamm. 2023, 20, 6. [Google Scholar] [CrossRef]
- Jiang, W.; He, F.; Ding, G.; Wu, J. Elamipretide reduces pyroptosis and improves functional recovery after spinal cord injury. CNS Neurosci. Ther. 2023, 29, 2843–2856. [Google Scholar] [CrossRef]
- Chu, T.C.; Ji, J.; Dagda, R.K.; Jiang, J.F.; Tyurina, Y.Y.; Kapralov, A.A.; Tyurin, V.A.; Yanamala, N.; Shrivastava, I.H.; Mohammadyani, D.; et al. Cardiolipin externalization to the outer mitochondrial membrane acts as an elimination signal for mitophagy in neuronal cells. Nat. Cell Biol. 2013, 15, 1197–1205. [Google Scholar] [CrossRef]
- Guo, W.-X.; Pye, Q.N.; Williamson, K.S.; Stewart, C.A.; Hensley, K.L.; Kotake, Y.; Floyd, R.A.; Broyles, R.H. Mitochondrial dysfunction in choline deficiency-induced apoptosis in cultured rat hepatocytes. Free Radic. Biol. Med. 2005, 39, 641–650. [Google Scholar] [CrossRef] [PubMed]
- Salido, M.; Gonzalez, J.L.; Vilches, J. Loss of mitochondrial membrane potential is inhibited by bombesin in etoposide-induced apoptosis in PC-3 prostate carcinoma cells. Mol. Cancer Ther. 2007, 6, 1292–1299. [Google Scholar] [CrossRef] [PubMed]
- Salvioli, S.; Ardizzoni, A.; Franceschi, C.; Cossarizza, A. JC-1, but not DiOC6(3) or rhodamine 123, is a reliable fluorescent probe to assess delta psi changes in intact cells: Implications for studies on mitochondrial functionality during apoptosis. FEBS Lett. 1997, 411, 77–82. [Google Scholar] [CrossRef] [PubMed]
- Wingrave, J.M.; Schaecher, K.E.; Sribnick, E.A.; Wilford, G.G.; Ray, S.K.; Hazen-Martin, D.J.; Hogan, E.L.; Banik, N.L. Early induction of secondary injury factors causing activation of calpain and mitochondria-mediated neuronal apoptosis following spinal cord injury in rats. J. Neurosci. Res. 2003, 73, 95–104. [Google Scholar] [CrossRef]
- Bayir, H.; Tyurin, V.A.; Tyurina, Y.Y.; Viner, R.; Ritov, V.; Amoscato, A.A.; Zhao, Q.; Zhang, X.J.; Janesko-Feldman, K.L.; Alexander, H.; et al. Selective early cardiolipin peroxidation after traumatic brain injury: An oxidative lipidomics analysis. Ann. Neurol. 2007, 62, 154–169. [Google Scholar] [CrossRef]
- Kagan, V.E.; Tyurin, V.A.; Jiang, J.; Tyurina, Y.Y.; Ritov, V.B.; Amoscato, A.A.; Osipov, A.N.; Belikova, N.A.; Kapralov, A.A.; Kini, V.; et al. Cytochrome c acts as a cardiolipin oxygenase required for release of proapoptotic factors. Nat. Chem. Biol. 2005, 1, 223–232. [Google Scholar] [CrossRef]
- Chicco, A.J.; Sparagna, G.C. Role of cardiolipin alterations in mitochondrial dysfunction and disease. Am. J. Physiol. Cell Physiol. 2007, 292, C33–C44. [Google Scholar] [CrossRef] [PubMed]
- Gonzalvez, F.; Gottlieb, E. Cardiolipin: Setting the beat of apoptosis. Apoptosis 2007, 12, 877–885. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Liu, Y.; Ma, B.; Zhou, M.; Zhao, X.; Fu, X.; Kan, S.; Hu, W.; Zhu, R. Mitochondrial regulatory mechanisms in spinal cord injury: A narrative review. Medicine 2022, 101, e31930. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, P.G.; Krishnamurthy, S.; Patel, S.P.; Pandya, J.D.; Rabchevsky, A.G. Temporal Characterization of Mitochondrial Bioenergetics after Spinal Cord Injury. J. Neurotrauma 2007, 24, 991–999. [Google Scholar] [CrossRef]
- Park, E.; Velumian, A.A.; Fehlings, M.G. The Role of Excitotoxicity in Secondary Mechanisms of Spinal Cord Injury: A Review with an Emphasis on the Implications for White Matter Degeneration. J. Neurotrauma 2004, 21, 754–774. [Google Scholar] [CrossRef] [PubMed]
- Szeto, H.H.; Birk, A.V. Serendipity and the Discovery of Novel Compounds That Restore Mitochondrial Plasticity. Clin. Pharmacol. Ther. 2014, 96, 672–683. [Google Scholar] [CrossRef] [PubMed]
- Siegel, M.P.; Kruse, S.E.; Percival, J.M.; Goh, J.; White, C.C.; Hopkins, H.C.; Kavanagh, T.J.; Szeto, H.H.; Rabinovitch, P.S.; Marcinek, D.J. Mitochondrial-targeted peptide rapidly improves mitochondrial energetics and skeletal muscle performance in aged mice. Aging Cell 2013, 12, 763–771. [Google Scholar] [CrossRef] [PubMed]
- Regan, R.; Choi, D. Glutamate neurotoxicity in spinal cord cell culture. Neuroscience 1991, 43, 585–591. [Google Scholar] [CrossRef]
- Zhou, L.; Li, F.; Xu, H.-B.; Luo, C.-X.; Wu, H.-Y.; Zhu, M.-M.; Lu, W.; Ji, X.; Zhou, Q.-G.; Zhu, D.-Y. Treatment of cerebral ischemia by disrupting ischemia-induced interaction of nNOS with PSD-95. Nat. Med. 2010, 16, 1439–1443, Erratum in Nat. Med. 2011, 17, 1153. [Google Scholar] [CrossRef]
- Bradbury, E.J.; Moon, L.D.F.; Popat, R.J.; King, V.R.; Bennett, G.S.; Patel, P.N.; Fawcett, J.W.; McMahon, S.B. Chondroitinase ABC promotes functional recovery after spinal cord injury. Nature 2002, 416, 636–640. [Google Scholar] [CrossRef]
- García-Alías, G.; Barkhuysen, S.; Buckle, M.; Fawcett, J.W. Chondroitinase ABC treatment opens a window of opportunity for task-specific rehabilitation. Nat. Neurosci. 2009, 12, 1145–1151. [Google Scholar] [CrossRef]
- Fouad, K.; Schnell, L.; Bunge, M.B.; Schwab, M.E.; Liebscher, T.; Pearse, D.D. Combining Schwann Cell Bridges and Olfactory-Ensheathing Glia Grafts with Chondroitinase Promotes Locomotor Recovery after Complete Transection of the Spinal Cord. J. Neurosci. 2005, 25, 1169–1178. [Google Scholar] [CrossRef]
- Rosenzweig, E.S.; Courtine, G.; Jindrich, D.L.; Brock, J.H.; Ferguson, A.R.; Strand, S.C.; Nout, Y.S.; Roy, R.R.; Miller, D.M.; Beattie, M.S.; et al. Extensive spontaneous plasticity of corticospinal projections after primate spinal cord injury. Nat. Neurosci. 2010, 13, 1505–1510. [Google Scholar] [CrossRef]
- Ohnishi, Y.; Yamamoto, M.; Sugiura, Y.; Setoyama, D.; Kishima, H. Rostro-caudal different energy metabolism leading to differences in degeneration in spinal cord injury. Brain Commun. 2021, 3, fcab058. [Google Scholar] [CrossRef] [PubMed]
- Liu, N.-K.; Zhang, Y.P.; Han, S.; Pei, J.; Xu, L.Y.; Lu, P.-H.; Shields, C.B.; Xu, X.-M. Annexin A1 Reduces Inflammatory Reaction and Tissue Damage Through Inhibition of Phospholipase A2 Activation in Adult Rats Following Spinal Cord Injury. J. Neuropathol. Exp. Neurol. 2007, 66, 932–943. [Google Scholar] [CrossRef] [PubMed]
- Essen Bioscience. Incucyte Live-Cell Analysis System—Technical Note. Available online: https://www.sartorius.com/download/1163270/incucyte-basic-analysis-software-guidelines-8000-0522-d00-data.pdf (accessed on 28 September 2024).
- Lou, J.; Lenke, L.G.; Ludwig, F.J.; O’Brien, M.F. Apoptosis as a mechanism of neuronal cell death following acute experimental spinal cord injury. Spinal Cord 1998, 36, 683–690. [Google Scholar] [CrossRef] [PubMed]
- Basso, D.M.; Fisher, L.C.; Anderson, A.J.; Jakeman, L.B.; Mctigue, D.M.; Popovich, P.G. Basso Mouse Scale for Locomotion Detects Differences in Recovery after Spinal Cord Injury in Five Common Mouse Strains. J. Neurotrauma 2006, 23, 635–659. [Google Scholar] [CrossRef] [PubMed]
- Liu, N.-K.; Zhang, Y.-P.; O’Connor, J.; Gianaris, A.; Oakes, E.; Lu, Q.-B.; Verhovshek, T.; Walker, C.L.; Shields, C.B.; Xu, X.-M. A bilateral head injury that shows graded brain damage and behavioral deficits in adultmice. Brain Res. 2013, 1499, 121–128. [Google Scholar] [CrossRef] [PubMed]
- Schucht, P.; Raineteau, O.; Schwab, M.E.; Fouad, K. Anatomical correlates of locomotor recovery following dorsal and ventral lesions of the rat spinal cord. Exp. Neurol. 2002, 176, 143–153. [Google Scholar] [CrossRef]
- Metz, G.A.; Merkler, D.; Dietz, V.; Schwab, M.E.; Fouad, K. Efficient testing of motor function in spinal cord injured rats. Brain Res. 2000, 883, 165–177. [Google Scholar] [CrossRef]
- Hamers, F.P.; Lankhorst, A.J.; van Laar, T.J.; Veldhuis, W.B.; Gispen, W.H. Automated Quantitative Gait Analysis During Overground Locomotion in the Rat: Its Application to Spinal Cord Contusion and Transection Injuries. J. Neurotrauma 2001, 18, 187–201. [Google Scholar] [CrossRef]
- Beare, J.E.; Morehouse, J.R.; DeVries, W.H.; Enzmann, G.U.; Burke, D.A.; Magnuson, D.S.; Whittemore, S.R. Gait Analysis in Normal and Spinal Contused Mice Using the TreadScan System. J. Neurotrauma 2009, 26, 2045–2056. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ravenscraft, B.; Lee, D.-H.; Dai, H.; Watson, A.L.; Aparicio, G.I.; Han, X.; Deng, L.-X.; Liu, N.-K. Mitochondrial Cardiolipin-Targeted Tetrapeptide, SS-31, Exerts Neuroprotective Effects Within In Vitro and In Vivo Models of Spinal Cord Injury. Int. J. Mol. Sci. 2025, 26, 3327. https://doi.org/10.3390/ijms26073327
Ravenscraft B, Lee D-H, Dai H, Watson AL, Aparicio GI, Han X, Deng L-X, Liu N-K. Mitochondrial Cardiolipin-Targeted Tetrapeptide, SS-31, Exerts Neuroprotective Effects Within In Vitro and In Vivo Models of Spinal Cord Injury. International Journal of Molecular Sciences. 2025; 26(7):3327. https://doi.org/10.3390/ijms26073327
Chicago/Turabian StyleRavenscraft, Baylen, Do-Hun Lee, Heqiao Dai, Abbie Lea Watson, Gabriela Inés Aparicio, Xianlin Han, Ling-Xiao Deng, and Nai-Kui Liu. 2025. "Mitochondrial Cardiolipin-Targeted Tetrapeptide, SS-31, Exerts Neuroprotective Effects Within In Vitro and In Vivo Models of Spinal Cord Injury" International Journal of Molecular Sciences 26, no. 7: 3327. https://doi.org/10.3390/ijms26073327
APA StyleRavenscraft, B., Lee, D.-H., Dai, H., Watson, A. L., Aparicio, G. I., Han, X., Deng, L.-X., & Liu, N.-K. (2025). Mitochondrial Cardiolipin-Targeted Tetrapeptide, SS-31, Exerts Neuroprotective Effects Within In Vitro and In Vivo Models of Spinal Cord Injury. International Journal of Molecular Sciences, 26(7), 3327. https://doi.org/10.3390/ijms26073327