
Luteinizing Hormone Regulates Testosterone Production, Leydig Cell Proliferation, Differentiation, and Circadian Rhythm During Spermatogenesis


Abstract
:1. Introduction
2. LH Affects LC Testosterone Secretion and Thus Spermatogenesis
2.1. Developmental Processes of FLCs and ALCs
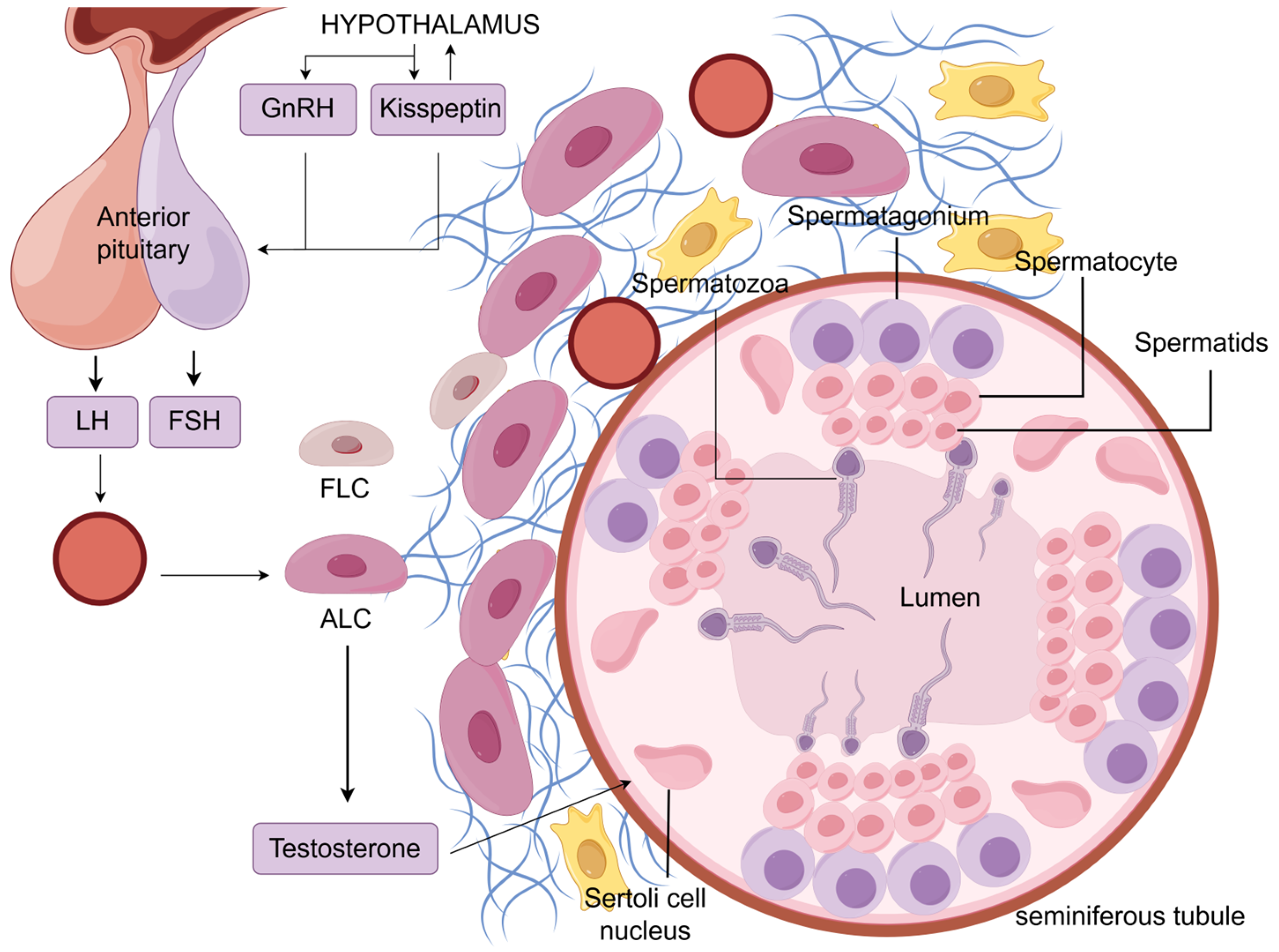
2.2. Hypothalamic–Pituitary–Gonadal Axis
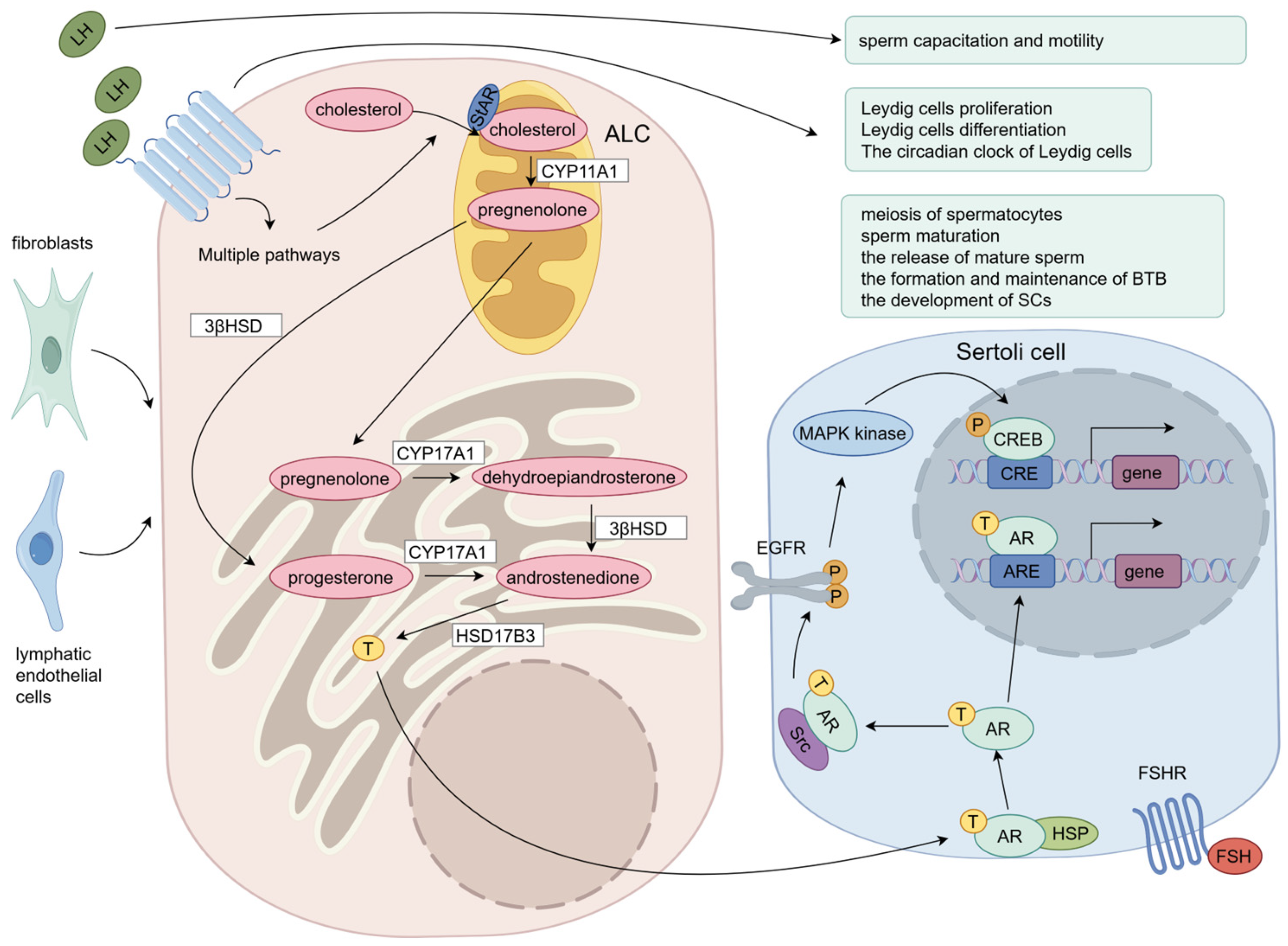
2.3. LH-Stimulated Testosterone Production in LCs
2.4. Testosterone Regulates Spermatogenesis
3. LH Regulates Proliferation, Differentiation, and the Circadian Clock of LCs
3.1. LH Regulates LC Proliferation
3.2. LH Regulates LC Differentiation
3.3. LH Regulates the Circadian Clock of LCs
4. LH Regulates Testosterone Secretion of LCs Through Multiple Signaling Pathways
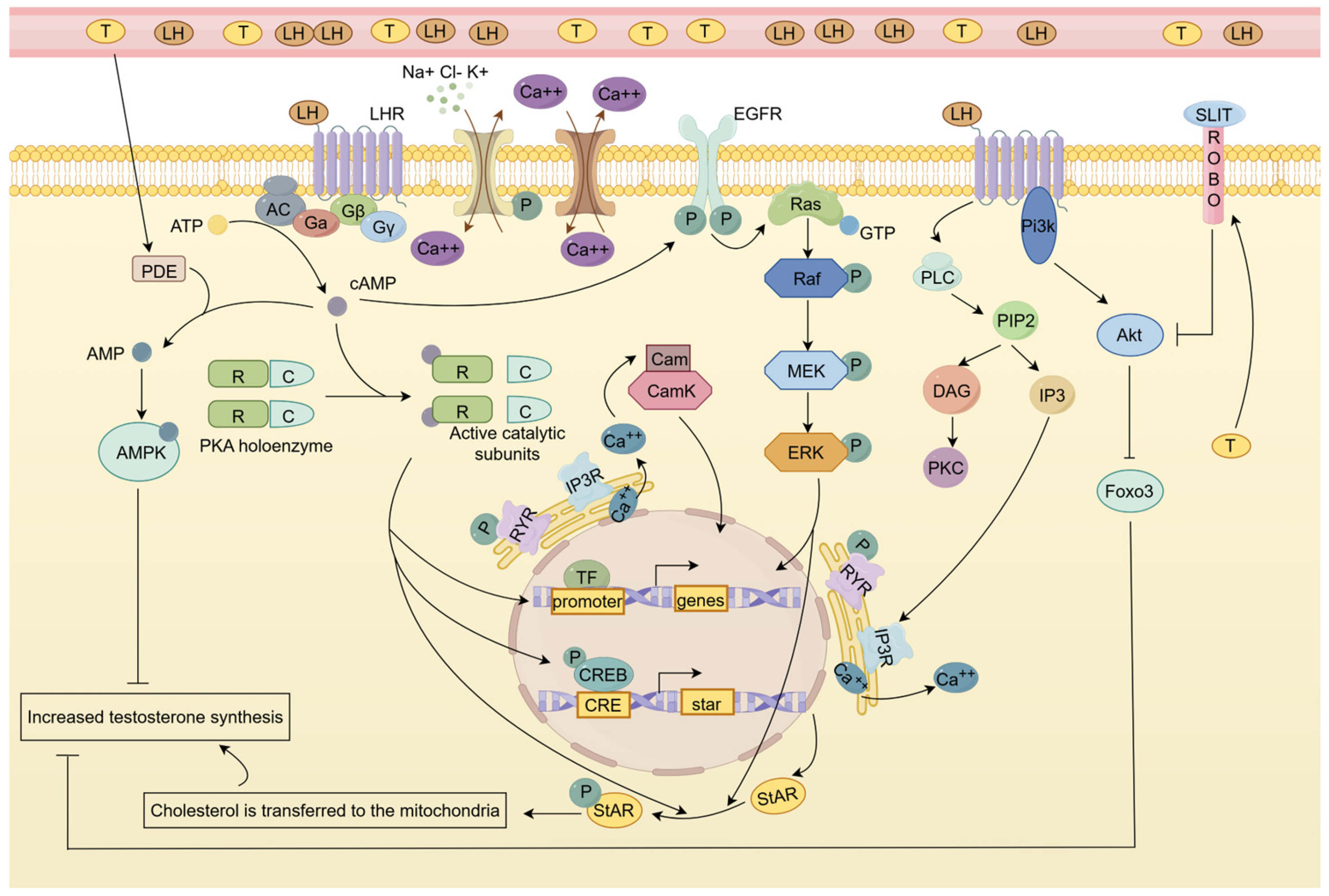
4.1. EGFR/MAPK Signaling Pathway Is Involved in LH-Induced LC Testosterone Synthesis
4.2. Calcium Signaling Pathway Is Involved in LH-Induced LC Testosterone Synthesis
4.3. cGMP-PKG Signaling Pathway Has a Bidirectional Effect on LH-Induced Testosterone Production
4.4. AMPK Signaling Pathway and Slit/Robo Pathway Have Feedback Regulatory Effects on LH-Induced Testosterone Production
5. Clinical Application of LH: Treatment of HH, Diagnosis of Gonadal-Related Diseases, and Monitoring of Sperm Quality
5.1. Treatment of Hypogonadotropic Hypogonadism (HH): By Rescuing the Broken HPG Axis
5.2. LH Level Can Be Used in the Diagnosis of Androgen Insensitivity Syndrome and Precocious Puberty
5.3. Sperm Quality Monitoring and Regulation
6. Conclusions and Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Fonseca, A.C.S.; Barreiro, M.; Tomé, A.; Vale-Fernandes, E. Male Reproductive Health—Study of a Sperm Donor Population. JBRA Assist. Reprod. 2022, 26, 247–254. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, A.; Baskaran, S.; Parekh, N.; Cho, C.-L.; Henkel, R.; Vij, S.; Arafa, M.; Panner Selvam, M.K.; Shah, R. Male Infertility. Lancet 2021, 397, 319–333. [Google Scholar] [CrossRef] [PubMed]
- Svingen, T.; Koopman, P. Building the Mammalian Testis: Origins, Differentiation, and Assembly of the Component Cell Populations. Genes Dev. 2013, 27, 2409–2426. [Google Scholar] [CrossRef]
- O’Donnell, L.; Smith, L.B.; Rebourcet, D. Sertoli Cells as Key Drivers of Testis Function. Semin. Cell Dev. Biol. 2022, 121, 2–9. [Google Scholar] [CrossRef]
- Haider, S.G. Cell Biology of Leydig Cells in the Testis. Int. Rev. Cytol. 2004, 233, 181–241. [Google Scholar] [CrossRef]
- Bhattacharya, I.; Dey, S. Emerging Concepts on Leydig Cell Development in Fetal and Adult Testis. Front. Endocrinol. 2022, 13, 1086276. [Google Scholar] [CrossRef]
- Sokwalla, S.M.R. Chapter 2—Reproductive Endocrine Physiology. In Subfertility; Elsevier: Amsterdam, The Netherlands, 2021; pp. 39–64. [Google Scholar]
- Neto, F.T.L.; Bach, P.V.; Najari, B.B.; Li, P.S.; Goldstein, M. Spermatogenesis in Humans and Its Affecting Factors. Semin. Cell Dev. Biol. 2016, 59, 10–26. [Google Scholar] [CrossRef]
- Walker, W.H. Regulation of Mammalian Spermatogenesis by miRNAs. Semin. Cell Dev. Biol. 2022, 121, 24–31. [Google Scholar] [CrossRef]
- Shen, L.; Yu, C.; Gao, F.; Hong, Y.-T. [PIWI/piRNA Complex-Mediated Regulation of Spermatogenesis]. Zhonghua Nan Ke Xue 2021, 27, 262–268. [Google Scholar]
- Xiong, B.; Jiang, Y.; Wang, Y.; Han, X.; Zhang, C.; Zhong, R.; Ge, W.; Han, B.; Ge, Z.; Huang, G.; et al. LncRNA8276 Primes Cell-Cell Adhesion for Regulation of Spermatogenesis. Andrology 2022, 10, 1687–1701. [Google Scholar] [CrossRef]
- Zhang, Z.; Miao, J.; Wang, Y. Mitochondrial Regulation in Spermatogenesis. Reproduction 2022, 163, R55–R69. [Google Scholar] [CrossRef] [PubMed]
- Mohammadi, A.; Shabani, R.; Bashiri, Z.; Rafiei, S.; Asgari, H.; Koruji, M. Therapeutic Potential of Exosomes in Spermatogenesis Regulation and Male Infertility. Biol. Cell 2024, 116, e2300127. [Google Scholar] [CrossRef] [PubMed]
- Xiong, Y.; Yu, C.; Zhang, Q. Ubiquitin-Proteasome System-Regulated Protein Degradation in Spermatogenesis. Cells 2022, 11, 1058. [Google Scholar] [CrossRef] [PubMed]
- Sofikitis, N.; Giotitsas, N.; Tsounapi, P.; Baltogiannis, D.; Giannakis, D.; Pardalidis, N. Hormonal Regulation of Spermatogenesis and Spermiogenesis. J. Steroid Biochem. Mol. Biol. 2008, 109, 323–330. [Google Scholar] [CrossRef]
- Sriraman, V.; Rao, V.S.; Sairam, M.R.; Rao, A.J. Effect of Deprival of LH on Leydig Cell Proliferation: Involvement of PCNA, Cyclin D3 and IGF-1. Mol. Cell. Endocrinol. 2000, 162, 113–120. [Google Scholar] [CrossRef]
- Sriraman, V.; Sairam, M.; Rao, A. Evaluation of Relative Roles of LH and FSH in Regulation of Differentiation of Leydig Cells Using an Ethane 1,2-Dimethylsulfonate-Treated Adult Rat Model. J. Endocrinol. 2003, 176, 151–161. [Google Scholar] [CrossRef]
- Baburski, A.Z.; Andric, S.A.; Kostic, T.S. Luteinizing Hormone Signaling Is Involved in Synchronization of Leydig Cell’s Clock and Is Crucial for Rhythm Robustness of Testosterone Production†. Biol. Reprod. 2019, 100, 1406–1415. [Google Scholar] [CrossRef]
- Vasquez, J.M.; Ben-Nun, I.; Greenblatt, R.B.; Mahesh, V.B.; Keel, B.A. Correlation between Follicle-Stimulating Hormone, Luteinizing Hormone, Prolactin, and Testosterone with Sperm Cell Concentration and Motility. Obs. Gynecol. 1986, 67, 86–90. [Google Scholar]
- Eblen, A.; Bao, S.; Lei, Z.M.; Nakajima, S.T.; Rao, C.V. The Presence of Functional Luteinizing Hormone/Chorionic Gonadotropin Receptors in Human Sperm. J. Clin. Endocrinol. Metab. 2001, 86, 2643–2648. [Google Scholar] [CrossRef]
- López-Torres, A.S.; González-González, M.E.; Mata-Martínez, E.; Larrea, F.; Treviño, C.L.; Chirinos, M. Luteinizing Hormone Modulates Intracellular Calcium, Protein Tyrosine Phosphorylation and Motility during Human Sperm Capacitation. Biochem. Biophys. Res. Commun. 2017, 483, 834–839. [Google Scholar] [CrossRef]
- Griswold, S.L.; Behringer, R.R. Fetal Leydig Cell Origin and Development. Sex. Dev. 2009, 3, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Kerr, J.B.; Knell, C.M. The Fate of Fetal Leydig Cells during the Development of the Fetal and Postnatal Rat Testis. Development 1988, 103, 535–544. [Google Scholar] [CrossRef] [PubMed]
- Brennan, J.; Capel, B. One Tissue, Two Fates: Molecular Genetic Events That Underlie Testis Versus Ovary Development. Nat. Rev. Genet. 2004, 5, 509–521. [Google Scholar] [CrossRef] [PubMed]
- Hall, P.F.; Irby, D.C.; Kretser, D.M.D. Conversion of Cholesterol to Androgens by Rat Testes: Comparison of Interstitial Cells and Seminiferous Tubules. Endocrinology 1969, 84, 488–496. [Google Scholar] [CrossRef]
- Brennan, J.; Tilmann, C.; Capel, B. Pdgfr-α Mediates Testis Cord Organization and Fetal Leydig Cell Development in the XY Gonad. Genes Dev. 2003, 17, 800–810. [Google Scholar] [CrossRef]
- Wen, Q.; Cheng, C.Y.; Liu, Y.-X. Development, Function and Fate of Fetal Leydig Cells. Semin. Cell Dev. Biol. 2016, 59, 89–98. [Google Scholar] [CrossRef]
- DeFalco, T.; Takahashi, S.; Capel, B. Two Distinct Origins for Leydig Cell Progenitors in the Fetal Testis. Dev. Biol. 2011, 352, 14–26. [Google Scholar] [CrossRef]
- Shima, Y.; Morohashi, K. Leydig Progenitor Cells in Fetal Testis. Mol. Cell. Endocrinol. 2017, 445, 55–64. [Google Scholar] [CrossRef]
- Clark, A.M.; Garland, K.K.; Russell, L.D. Desert Hedgehog (Dhh) Gene Is Required in the Mouse Testis for Formation of Adult-Type Leydig Cells and Normal Development of Peritubular Cells and Seminiferous Tubules. Biol. Reprod. 2000, 63, 1825–1838. [Google Scholar] [CrossRef]
- Yao, H.H.-C.; Whoriskey, W.; Capel, B. Desert Hedgehog/Patched 1 Signaling Specifies Fetal Leydig Cell Fate in Testis Organogenesis. Genes Dev. 2002, 16, 1433–1440. [Google Scholar] [CrossRef]
- Kitamura, K.; Yanazawa, M.; Sugiyama, N.; Miura, H.; Iizuka-Kogo, A.; Kusaka, M.; Omichi, K.; Suzuki, R.; Kato-Fukui, Y.; Kamiirisa, K.; et al. Mutation of ARX Causes Abnormal Development of Forebrain and Testes in Mice and X-Linked Lissencephaly with Abnormal Genitalia in Humans. Nat. Genet. 2002, 32, 359–369. [Google Scholar] [CrossRef] [PubMed]
- Inoue, M.; Baba, T.; Morohashi, K. Recent Progress in Understanding the Mechanisms of Leydig Cell Differentiation. Mol. Cell. Endocrinol. 2018, 468, 39–46. [Google Scholar] [CrossRef] [PubMed]
- Teerds, K.J.; Huhtaniemi, I.T. Morphological and Functional Maturation of Leydig Cells: From Rodent Models to Primates. Hum. Reprod. Update 2015, 21, 310–328. [Google Scholar] [CrossRef] [PubMed]
- Kaftanovskaya, E.M.; Lopez, C.; Ferguson, L.; Myhr, C.; Agoulnik, A.I. Genetic Ablation of Androgen Receptor Signaling in Fetal Leydig Cell Lineage Affects Leydig Cell Functions in Adult Testis. FASEB J. 2015, 29, 2327–2337. [Google Scholar] [CrossRef]
- Shima, Y.; Matsuzaki, S.; Miyabayashi, K.; Otake, H.; Baba, T.; Kato, S.; Huhtaniemi, I.; Morohashi, K. Fetal Leydig Cells Persist as an Androgen-Independent Subpopulation in the Postnatal Testis. Mol. Endocrinol. 2015, 29, 1581–1593. [Google Scholar] [CrossRef]
- Chen, H.; Wang, Y.; Ge, R.; Zirkin, B.R. Leydig Cell Stem Cells: Identification, Proliferation and Differentiation. Mol. Cell. Endocrinol. 2017, 445, 65–73. [Google Scholar] [CrossRef]
- Chen, H.; Stanley, E.; Jin, S.; Zirkin, B.R. Stem Leydig Cells: From Fetal to Aged Animals. Birth Defects Res. Part C Embryo Today Rev. 2010, 90, 272–283. [Google Scholar] [CrossRef]
- O’Shaughnessy, P.J.; Fleming, L.M.; Jackson, G.; Hochgeschwender, U.; Reed, P.; Baker, P.J. Adrenocorticotropic Hormone Directly Stimulates Testosterone Production by the Fetal and Neonatal Mouse Testis. Endocrinology 2003, 144, 3279–3284. [Google Scholar] [CrossRef]
- Kaprara, A.; Huhtaniemi, I.T. The Hypothalamus-Pituitary-Gonad Axis: Tales of Mice and Men. Metabolism 2018, 86, 3–17. [Google Scholar] [CrossRef]
- Xie, Q.; Kang, Y.; Zhang, C.; Xie, Y.; Wang, C.; Liu, J.; Yu, C.; Zhao, H.; Huang, D. The Role of Kisspeptin in the Control of the Hypothalamic-Pituitary-Gonadal Axis and Reproduction. Front. Endocrinol. 2022, 13, 925206. [Google Scholar] [CrossRef]
- Pinilla, L.; Aguilar, E.; Dieguez, C.; Millar, R.P.; Tena-Sempere, M. Kisspeptins and Reproduction: Physiological Roles and Regulatory Mechanisms. Physiol. Rev. 2012, 92, 1235–1316. [Google Scholar] [CrossRef] [PubMed]
- Bliss, S.P.; Navratil, A.M.; Xie, J.; Roberson, M.S. GnRH Signaling, the Gonadotrope and Endocrine Control of Fertility. Front. Neuroendocr. 2010, 31, 322–340. [Google Scholar] [CrossRef] [PubMed]
- Loosfelt, H.; Misrahi, M.; Atger, M.; Salesse, R.; Vu Hai-Luu Thi, M.T.; Jolivet, A.; Guiochon-Mantel, A.; Sar, S.; Jallal, B.; Garnier, J.; et al. Cloning and Sequencing of Porcine LH-hCG Receptor cDNA: Variants Lacking Transmembrane Domain. Science 1989, 245, 525–528. [Google Scholar] [CrossRef] [PubMed]
- Kremer, H.; Mariman, E.; Otten, B.J.; Moll, G.W.; Stoellnga, G.B.A.; Wit, J.M.; Jansen, M.; Drop, S.L.; Faas, B.; Ropers, H.-H.; et al. Cosegregation of Missense Mutations of the Luteinizing Hormone Receptor Gene with Familial Male-Limited Precocious Puberty. Hum. Mol. Genet. 1993, 2, 1779–1783. [Google Scholar] [CrossRef]
- Miller, W.L.; Auchus, R.J. The Molecular Biology, Biochemistry, and Physiology of Human Steroidogenesis and Its Disorders. Endocr. Rev. 2011, 32, 81–151. [Google Scholar] [CrossRef]
- Papadopoulos, V.; Miller, W.L. Role of Mitochondria in Steroidogenesis. Best Pract. Res. Clin. Endocrinol. Metab. 2012, 26, 771–790. [Google Scholar] [CrossRef]
- Tugaeva, K.V.; Sluchanko, N.N. Steroidogenic Acute Regulatory Protein: Structure, Functioning, and Regulation. Biochemistry 2019, 84, 233–253. [Google Scholar] [CrossRef]
- Epstein, L.F.; Orme-Johnson, N.R. Acute Action of Luteinizing Hormone on Mouse Leydig Cells: Accumulation of Mitochondrial Phosphoproteins and Stimulation of Testosterone Synthesis. Mol. Cell. Endocrinol. 1991, 81, 113–126. [Google Scholar] [CrossRef]
- Clark, B.J.; Wells, J.; King, S.R.; Stocco, D.M. The Purification, Cloning, and Expression of a Novel Luteinizing Hormone-Induced Mitochondrial Protein in MA-10 Mouse Leydig Tumor Cells. Characterization of the Steroidogenic Acute Regulatory Protein (StAR). J. Biol. Chem. 1994, 269, 28314–28322. [Google Scholar] [CrossRef]
- Galano, M.; Li, Y.; Li, L.; Sottas, C.; Papadopoulos, V. Role of Constitutive STAR in Leydig Cells. Int. J. Mol. Sci. 2021, 22, 2021. [Google Scholar] [CrossRef]
- Bose, H.S.; Whittal, R.M.; Ran, Y.; Bose, M.; Baker, B.Y.; Miller, W.L. StAR-like Activity and Molten Globule Behavior of StARD6, A Male Germ-Line Protein. Biochemistry 2008, 47, 2277–2288. [Google Scholar] [CrossRef]
- Munell, F.; Suárez-Quian, C.A.; Selva, D.M.; Tirado, O.M.; Reventós, J. Androgen-Binding Protein and Reproduction: Where Do We Stand? J. Androl. 2002, 23, 598–609. [Google Scholar] [CrossRef] [PubMed]
- O’Hara, L.; Smith, L.B. Androgen Receptor Roles in Spermatogenesis and Infertility. Best Pract. Res. Clin. Endocrinol. Metab. 2015, 29, 595–605. [Google Scholar] [CrossRef] [PubMed]
- Naamneh Elzenaty, R.; du Toit, T.; Flück, C.E. Basics of Androgen Synthesis and Action. Best Pract. Res. Clin. Endocrinol. Metab. 2022, 36, 101665. [Google Scholar] [CrossRef] [PubMed]
- Heinlein, C.A.; Chang, C. Androgen Receptor (AR) Coregulators: An Overview. Endocr. Rev. 2002, 23, 175–200. [Google Scholar] [CrossRef]
- Cheng, J.; Watkins, S.C.; Walker, W.H. Testosterone Activates Mitogen-Activated Protein Kinase via Src Kinase and the Epidermal Growth Factor Receptor in Sertoli Cells. Endocrinology 2007, 148, 2066–2074. [Google Scholar] [CrossRef]
- Walker, W.H. Non-Classical Actions of Testosterone and Spermatogenesis. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2010, 365, 1557–1569. [Google Scholar] [CrossRef]
- Shang, Y.; Myers, M.; Brown, M. Formation of the Androgen Receptor Transcription Complex. Mol. Cell 2002, 9, 601–610. [Google Scholar] [CrossRef]
- Pillerová, M.; Borbélyová, V.; Hodosy, J.; Riljak, V.; Renczés, E.; Frick, K.M.; Tóthová, Ľ. On the Role of Sex Steroids in Biological Functions by Classical and Non-Classical Pathways. An Update. Front. Neuroendocrinol. 2021, 62, 100926. [Google Scholar] [CrossRef]
- Bartlett, J.M.S.; Kerr, J.B.; Sharpe, R.M. The Effect of Selective Destruction and Regeneration of Rat Leydig Cells on the Intratesticular Distribution of Testosterone and Morphology of the Seminiferous Epithelium. J. Androl. 1986, 7, 240–253. [Google Scholar] [CrossRef]
- O’Donnell, L.; McLachlan, R.I.; Wreford, N.G.; Robertson, D.M. Testosterone Promotes the Conversion of Round Spermatids between Stages VII and VIII of the Rat Spermatogenic Cycle. Endocrinology 1994, 135, 2608–2614. [Google Scholar] [CrossRef] [PubMed]
- Stanton, P.G.; Sluka, P.; Foo, C.F.H.; Stephens, A.N.; Smith, A.I.; McLachlan, R.I.; O’Donnell, L. Proteomic Changes in Rat Spermatogenesis in Response to in Vivo Androgen Manipulation; Impact on Meiotic Cells. PLoS ONE 2012, 7, e41718. [Google Scholar] [CrossRef] [PubMed]
- Meng, J.; Holdcraft, R.W.; Shima, J.E.; Griswold, M.D.; Braun, R.E. Androgens Regulate the Permeability of the Blood-Testis Barrier. Proc. Natl. Acad. Sci. USA 2005, 102, 16696–16700. [Google Scholar] [CrossRef] [PubMed]
- O’Donnell, L.; Pratis, K.; Wagenfeld, A.; Gottwald, U.; Müller, J.; Leder, G.; McLachlan, R.I.; Stanton, P.G. Transcriptional Profiling of the Hormone-Responsive Stages of Spermatogenesis Reveals Cell-, Stage-, and Hormone-Specific Events. Endocrinology 2009, 150, 5074–5084. [Google Scholar] [CrossRef]
- Guo, J.; Nie, X.; Giebler, M.; Mlcochova, H.; Wang, Y.; Grow, E.J.; DonorConnect; Kim, R.; Tharmalingam, M.; Matilionyte, G.; et al. The Dynamic Transcriptional Cell Atlas of Testis Development during Human Puberty. Cell Stem Cell 2020, 26, 262–276.e4. [Google Scholar] [CrossRef]
- Sriraman, V.; Anbalagan, M.; Rao, A.J. Hormonal Regulation of Leydig Cell Proliferation and Differentiation in Rodent Testis: A Dynamic Interplay between Gonadotrophins and Testicular Factors. Reprod. BioMed. Online 2005, 11, 507–518. [Google Scholar] [CrossRef]
- Teerds, K.J.; de Rooij, D.G.; Rommerts, F.F.G.; van den Hurk, R.; Wensing, C.J.G. Proliferation and Differentiation of Possible Leydig Cell Precursors after Destruction of the Existing Leydig Cells with Ethane Dimethyl Sulphonate: The Role of LH/Human Chorionic Gonadotrophin. J. Endocrinol. 1989, 122, 689-NP. [Google Scholar] [CrossRef]
- Lei, Z.M.; Mishra, S.; Zou, W.; Xu, B.; Foltz, M.; Li, X.; Rao, C.V. Targeted Disruption of Luteinizing Hormone/Human Chorionic Gonadotropin Receptor Gene. Mol. Endocrinol. 2001, 15, 184–200. [Google Scholar] [CrossRef]
- Verhagen, I.; Ramaswamy, S.; Teerds, K.J.; Keijer, J.; Plant, T.M. Time Course and Role of Luteinizing Hormone and Follicle-Stimulating Hormone in the Expansion of the Leydig Cell Population at the Time of Puberty in the Rhesus Monkey (Macaca mulatta). Andrology 2014, 2, 924–930. [Google Scholar] [CrossRef]
- Shiraishi, K.; Ascoli, M. Lutropin/Choriogonadotropin Stimulate the Proliferation of Primary Cultures of Rat Leydig Cells through a Pathway That Involves Activation of the Extracellularly Regulated Kinase 1/2 Cascade. Endocrinology 2007, 148, 3214–3225. [Google Scholar] [CrossRef]
- Anbalagan, M.; Rao, A.J. Collagen IV-Mediated Signalling Is Involved in Progenitor Leydig Cell Proliferation. Reprod. BioMed. Online 2004, 9, 391–403. [Google Scholar] [CrossRef] [PubMed]
- Umehara, T.; Kawashima, I.; Kawai, T.; Hoshino, Y.; Morohashi, K.; Shima, Y.; Zeng, W.; Richards, J.S.; Shimada, M. Neuregulin 1 Regulates Proliferation of Leydig Cells to Support Spermatogenesis and Sexual Behavior in Adult Mice. Endocrinology 2016, 157, 4899–4913. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Yan, H.; Yu, Y.; Zou, C.; Tian, L.; Xin, X.; Zhang, S.; Li, Z.; Ma, F.; Ge, R.-S. Bisphenol B Stimulates Leydig Cell Proliferation but Inhibits Maturation in Late Pubertal Rats. Food Chem. Toxicol. 2021, 153, 112248. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Wang, Z.; Jiang, Z.; Guo, J.; Zhang, Y.; Li, C.; Chung, J.; Folmer, J.; Liu, J.; Lian, Q.; et al. Regulation of Seminiferous Tubule-Associated Stem Leydig Cells in Adult Rat Testes. Proc. Natl. Acad. Sci. USA 2016, 113, 2666–2671. [Google Scholar] [CrossRef]
- Arora, H.; Qureshi, R.; Khodamoradi, K.; Seetharam, D.; Parmar, M.; Van Booven, D.J.; Issa, I.C.; Sackstein, R.; Lamb, D.; Hare, J.M.; et al. Leptin Secreted from Testicular Microenvironment Modulates Hedgehog Signaling to Augment the Endogenous Function of Leydig Cells. Cell Death Dis. 2022, 13, 208. [Google Scholar] [CrossRef]
- Li, X.; Zhu, Q.; Wen, Z.; Yuan, K.; Su, Z.; Wang, Y.; Zhong, Y.; Ge, R.-S. Androgen and Luteinizing Hormone Stimulate the Function of Rat Immature Leydig Cells Through Different Transcription Signals. Front. Endocrinol. 2021, 12, 599149. [Google Scholar] [CrossRef]
- Griffin, D.K.; Ellis, P.J.; Dunmore, B.; Bauer, J.; Abel, M.H.; Affara, N.A. Transcriptional Profiling of Luteinizing Hormone Receptor-Deficient Mice Before and after Testosterone Treatment Provides Insight into the Hormonal Control of Postnatal Testicular Development and Leydig Cell Differentiation1. Biol. Reprod. 2010, 82, 1139–1150. [Google Scholar] [CrossRef]
- Koronowski, K.B.; Sassone-Corsi, P. Communicating Clocks Shape Circadian Homeostasis. Science 2021, 371, eabd0951. [Google Scholar] [CrossRef]
- Marinkovic, D.Z.; Medar, M.L.J.; Becin, A.P.; Andric, S.A.; Kostic, T.S. Growing Up Under Constant Light: A Challenge to the Endocrine Function of the Leydig Cells. Front. Endocrinol. 2021, 12, 653602. [Google Scholar] [CrossRef]
- Alvarez, J.D.; Hansen, A.; Ord, T.; Bebas, P.; Chappell, P.E.; Giebultowicz, J.M.; Williams, C.; Moss, S.; Sehgal, A. The Circadian Clock Protein BMAL1 Is Necessary for Fertility and Proper Testosterone Production in Mice. J. Biol. Rhythm. 2008, 23, 26–36. [Google Scholar] [CrossRef]
- Li, C.; Zhang, L.; Ma, T.; Gao, L.; Yang, L.; Wu, M.; Pang, Z.; Wang, X.; Yao, Q.; Xiao, Y.; et al. Bisphenol A Attenuates Testosterone Production in Leydig Cells via the Inhibition of NR1D1 Signaling. Chemosphere 2021, 263, 128020. [Google Scholar] [CrossRef] [PubMed]
- Baburski, A.Z.; Sokanovic, S.J.; Bjelic, M.M.; Radovic, S.M.; Andric, S.A.; Kostic, T.S. Circadian Rhythm of the Leydig Cells Endocrine Function Is Attenuated during Aging. Exp. Gerontol. 2016, 73, 5–13. [Google Scholar] [CrossRef] [PubMed]
- Xiao, Y.; Zhao, L.; Li, W.; Wang, X.; Ma, T.; Yang, L.; Gao, L.; Li, C.; Zhang, M.; Yang, D.; et al. Circadian Clock Gene BMAL1 Controls Testosterone Production by Regulating Steroidogenesis-Related Gene Transcription in Goat Leydig Cells. J. Cell. Physiol. 2021, 236, 6706–6725. [Google Scholar] [CrossRef]
- Baburski, A.Z.; Sokanovic, S.J.; Andric, S.A.; Kostic, T.S. Aging Has the Opposite Effect on cAMP and cGMP Circadian Variations in Rat Leydig Cells. J. Comp. Physiol. B 2017, 187, 613–623. [Google Scholar] [CrossRef]
- Gao, L.; Gao, D.; Zhang, J.; Li, C.; Wu, M.; Xiao, Y.; Yang, L.; Ma, T.; Wang, X.; Zhang, M.; et al. Age-Related Endoplasmic Reticulum Stress Represses Testosterone Synthesis via Attenuation of the Circadian Clock in Leydig Cells. Theriogenology 2022, 189, 137–149. [Google Scholar] [CrossRef]
- He, M.; Liu, K.; Cao, J.; Chen, Q. An Update on the Role and Potential Mechanisms of Clock Genes Regulating Spermatogenesis: A Systematic Review of Human and Animal Experimental Studies. Rev. Endocr. Metab. Disord. 2023, 24, 585–610. [Google Scholar] [CrossRef]
- Dufau, M.L.; Winters, C.A.; Hattori, M.; Aquilano, D.; Barañao, J.L.S.; Nozu, K.; Baukal, A.; Catt, K.J. Hormonal Regulation of Androgen Production by the Leydig Cell. J. Steroid Biochem. 1984, 20, 161–173. [Google Scholar] [CrossRef]
- Manna, P.R.; Huhtaniemi, I.T.; Stocco, D.M. Mechanisms of Protein Kinase C Signaling in the Modulation of 3′,5′-Cyclic Adenosine Monophosphate-Mediated Steroidogenesis in Mouse Gonadal Cells. Endocrinology 2009, 150, 3308–3317. [Google Scholar] [CrossRef]
- Casarini, L.; Lispi, M.; Longobardi, S.; Milosa, F.; La Marca, A.; Tagliasacchi, D.; Pignatti, E.; Simoni, M. LH and hCG Action on the Same Receptor Results in Quantitatively and Qualitatively Different Intracellular Signalling. PLoS ONE 2012, 7, e46682. [Google Scholar] [CrossRef]
- Riccetti, L.; De Pascali, F.; Gilioli, L.; Potì, F.; Giva, L.B.; Marino, M.; Tagliavini, S.; Trenti, T.; Fanelli, F.; Mezzullo, M.; et al. Human LH and hCG Stimulate Differently the Early Signalling Pathways but Result in Equal Testosterone Synthesis in Mouse Leydig Cells in Vitro. Reprod. Biol. Endocrinol. 2017, 15, 2. [Google Scholar] [CrossRef]
- Clem, B.F.; Hudson, E.A.; Clark, B.J. Cyclic Adenosine 3′,5′-Monophosphate (cAMP) Enhances cAMP-Responsive Element Binding (CREB) Protein Phosphorylation and Phospho-CREB Interaction with the Mouse Steroidogenic Acute Regulatory Protein Gene Promoter. Endocrinology 2005, 146, 1348–1356. [Google Scholar] [CrossRef] [PubMed]
- Manna, P.R.; Dyson, M.T.; Eubank, D.W.; Clark, B.J.; Lalli, E.; Sassone-Corsi, P.; Zeleznik, A.J.; Stocco, D.M. Regulation of Steroidogenesis and the Steroidogenic Acute Regulatory Protein by a Member of the cAMP Response-Element Binding Protein Family. Mol. Endocrinol. 2002, 16, 184–199. [Google Scholar] [CrossRef] [PubMed]
- Manna, P.R.; Dyson, M.T.; Stocco, D.M. Regulation of the Steroidogenic Acute Regulatory Protein Gene Expression: Present and Future Perspectives. Mol. Hum. Reprod. 2009, 15, 321–333. [Google Scholar] [CrossRef]
- Manna, P.R.; Stocco, D.M. Crosstalk of CREB and Fos/Jun on a Single Cis-Element: Transcriptional Repression of the Steroidogenic Acute Regulatory Protein Gene. J. Mol. Endocrinol. 2007, 39, 261–277. [Google Scholar] [CrossRef]
- Duan, H.; Jefcoate, C.R. The Predominant cAMP-Stimulated 3.5 Kb StAR mRNA Contains Specific Sequence Elements in the Extended 3′UTR That Confer High Basal Instability. J. Mol. Endocrinol. 2007, 38, 159–179. [Google Scholar] [CrossRef]
- De Mattos, K.; Viger, R.S.; Tremblay, J.J. Transcription Factors in the Regulation of Leydig Cell Gene Expression and Function. Front. Endocrinol. 2022, 13, 881309. [Google Scholar] [CrossRef]
- Xu, B.; Yang, W.-H.; Gerin, I.; Hu, C.-D.; Hammer, G.D.; Koenig, R.J. Dax-1 and Steroid Receptor RNA Activator (SRA) Function as Transcriptional Coactivators for Steroidogenic Factor 1 in Steroidogenesis. Mol. Cell. Biol. 2009, 29, 1719–1734. [Google Scholar] [CrossRef]
- Dyson, M.T.; Jones, J.K.; Kowalewski, M.P.; Manna, P.R.; Alonso, M.; Gottesman, M.E.; Stocco, D.M. Mitochondrial A-Kinase Anchoring Protein 121 Binds Type II Protein Kinase A and Enhances Steroidogenic Acute Regulatory Protein-Mediated Steroidogenesis in MA-10 Mouse Leydig Tumor Cells1. Biol. Reprod. 2008, 78, 267–277. [Google Scholar] [CrossRef]
- Hirakawa, T.; Ascoli, M. The Lutropin/Choriogonadotropin Receptor-Induced Phosphorylation of the Extracellular Signal-Regulated Kinases in Leydig Cells Is Mediated by a Protein Kinase A-Dependent Activation of Ras. Mol. Endocrinol. 2003, 17, 2189–2200. [Google Scholar] [CrossRef]
- Shiraishi, K.; Ascoli, M. Activation of the Lutropin/Choriogonadotropin Receptor in MA-10 Cells Stimulates Tyrosine Kinase Cascades That Activate Ras and the Extracellular Signal Regulated Kinases (ERK1/2). Endocrinology 2006, 147, 3419–3427. [Google Scholar] [CrossRef]
- Shiraishi, K.; Ascoli, M. A Co-Culture System Reveals the Involvement of Intercellular Pathways as Mediators of the Lutropin Receptor (LHR)-Stimulated ERK1/2 Phosphorylation in Leydig Cells. Exp. Cell Res. 2008, 314, 25–37. [Google Scholar] [CrossRef] [PubMed]
- Johnson, G.P.; Jonas, K.C. Mechanistic Insight into How Gonadotropin Hormone Receptor Complexes Direct Signaling†. Biol. Reprod. 2020, 102, 773–783. [Google Scholar] [CrossRef] [PubMed]
- Jamnongjit, M.; Gill, A.; Hammes, S.R. Epidermal Growth Factor Receptor Signaling Is Required for Normal Ovarian Steroidogenesis and Oocyte Maturation. Proc. Natl. Acad. Sci. USA 2005, 102, 16257–16262. [Google Scholar] [CrossRef]
- Evaul, K.; Hammes, S.R. Cross-Talk between G Protein-Coupled and Epidermal Growth Factor Receptors Regulates Gonadotropin-Mediated Steroidogenesis in Leydig Cells. J. Biol. Chem. 2008, 283, 27525–27533. [Google Scholar] [CrossRef]
- Martinat, N.; Crépieux, P.; Reiter, E.; Guillou, F. Extracellular Signal-Regulated Kinases (ERK) 1, 2 Are Required for Luteinizing Hormone (LH)-Induced Steroidogenesis in Primary Leydig Cells and Control Steroidogenic Acute Regulatory (StAR) Expression. Reprod. Nutr. Dev. 2005, 45, 101–108. [Google Scholar] [CrossRef]
- Matzkin, M.E.; Yamashita, S.; Ascoli, M. The ERK1/2 Pathway Regulates Testosterone Synthesis by Coordinately Regulating the Expression of Steroidogenic Genes in Leydig Cells. Mol. Cell. Endocrinol. 2013, 370, 130–137. [Google Scholar] [CrossRef]
- de Mattos, K.; Dumas, F.-O.; Campolina-Silva, G.H.; Belleannée, C.; Viger, R.S.; Tremblay, J.J. ERK5 Cooperates With MEF2C to Regulate Nr4a1 Transcription in MA-10 and MLTC-1 Leydig Cells. Endocrinology 2023, 164, bqad120. [Google Scholar] [CrossRef]
- Luan, S.; Wang, C. Calcium Signaling Mechanisms Across Kingdoms. Annu. Rev. Cell Dev. Biol. 2021, 37, 311–340. [Google Scholar] [CrossRef]
- Costa, R.R.; Varanda, W.A. Intracellular Calcium Changes in Mice Leydig Cells Are Dependent on Calcium Entry through T-Type Calcium Channels: T-Type Calcium Channels in Leydig Cells. J. Physiol. 2007, 585, 339–349. [Google Scholar] [CrossRef]
- Costa, R.R.; Varanda, W.A.; Franci, C.R. A Calcium-Induced Calcium Release Mechanism Supports Luteinizing Hormone-Induced Testosterone Secretion in Mouse Leydig Cells. Am. J. Physiol.-Cell Physiol. 2010, 299, C316–C323. [Google Scholar] [CrossRef]
- Sullivan, M.H.F.; Cooke, B.A. The Role of Ca2+ in Steroidogenesis in Leydig Cells. Stimulation of Intracellular Free Ca2+ by Lutropin (LH), Luliberin (LHRH) Agonist and Cyclic AMP. Biochem. J. 1986, 236, 45–51. [Google Scholar] [CrossRef] [PubMed]
- Costa, R.R.; Dos Reis, R.I.; Aguiar, J.F.; Varanda, W.A. Luteinizing Hormone (LH) Acts through PKA and PKC to Modulate T-Type Calcium Currents and Intracellular Calcium Transients in Mice Leydig Cells. Cell Calcium 2011, 49, 191–199. [Google Scholar] [CrossRef] [PubMed]
- Martin, L.J.; Boucher, N.; Brousseau, C.; Tremblay, J.J. The Orphan Nuclear Receptor NUR77 Regulates Hormone-Induced StAR Transcription in Leydig Cells through Cooperation with Ca2+/Calmodulin-Dependent Protein Kinase I. Mol. Endocrinol. 2008, 22, 2021–2037. [Google Scholar] [CrossRef]
- Abdou, H.S.; Villeneuve, G.; Tremblay, J.J. The Calcium Signaling Pathway Regulates Leydig Cell Steroidogenesis through a Transcriptional Cascade Involving the Nuclear Receptor NR4A1 and the Steroidogenic Acute Regulatory Protein. Endocrinology 2013, 154, 511–520. [Google Scholar] [CrossRef]
- De Mattos, K.; Pierre, K.J.; Tremblay, J.J. Hormones and Signaling Pathways Involved in the Stimulation of Leydig Cell Steroidogenesis. Endocrines 2023, 4, 573–594. [Google Scholar] [CrossRef]
- Abdou, H.S.; Robert, N.M.; Tremblay, J.J. Calcium-Dependent Nr4a1 Expression in Mouse Leydig Cells Requires Distinct AP1/CRE and MEF2 Elements. J. Mol. Endocrinol. 2016, 56, 151–161. [Google Scholar] [CrossRef]
- Pandey, A.K.; Li, W.; Yin, X.; Stocco, D.M.; Grammas, P.; Wang, X. Blocking L-Type Calcium Channels Reduced the Threshold of cAMP-Induced Steroidogenic Acute Regulatory Gene Expression in MA-10 Mouse Leydig Cells. J. Endocrinol. 2010, 204, 67–74. [Google Scholar] [CrossRef]
- Matzkin, M.E.; Lauf, S.; Spinnler, K.; Rossi, S.P.; Köhn, F.M.; Kunz, L.; Calandra, R.S.; Frungieri, M.B.; Mayerhofer, A. The Ca2+-Activated, Large Conductance K+-Channel (BKCa) Is a Player in the LH/hCG Signaling Cascade in Testicular Leydig Cells. Mol. Cell. Endocrinol. 2013, 367, 41–49. [Google Scholar] [CrossRef]
- Bi, X.; Liu, J.; Xu, S.; Wang, Y.; Wu, X. Testicular STAC3 Regulates Leydig Cell Steroidogenesis through Potentiating Mitochondrial Membrane Potential and StAR Processing. Cell Tissue Res. 2021, 384, 195–209. [Google Scholar] [CrossRef]
- Ramnath, H.I.; Peterson, S.; Michael, A.E.; Stocco, D.M.; Cooke, B.A. Modulation of Steroidogenesis by Chloride Ions in MA-10 Mouse Tumor Leydig Cells: Roles of Calcium, Protein Synthesis, and the Steroidogenic Acute Regulatory Protein. Endocrinology 1997, 138, 2308–2314. [Google Scholar] [CrossRef]
- Cooke, B.A.; Ashford, L.; Abayasekara, D.R.E.; Choi, M. The Role of Chloride Ions in the Regulation of Steroidogenesis in Rat Leydig Cells and Adrenal Cells p. J. Steroid Biochem. Mol. Biol. 1999, 69, 359–365. [Google Scholar] [CrossRef] [PubMed]
- Gambaryan, S. The Role of NO/sGC/cGMP/PKG Signaling Pathway in Regulation of Platelet Function. Cells 2022, 11, 3704. [Google Scholar] [CrossRef] [PubMed]
- Valenti, S.; Cuttica, C.M.; Fazzuoli, L.; Giordano, G.; Giusti, M. Biphasic Effect of Nitric Oxide on Testosterone and Cyclic GMP Production by Purified Rat Leydig Cells Cultured in Vitro. Int. J. Androl. 1999, 22, 336–341. [Google Scholar] [CrossRef] [PubMed]
- Andric, S.A.; Janjic, M.M.; Stojkov, N.J.; Kostic, T.S. Protein Kinase G-Mediated Stimulation of Basal Leydig Cell Steroidogenesis. Am. J. Physiol.-Endocrinol. Metab. 2007, 293, E1399–E1408. [Google Scholar] [CrossRef]
- Sokanovic, S.J.; Baburski, A.Z.; Kojic, Z.; Medar, M.L.J.; Andric, S.A.; Kostic, T.S. Aging-Related Increase of cGMP Disrupts Mitochondrial Homeostasis in Leydig Cells. J. Gerontol. Ser. A 2021, 76, 177–186. [Google Scholar] [CrossRef]
- Andric, S.A.; Janjic, M.M.; Stojkov, N.J.; Kostic, T.S. Testosterone-Induced Modulation of Nitric Oxide-cGMP Signaling Pathway and Androgenesis in the Rat Leydig Cells1. Biol. Reprod. 2010, 83, 434–442. [Google Scholar] [CrossRef]
- Vasta, V.; Shimizu-Albergine, M.; Beavo, J.A. Modulation of Leydig Cell Function by Cyclic Nucleotide Phosphodiesterase 8A. Proc. Natl. Acad. Sci. USA 2006, 103, 19925–19930. [Google Scholar] [CrossRef]
- Shimizu-Albergine, M.; Tsai, L.-C.L.; Patrucco, E.; Beavo, J.A. cAMP-Specific Phosphodiesterases 8A and 8B, Essential Regulators of Leydig Cell Steroidogenesis. Mol. Pharmacol. 2012, 81, 556–566. [Google Scholar] [CrossRef]
- Tartarin, P.; Guibert, E.; Touré, A.; Ouiste, C.; Leclerc, J.; Sanz, N.; Brière, S.; Dacheux, J.-L.; Delaleu, B.; McNeilly, J.R.; et al. Inactivation of AMPKα1 Induces Asthenozoospermia and Alters Spermatozoa Morphology. Endocrinology 2012, 153, 3468–3481. [Google Scholar] [CrossRef]
- Abdou, H.S.; Bergeron, F.; Tremblay, J.J. A Cell-Autonomous Molecular Cascade Initiated by AMP-Activated Protein Kinase Represses Steroidogenesis. Mol. Cell. Biol. 2014, 34, 4257–4271. [Google Scholar] [CrossRef]
- Demmouche, Z.B.; Tremblay, J.J. Comprehensive and Quantitative Analysis of the Changes in Proteomic and Phosphoproteomic Profiles during Stimulation and Repression of Steroidogenesis in MA-10 Leydig Cells. Int. J. Mol. Sci. 2022, 23, 12846. [Google Scholar] [CrossRef] [PubMed]
- Walker, W.H.; Girardet, C.; Habener, J.F. Alternative Exon Splicing Controls a Translational Switch from Activator to Repressor Isoforms of Transcription Factor CREB during Spermatogenesis. J. Biol. Chem. 1996, 271, 20145–20150. [Google Scholar] [CrossRef] [PubMed]
- Blockus, H.; Chédotal, A. Slit-Robo Signaling. Development 2016, 143, 3037–3044. [Google Scholar] [CrossRef] [PubMed]
- Martinot, E.; Boerboom, D. Slit/Robo Signaling Regulates Leydig Cell Steroidogenesis. Cell Commun. Signal. 2021, 19, 8. [Google Scholar] [CrossRef]
- Butler, G.; Srirangalingam, U.; Faithfull, J.; Sangster, P.; Senniappan, S.; Mitchell, R. Klinefelter Syndrome: Going beyond the Diagnosis. Arch. Dis. Child. 2023, 108, 166–171. [Google Scholar] [CrossRef]
- Ulloa-Aguirre, A.; Lira-Albarrán, S. Clinical Applications of Gonadotropins in the Male. Prog. Mol. Biol. Transl. Sci. 2016, 143, 121–174. [Google Scholar] [CrossRef]
- Szeliga, A.; Kunicki, M.; Maciejewska-Jeske, M.; Rzewuska, N.; Kostrzak, A.; Meczekalski, B.; Bala, G.; Smolarczyk, R.; Adashi, E.Y. The Genetic Backdrop of Hypogonadotropic Hypogonadism. Int. J. Mol. Sci. 2021, 22, 13241. [Google Scholar] [CrossRef]
- Stamou, M.I.; Georgopoulos, N.A. Kallmann Syndrome: Phenotype and Genotype of Hypogonadotropic Hypogonadism. Metabolism 2018, 86, 124–134. [Google Scholar] [CrossRef]
- Casarini, L.; Santi, D.; Brigante, G.; Simoni, M. Two Hormones for One Receptor: Evolution, Biochemistry, Actions, and Pathophysiology of LH and hCG. Endocr. Rev. 2018, 39, 549–592. [Google Scholar] [CrossRef]
- Pitteloud, N.; Dwyer, A. Hormonal Control of Spermatogenesis in Men: Therapeutic Aspects in Hypogonadotropic Hypogonadism. Ann. D’endocrinologie 2014, 75, 98–100. [Google Scholar] [CrossRef]
- Main, K.M.; Schmidt, I.M.; Toppari, J.; Skakkebaek, N.E. Early Postnatal Treatment of Hypogonadotropic Hypogonadism with Recombinant Human FSH and LH. Eur. J. Endocrinol. 2002, 146, 75–79. [Google Scholar] [CrossRef] [PubMed]
- Bougnères, P.; François, M.; Pantalone, L.; Rodrigue, D.; Bouvattier, C.; Demesteere, E.; Roger, D.; Lahlou, N. Effects of an Early Postnatal Treatment of Hypogonadotropic Hypogonadism with a Continuous Subcutaneous Infusion of Recombinant Follicle-Stimulating Hormone and Luteinizing Hormone. J. Clin. Endocrinol. Metab. 2008, 93, 2202–2205. [Google Scholar] [CrossRef] [PubMed]
- Avril, T.; Hennocq, Q.; Lambert, A.-S.; Leger, J.; Simon, D.; Martinerie, L.; Bouvattier, C. Gonadotropin Administration to Mimic Mini-Puberty in Hypogonadotropic Males: Pump or Injections? Endocr. Connect. 2023, 12, e220252. [Google Scholar] [CrossRef] [PubMed]
- Rohayem, J.; Hauffa, B.P.; Zacharin, M.; Kliesch, S.; Zitzmann, M.; the “German Adolescent Hypogonadotropic Hypogonadism Study Group”. Testicular Growth and Spermatogenesis: New Goals for Pubertal Hormone Replacement in Boys with Hypogonadotropic Hypogonadism?—A Multicentre Prospective Study of hCG/rFSH Treatment Outcomes during Adolescence. Clin. Endocrinol. 2017, 86, 75–87. [Google Scholar] [CrossRef]
- Young, J.; Xu, C.; Papadakis, G.E.; Acierno, J.S.; Maione, L.; Hietamäki, J.; Raivio, T.; Pitteloud, N. Clinical Management of Congenital Hypogonadotropic Hypogonadism. Endocr. Rev. 2019, 40, 669–710. [Google Scholar] [CrossRef]
- Han, T.S.; Bouloux, P.M.G. What Is the Optimal Therapy for Young Males with Hypogonadotropic Hypogonadism? Clin. Endocrinol. 2009, 72, 731–737. [Google Scholar] [CrossRef]
- Park, S.-J.; Kim, T.-S.; Park, C.-K.; Lee, S.-H.; Kim, J.-M.; Lee, K.-S.; Lee, I.; Park, J.-W.; Lawson, M.A.; Lee, D.-S. hCG-Induced Endoplasmic Reticulum Stress Triggers Apoptosis and Reduces Steroidogenic Enzyme Expression through Activating Transcription Factor 6 in Leydig Cells of the Testis. J. Mol. Endocrinol. 2013, 50, 151–166. [Google Scholar] [CrossRef]
- Delli Paoli, E.; Di Chiano, S.; Paoli, D.; Lenzi, A.; Lombardo, F.; Pallotti, F. Androgen Insensitivity Syndrome: A Review. J. Endocrinol. Investig. 2023, 46, 2237–2245. [Google Scholar] [CrossRef]
- Batista, R.L.; Costa, E.M.F.; Rodrigues, A.D.S.; Gomes, N.L.; Faria, J.A.; Nishi, M.Y.; Arnhold, I.J.P.; Domenice, S.; Mendonca, B.B.D. Androgen Insensitivity Syndrome: A Review. Arch. Endocrinol. Metab. 2018, 62, 227–235. [Google Scholar] [CrossRef]
- Liu, Q.; Yin, X.; Li, P. Clinical, Hormonal and Genetic Characteristics of Androgen Insensitivity Syndrome in 39 Chinese Patients. Reprod. Biol. Endocrinol. 2020, 18, 34. [Google Scholar] [CrossRef]
- Banerjee, S.; Bajpai, A. Precocious Puberty. Indian J. Pediatr. 2023, 90, 582–589. [Google Scholar] [CrossRef] [PubMed]
- Fudge, E. Precocious Puberty. In Endocrinology and Diabetes; Bandeira, F., Gharib, H., Golbert, A., Griz, L., Faria, M., Eds.; Springer: New York, NY, USA, 2014; pp. 219–233. ISBN 978-1-4614-8683-1. [Google Scholar]
- Latronico, A.C.; Brito, V.N.; Carel, J.-C. Causes, Diagnosis, and Treatment of Central Precocious Puberty. Lancet Diabetes Endocrinol. 2016, 4, 265–274. [Google Scholar] [CrossRef] [PubMed]
- Cheuiche, A.V.; Da Silveira, L.G.; De Paula, L.C.P.; Lucena, I.R.S.; Silveiro, S.P. Diagnosis and Management of Precocious Sexual Maturation: An Updated Review. Eur. J. Pediatr. 2021, 180, 3073–3087. [Google Scholar] [CrossRef]
- Yüce, Ö.; Bideci, A.; Çelik, N.; Çamurdan, O.; Cinaz, P. Diagnostic Value of Urinary Luteinizing Hormone Levels in the Monitoring of Precocious Puberty Treatment. Arch. Endocrinol. Metab. 2020, 64, 121–127. [Google Scholar] [CrossRef]
- Roldan, E.R.S. Assessments of Sperm Quality Integrating Morphology, Swimming Patterns, Bioenergetics and Cell Signalling. Theriogenology 2020, 150, 388–395. [Google Scholar] [CrossRef]
- Zhao, W.; Jing, J.; Shao, Y.; Zeng, R.; Wang, C.; Yao, B.; Hang, D. Circulating Sex Hormone Levels in Relation to Male Sperm Quality. BMC Urol. 2020, 20, 101. [Google Scholar] [CrossRef]
- Sun, L.-P.; Du, Q.-Z.; Song, Y.-P.; Yu, J.-N.; Wang, S.-J.; Sang, L.; Song, L.-W.; Yue, Y.-M.; Lian, Y.-Z.; Zhang, S.-L.; et al. Polymorphisms in Luteinizing Hormone Receptor and Hypothalamic Gonadotropin-Releasing Hormone Genes and Their Effects on Sperm Quality Traits in Chinese Holstein Bulls. Mol. Biol. Rep. 2012, 39, 7117–7123. [Google Scholar] [CrossRef]
- Pakarainen, T.; Zhang, F.-P.; Mäkelä, S.; Poutanen, M.; Huhtaniemi, I. Testosterone Replacement Therapy Induces Spermatogenesis and Partially Restores Fertility in Luteinizing Hormone Receptor Knockout Mice. Endocrinology 2005, 146, 596–606. [Google Scholar] [CrossRef]
- Sun, L.P.; Song, Y.P.; Liu, J.J.; Liu, X.R.; Guo, A.Z.; Yang, L.G. Differential Expression of Luteinizing Hormone Receptor, Androgen Receptor and Heat-Shock Protein 70 in the Testis of Long-Distance Transported Mice. Genet. Mol. Res. 2015, 14, 9985–9993. [Google Scholar] [CrossRef]
- Sherman, B.M.; Korenman, S.G. Hormonal Characteristics of the Human Menstrual Cycle throughout Reproductive Life. J. Clin. Investig. 1975, 55, 699–706. [Google Scholar] [CrossRef]
- Choi, Y.S.; Song, J.E.; Kong, B.S.; Hong, J.W.; Novelli, S.; Lee, E.J. The Role of Foxo3 in Leydig Cells. Yonsei Med. J. 2015, 56, 1590–1596. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
| Role | Signaling Pathway | Targets | Species/Cell Lines | References |
|---|---|---|---|---|
| Testosterone production | The ERK pathway | StAR, Hsd3b6, Cyp17a1, and Hsd17b3 | Rat | [100,101,102,106] |
| The JNK pathway | c-Jun | Rat | [95] | |
| The ERK5 pathway | StAR and Nr4a1 | MA-10 LCs | [108] | |
| The calcium signaling pathway | STAC3, CaMK, Dax-1 | MA-10 LCs, hamster | [110,111,118,119] | |
| The cGMP-PKG pathway | StAR | Mouse, rat | [124,125,126,127] | |
| The PI3k/AKT pathway | FOXO3 | LC lines R2C | [163] | |
| The AMPK signaling pathway | 3βHSD, CYP17, and STAR | Mouse, MA-10 LCs | [128,129,130,131,132,133] | |
| The Slit/Robo pathway | CREB, AKT | Mouse | [134,135] | |
| LCs proliferation | The ERK pathway | PCNA, D-cyclins D3 | Mouse, rat | [71,72,73,74] |
| The PI3k/AKT pathway | Mouse, rat | [74] | ||
| LCs differentiation | Unknown | Scarb1, Cyp11a1, Cyp17a1, and Srd5a1, Insl3 | Mouse, rat | [68,69,77,78] |
| The circadian clock of LCs | Unknown | Per1/PER1, Dec1/2 and Rorb et al. | Rat | [18] |
| Sperm motility | Unknown | Unknown | Human, Holstein bulls | [19,159] |
| sperm concentration | Unknown | Unknown | Human, Holstein bulls, rat | [19,159] |
| Sperm morphology | Unknown | Unknown | Rat | [160] |
| Sperm capacity | The cAMP/PKA pathway | Unknown | Rat, human | [20,21,162] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lei, T.; Yang, Y.; Yang, W.-X. Luteinizing Hormone Regulates Testosterone Production, Leydig Cell Proliferation, Differentiation, and Circadian Rhythm During Spermatogenesis. Int. J. Mol. Sci. 2025, 26, 3548. https://doi.org/10.3390/ijms26083548
Lei T, Yang Y, Yang W-X. Luteinizing Hormone Regulates Testosterone Production, Leydig Cell Proliferation, Differentiation, and Circadian Rhythm During Spermatogenesis. International Journal of Molecular Sciences. 2025; 26(8):3548. https://doi.org/10.3390/ijms26083548
Chicago/Turabian StyleLei, Tian, Yu Yang, and Wan-Xi Yang. 2025. "Luteinizing Hormone Regulates Testosterone Production, Leydig Cell Proliferation, Differentiation, and Circadian Rhythm During Spermatogenesis" International Journal of Molecular Sciences 26, no. 8: 3548. https://doi.org/10.3390/ijms26083548
APA StyleLei, T., Yang, Y., & Yang, W.-X. (2025). Luteinizing Hormone Regulates Testosterone Production, Leydig Cell Proliferation, Differentiation, and Circadian Rhythm During Spermatogenesis. International Journal of Molecular Sciences, 26(8), 3548. https://doi.org/10.3390/ijms26083548