
Unveiling the Forensic Potential of Oral and Nasal Microbiota in Post-Mortem Interval Estimation

 ,
, 
Abstract
1. Introduction
2. Results
2.1. Overall Characterization of 16S rRNA Sequencing Data
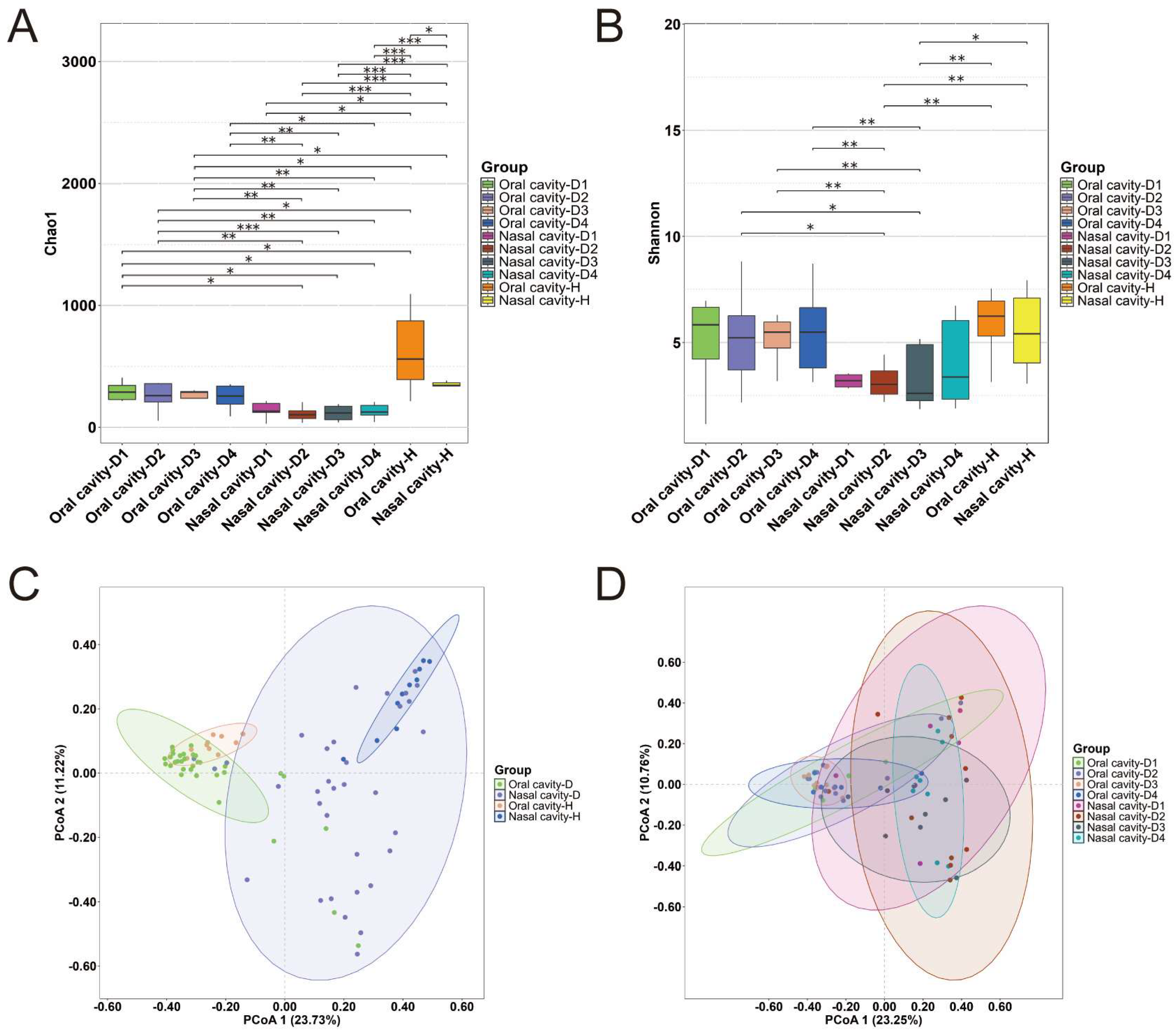
2.2. Microbial Diversity
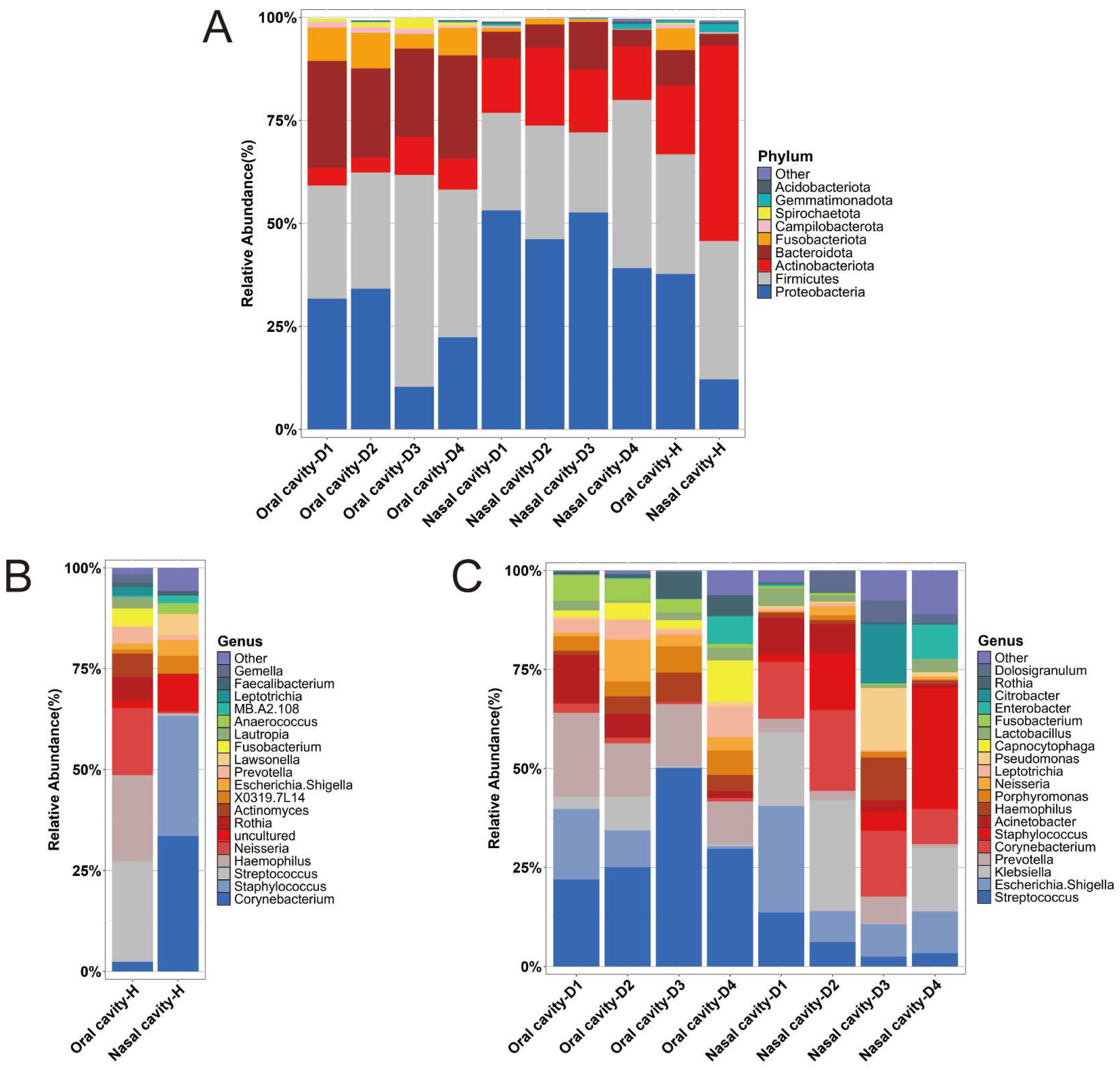
2.3. Microbial Abundance
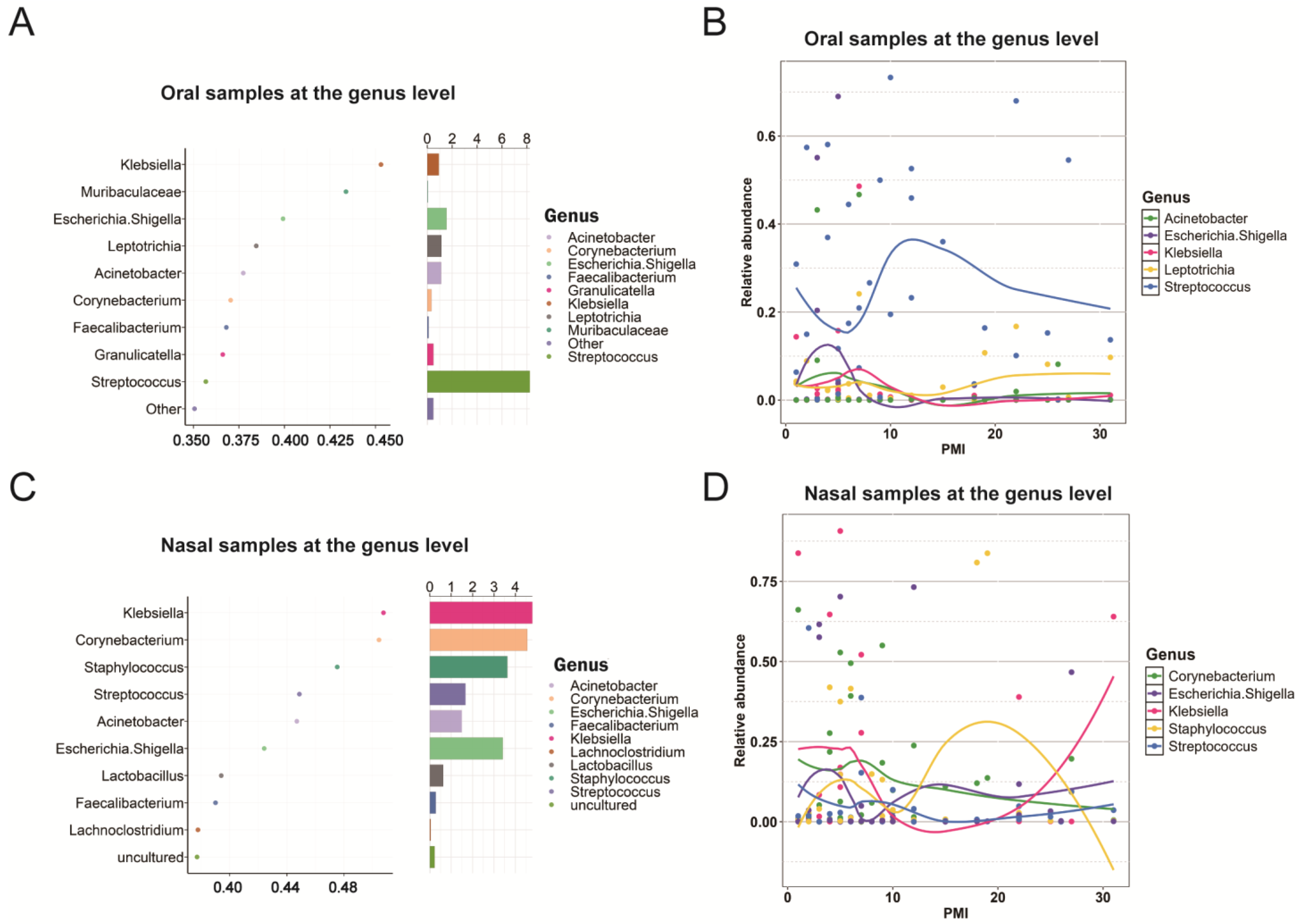
2.4. Biomarkers Associated with PMIs
2.5. Comparison of Samples from Frozen and Unfrozen Cadavers
3. Discussion
4. Materials and Methods
4.1. Sample Collection
4.2. DNA Extraction, Amplification and Sequencing
4.3. Data Analysis
4.4. Random Forest and LOESS Regression Modeling
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Henssge, C.; Madea, B. Estimation of the time since death. Forensic Sci. Int. 2007, 165, 182–184. [Google Scholar]
- Roy, D.; Tomo, S.; Purohit, P.; Setia, P. Microbiome in Death and Beyond: Current Vistas and Future Trends. Front. Ecol. Evol. 2021, 9, 630397. [Google Scholar]
- Tozzo, P.; Amico, I.; Delicati, A.; Toselli, F.; Caenazzo, L. Post-Mortem Interval and Microbiome Analysis through 16S rRNA Analysis: A Systematic Review. Diagnostics 2022, 12, 2641. [Google Scholar] [CrossRef]
- Liu, R.; Gu, Y.; Shen, M.; Li, H.; Zhang, K.; Wang, Q.; Wei, X.; Zhang, H.; Wu, D.; Yu, K.; et al. Predicting postmortem interval based on microbial community sequences and machine learning algorithms. Environ. Microbiol. 2020, 22, 2273–2291. [Google Scholar]
- Matuszewski, S. Post-Mortem Interval Estimation Based on Insect Evidence: Current Challenges. Insects 2021, 12, 314. [Google Scholar] [CrossRef] [PubMed]
- Sangwan, A.; Singh, S.P.; Singh, P.; Gupta, O.P.; Manas, A.; Gupta, S. Role of molecular techniques in PMI estimation: An update. J. Forensic Leg. Med. 2021, 83, 102251. [Google Scholar] [PubMed]
- Tozzo, P.; Scrivano, S.; Sanavio, M.; Caenazzo, L. The Role of DNA Degradation in the Estimation of Post-Mortem Interval: A Systematic Review of the Current Literature. Int. J. Mol. Sci. 2020, 21, 3540. [Google Scholar] [CrossRef]
- Maiese, A.; Scatena, A.; Costantino, A.; Di Paolo, M.; La Russa, R.; Turillazzi, E.; Frati, P.; Fineschi, V. MicroRNAs as Useful Tools to Estimate Time Since Death. A Systematic Review of Current Literature. Diagnostics 2021, 11, 64. [Google Scholar] [CrossRef]
- Wang, Q.; Zhang, Y.; Lin, H.; Zha, S.; Fang, R.; Wei, X.; Fan, S.; Wang, Z. Estimation of the late postmortem interval using FTIR spectroscopy and chemometrics in human skeletal remains. Forensic Sci. Int. 2017, 281, 113–120. [Google Scholar]
- Schmidt, V.M.; Zelger, P.; Woess, C.; Pallua, A.K.; Arora, R.; Degenhart, G.; Brunner, A.; Zelger, B.; Schirmer, M.; Rabl, W.; et al. Application of Micro-Computed Tomography for the Estimation of the Post-Mortem Interval of Human Skeletal Remains. Biology 2022, 11, 1105. [Google Scholar] [CrossRef]
- Hyde, E.R.; Haarmann, D.P.; Lynne, A.M.; Bucheli, S.R.; Petrosino, J.F. The living dead: Bacterial community structure of a cadaver at the onset and end of the bloat stage of decomposition. PLoS ONE 2013, 8, e77733. [Google Scholar] [CrossRef]
- Metcalf, J.L.; Wegener Parfrey, L.; Gonzalez, A.; Lauber, C.L.; Knights, D.; Ackermann, G.; Humphrey, G.C.; Gebert, M.J.; Van Treuren, W.; Berg-Lyons, D.; et al. A microbial clock provides an accurate estimate of the postmortem interval in a mouse model system. eLife 2013, 2, e01104. [Google Scholar]
- Cláudia-Ferreira, A.; Barbosa, D.J.; Saegeman, V.; Fernández-Rodríguez, A.; Dinis-Oliveira, R.J.; Freitas, A.R.; on behalf of the MR CLEAN Registry Investigators. The Future Is Now: Unraveling the Expanding Potential of Human (Necro)Microbiome in Forensic Investigations. Microorganisms 2023, 11, 2509. [Google Scholar] [CrossRef]
- Pechal, J.L.; Crippen, T.L.; Benbow, M.E.; Tarone, A.M.; Dowd, S.; Tomberlin, J.K. The potential use of bacterial community succession in forensics as described by high throughput metagenomic sequencing. Int. J. Leg. Med. 2014, 128, 193–205. [Google Scholar]
- Moitas, B.; Caldas, I.M.; Sampaio-Maia, B. Microbiology and postmortem interval: A systematic review. Forensic Sci. Med. Pathol. 2024, 20, 696–715. [Google Scholar] [PubMed]
- Wang, Z.; Zhang, F.; Wang, L.; Yuan, H.; Guan, D.; Zhao, R. Advances in artificial intelligence-based microbiome for PMI estimation. Front. Microbiol. 2022, 13, 1034051. [Google Scholar] [CrossRef] [PubMed]
- Li, N.; Liang, X.R.; Zhou, S.D.; Dang, L.H.; Li, J.; An, G.S.; Ren, K.; Jin, Q.Q.; Liang, X.H.; Cao, J.; et al. Exploring postmortem succession of rat intestinal microbiome for PMI based on machine learning algorithms and potential use for humans. Forensic Sci. Int. Genet. 2023, 66, 102904. [Google Scholar]
- Yang, F.; Zhang, X.; Hu, S.; Nie, H.; Gui, P.; Zhong, Z.; Guo, Y.; Zhao, X. Changes in Microbial Communities Using Pigs as a Model for Postmortem Interval Estimation. Microorganisms 2023, 11, 2811. [Google Scholar] [CrossRef]
- Janaway, R.C.; Percival, S.L.; Wilson, A.S. Decomposition of Human Remains. In Microbiology and Aging; Humana Press: Totowa, NJ, USA, 2009; pp. 313–334. [Google Scholar]
- Zhao, X.; Zhong, Z.; Hua, Z. Estimation of the post-mortem interval by modelling the changes in oral bacterial diversity during decomposition. J. Appl. Microbiol. 2022, 133, 3451–3464. [Google Scholar] [CrossRef]
- Adserias-Garriga, J.; Quijada, N.M.; Hernandez, M.; Rodriguez Lazaro, D.; Steadman, D.; Garcia-Gil, L.J. Dynamics of the oral microbiota as a tool to estimate time since death. Mol. Oral. Microbiol. 2017, 32, 511–516. [Google Scholar]
- Dong, K.; Xin, Y.; Cao, F.; Huang, Z.; Sun, J.; Peng, M.; Liu, W.; Shi, P. Succession of oral microbiota community as a tool to estimate postmortem interval. Sci. Rep. 2019, 9, 13063. [Google Scholar] [CrossRef]
- Johnson, H.R.; Trinidad, D.D.; Guzman, S.; Khan, Z.; Parziale, J.V.; DeBruyn, J.M.; Lents, N.H. A Machine Learning Approach for Using the Postmortem Skin Microbiome to Estimate the Postmortem Interval. PLoS ONE 2016, 11, e0167370. [Google Scholar] [CrossRef] [PubMed]
- Iancu, L.; Bonicelli, A.; Procopio, N. Decomposition in an extreme cold environment and associated microbiome-prediction model implications for the postmortem interval estimation. Front. Microbiol. 2024, 15, 1392716. [Google Scholar] [CrossRef]
- Sguazzi, G.; Mickleburgh, H.L.; Ghignone, S.; Voyron, S.; Reno, F.; Migliario, M.; Sellitto, F.; Lovisolo, F.; Camurani, G.; Ogbanga, N.; et al. Microbial DNA in human nucleic acid extracts: Recoverability of the microbiome in DNA extracts stored frozen long-term and its potential and ethical implications for forensic investigation. Forensic Sci. Int. Genet. 2022, 59, 102686. [Google Scholar] [CrossRef] [PubMed]
- Ogbanga, N.; Nelson, A.; Gino, S.; Wescott, D.J.; Mickleburgh, H.L.; Gocha, T.P.; Procopio, N. The impact of freezing on the post-mortem human microbiome. Front. Ecol. Evol. 2023, 11, 1151001. [Google Scholar] [CrossRef]
- Preiswerk, D.; Walser, J.C.; Ebert, D. Temporal dynamics of microbiota before and after host death. ISME J. 2018, 12, 2076–2085. [Google Scholar] [CrossRef] [PubMed]
- Lemon, K.P.; Klepac-Ceraj, V.; Schiffer, H.K.; Brodie, E.L.; Lynch, S.V.; Kolter, R. Comparative analyses of the bacterial microbiota of the human nostril and oropharynx. mBio 2010, 1, 10–1128. [Google Scholar] [CrossRef]
- Bassis, C.M.; Tang, A.L.; Young, V.B.; Pynnonen, M.A. The nasal cavity microbiota of healthy adults. Microbiome 2014, 2, 27. [Google Scholar] [CrossRef]
- Hyde, E.R.; Haarmann, D.P.; Petrosino, J.F.; Lynne, A.M.; Bucheli, S.R. Initial insights into bacterial succession during human decomposition. Int. J. Leg. Med. 2015, 129, 661–671. [Google Scholar] [CrossRef]
- Huang, X.; Zeng, J.; Li, S.; Chen, J.; Wang, H.; Li, C.; Zhang, S. 16S rRNA, metagenomics and 2bRAD-M sequencing to decode human thanatomicrobiome. Sci. Data 2024, 11, 736. [Google Scholar] [CrossRef]
- Metcalf, J.L.; Xu, Z.Z.; Weiss, S.; Lax, S.; Van Treuren, W.; Hyde, E.R.; Song, S.J.; Amir, A.; Larsen, P.; Sangwan, N.; et al. Microbial community assembly and metabolic function during mammalian corpse decomposition. Science 2016, 351, 158–162. [Google Scholar] [CrossRef] [PubMed]
- Hyde, E.R.; Metcalf, J.L.; Bucheli, S.R.; Lynne, A.M.; Knight, R. Microbial communities associated with decomposing corpses. In Forensic Microbiology; Wiley: Hoboken, NJ, USA, 2017; pp. 245–273. [Google Scholar]
- Baker, J.L.; Mark Welch, J.L.; Kauffman, K.M.; McLean, J.S.; He, X. The oral microbiome: Diversity, biogeography and human health. Nat. Rev. Microbiol. 2024, 22, 89–104. [Google Scholar] [CrossRef] [PubMed]
- Pechal, J.L.; Schmidt, C.J.; Jordan, H.R.; Benbow, M.E. A large-scale survey of the postmortem human microbiome, and its potential to provide insight into the living health condition. Sci. Rep. 2018, 8, 5724. [Google Scholar] [CrossRef]
- Braga, R.M.; Dourado, M.N.; Araújo, W.L. Microbial interactions: Ecology in a molecular perspective. Braz. J. Microbiol. 2016, 47, 86–98. [Google Scholar] [CrossRef] [PubMed]
- Callahan, B.J.; McMurdie, P.J.; Rosen, M.J.; Han, A.W.; Johnson, A.J.A.; Holmes, S.P. DADA2: High-resolution sample inference from Illumina amplicon data. Nat. Methods 2016, 13, 581–583. [Google Scholar] [CrossRef]
- Bolyen, E.; Rideout, J.R.; Dillon, M.R.; Bokulich, N.A.; Abnet, C.C.; Al-Ghalith, G.A.; Alexander, H.; Alm, E.J.; Arumugam, M.; Asnicar, F.; et al. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat. Biotechnol. 2019, 37, 852–857. [Google Scholar]
- Chao, A.; Bunge, J. Estimating the number of species in a stochastic abundance model. Biometrics 2002, 58, 531–539. [Google Scholar] [CrossRef]
- Hill, T.C.; Walsh, K.A.; Harris, J.A.; Moffett, B.F. Using ecological diversity measures with bacterial communities. FEMS Microbiol. Ecol. 2003, 43, 1–11. [Google Scholar]
- Liaw, A.; Wiener, M.C. Classification and Regression by randomForest. R J. 2007, 2–3, 18–22. [Google Scholar]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Group | Individual | PMI (Days) | Age (Years) | Gender | Cause of Death | Frozen | Season of Death |
|---|---|---|---|---|---|---|---|
| D1 | S06 | 1 | 41 | M | Craniocerebral injury | No | Spring |
| D1 | S08 | 1 | 47 | M | Coronary heart disease | No | Spring |
| D1 | S09 | 2 | 37 | F | Craniocerebral injury | No | Spring |
| D1 | S10 | 2 | 48 | M | Coronary heart disease | No | Spring |
| D1 | S12 | 3 | 47 | M | Drowning | No | Spring |
| D1 | S40 | 3 | 60 | F | Drug intoxication | Yes | Summer |
| D2 | S17 | 4 | 49 | M | Coronary heart disease | No | Spring |
| D2 | S31 | 4 | 19 | F | Traffic injury | Yes | Summer |
| D2 | S05 | 5 | 80 | M | Stomach tumor and lung infection | No | Winter |
| D2 | S16 | 5 | 30 | F | Craniocerebral injury | Yes | Spring |
| D2 | S30 | 5 | 51 | M | Traffic injury | Yes | Summer |
| D2 | S24 | 5 | 65 | M | Coronary heart disease | Yes | Spring |
| D2 | S22 | 6 | 27 | M | Sudden death | Yes | Spring |
| D2 | S33 | 6 | 42 | M | Electric shock | Yes | Summer |
| D2 | S25 | 7 | 62 | F | Septic shock and multiple organ failure | Yes | Spring |
| D2 | S28 | 7 | 57 | M | Sudden death | Yes | Spring |
| D2 | S35 | 7 | 78 | M | Aortoclasia | Yes | Summer |
| D3 | S20 | 8 | 54 | M | Hanging | Yes | Spring |
| D3 | S21 | 9 | 80 | M | Traffic injury | No | Spring |
| D3 | S01 | 9 | 54 | M | Craniocerebral injury | Yes | Winter |
| D3 | S19 | 10 | 55 | M | Acute pulmonary embolism | No | Spring |
| D3 | S23 | 10 | 71 | M | Traffic injury | Yes | Spring |
| D3 | S18 | 12 | 51 | M | Traumatic shock | No | Spring |
| D3 | S14 | 12 | 23 | F | Injury by fall from height | Yes | Spring |
| D3 | S38 | 12 | 51 | M | Unknown | Yes | Summer |
| D3 | S02 | 15 | 64 | M | Sudden cardiac death | Yes | Winter |
| D4 | S27 | 18 | 46 | M | Traffic injury | Yes | Spring |
| D4 | S32 | 19 | 45 | M | Traffic injury | Yes | Summer |
| D4 | S04 | 22 | 87 | F | Coronary heart disease | Yes | Winter |
| D4 | S07 | 22 | 65 | F | Coronary heart disease | Yes | Spring |
| D4 | S11 | 25 | 28 | M | Craniocerebral injury | Yes | Spring |
| D4 | S34 | 26 | 74 | M | Traffic injury | Yes | Autumn |
| D4 | S39 | 27 | 42 | M | Trauma | Yes | Summer |
| D4 | S13 | 31 | 57 | M | Coronary heart disease | No | Spring |
| H | 1 | - | 20 | M | - | - | - |
| H | 2 | - | 24 | F | - | - | - |
| H | 3 | - | 28 | M | - | - | - |
| H | 4 | - | 27 | M | - | - | - |
| H | 5 | - | 26 | F | - | - | - |
| H | 6 | - | 34 | F | - | - | - |
| H | 7 | - | 28 | M | - | - | - |
| H | 8 | - | 31 | M | - | - | - |
| H | 9 | - | 23 | F | - | - | - |
| H | 10 | - | 22 | F | - | - | - |
| Group | Oral Cavity-H | Oral Cavity-D | Nasal Cavity-H | Nasal Cavity-D |
|---|---|---|---|---|
| Alpha diversity | Higher in healthy individual group than cadaver group; higher in oral group than nasal group | |||
| Beta diversity | Oral vs. nasal: significant clustering Healthy individual vs. cadaver: no significant clustering Frozen vs. unfrozen: no significant clustering | |||
| Dominant phyla | Proteobacteria, Firmicutes, and Actinobacteriota | Proteobacteria, Firmicutes, and Bacteroidota | Proteobacteria, Firmicutes, and Actinobacteriota | Proteobacteria, Firmicutes, and Actinobacteriota |
| Frozen vs. unfrozen: frozen samples exhibited a higher relative abundance of Proteobacteria and a lower relative abundance of Actinobacteriota compared to unfrozen samples | ||||
| Dominant genera | Streptococcus and Haemophilus | Streptococcus and Prevotella | Corynebacterium and Staphylococcus | Variation across different PMI stages |
| Frozen vs. unfrozen: the dominant genera in the nasal group showed substantial differences between frozen and unfrozen samples | ||||
| PMI-related biomarkers (phylum level) | - | Firmicutes, Proteobacteria, Bacteroidota, Actinobacteriota, and Fusobacteriota Key turning points were observed on days 5, 12, and 22 | - | Firmicutes, Proteobacteria, Bacteroidota, Actinobacteriota, and Fusobacteriota Similar turning points and the trends for Proteobacteria and Firmicutes were reversed compared to the oral group |
| - | Frozen vs. unfrozen: shared the same biomarkers: Firmicutes, Proteobacteria, Bacteroidota, Actinobacteriota, and Fusobacteriota | - | Frozen vs. unfrozen: shared the same biomarkers: Firmicutes, Proteobacteria, Bacteroidota, and Actinobacteriota | |
| PMI-related biomarkers (genus level) | - | Streptococcus, Escherichia-Shigella, Acinetobacter, Klebsiella, and Leptotrichia | - | Klebsiella, Corynebacterium, Staphylococcus, Escherichia-Shigella, and Streptococcus |
| - | Frozen vs. unfrozen: different biomarkers | - | Frozen vs. unfrozen: different biomarkers except for Klebsiella | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, J.; Wei, Q.; Yang, F.; Liu, Y.; Zhao, Y.; Zhang, H.; Huang, X.; Zeng, J.; Wang, X.; Zhang, S. Unveiling the Forensic Potential of Oral and Nasal Microbiota in Post-Mortem Interval Estimation. Int. J. Mol. Sci. 2025, 26, 3432. https://doi.org/10.3390/ijms26073432
Chen J, Wei Q, Yang F, Liu Y, Zhao Y, Zhang H, Huang X, Zeng J, Wang X, Zhang S. Unveiling the Forensic Potential of Oral and Nasal Microbiota in Post-Mortem Interval Estimation. International Journal of Molecular Sciences. 2025; 26(7):3432. https://doi.org/10.3390/ijms26073432
Chicago/Turabian StyleChen, Ji, Qi Wei, Fan Yang, Yanan Liu, Yurong Zhao, Han Zhang, Xin Huang, Jianye Zeng, Xiang Wang, and Suhua Zhang. 2025. "Unveiling the Forensic Potential of Oral and Nasal Microbiota in Post-Mortem Interval Estimation" International Journal of Molecular Sciences 26, no. 7: 3432. https://doi.org/10.3390/ijms26073432
APA StyleChen, J., Wei, Q., Yang, F., Liu, Y., Zhao, Y., Zhang, H., Huang, X., Zeng, J., Wang, X., & Zhang, S. (2025). Unveiling the Forensic Potential of Oral and Nasal Microbiota in Post-Mortem Interval Estimation. International Journal of Molecular Sciences, 26(7), 3432. https://doi.org/10.3390/ijms26073432