
Activation of Small Conductance Ca2+-Activated K+ Channels Suppresses Electrical and Calcium Alternans in Atrial Myocytes


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
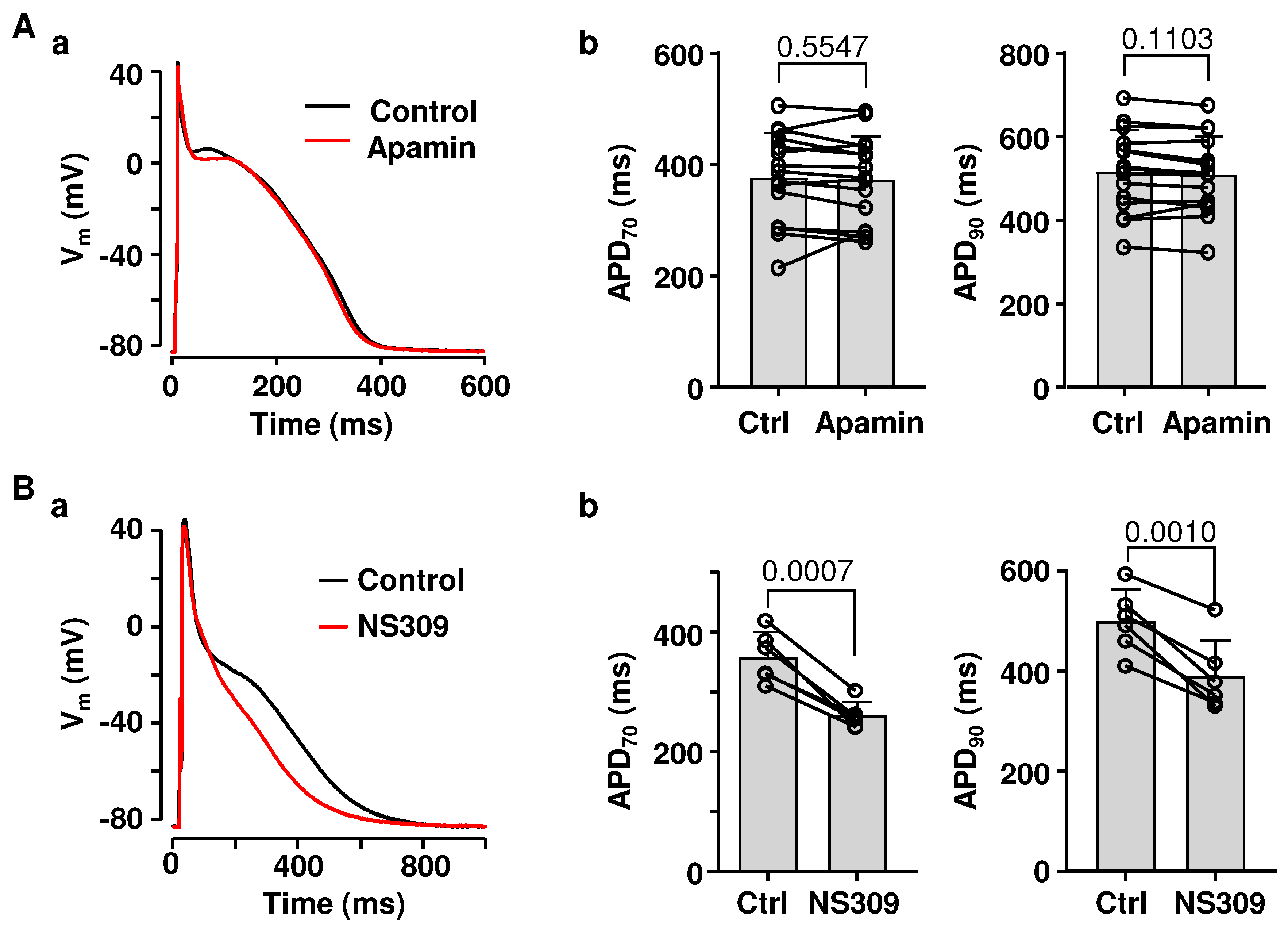
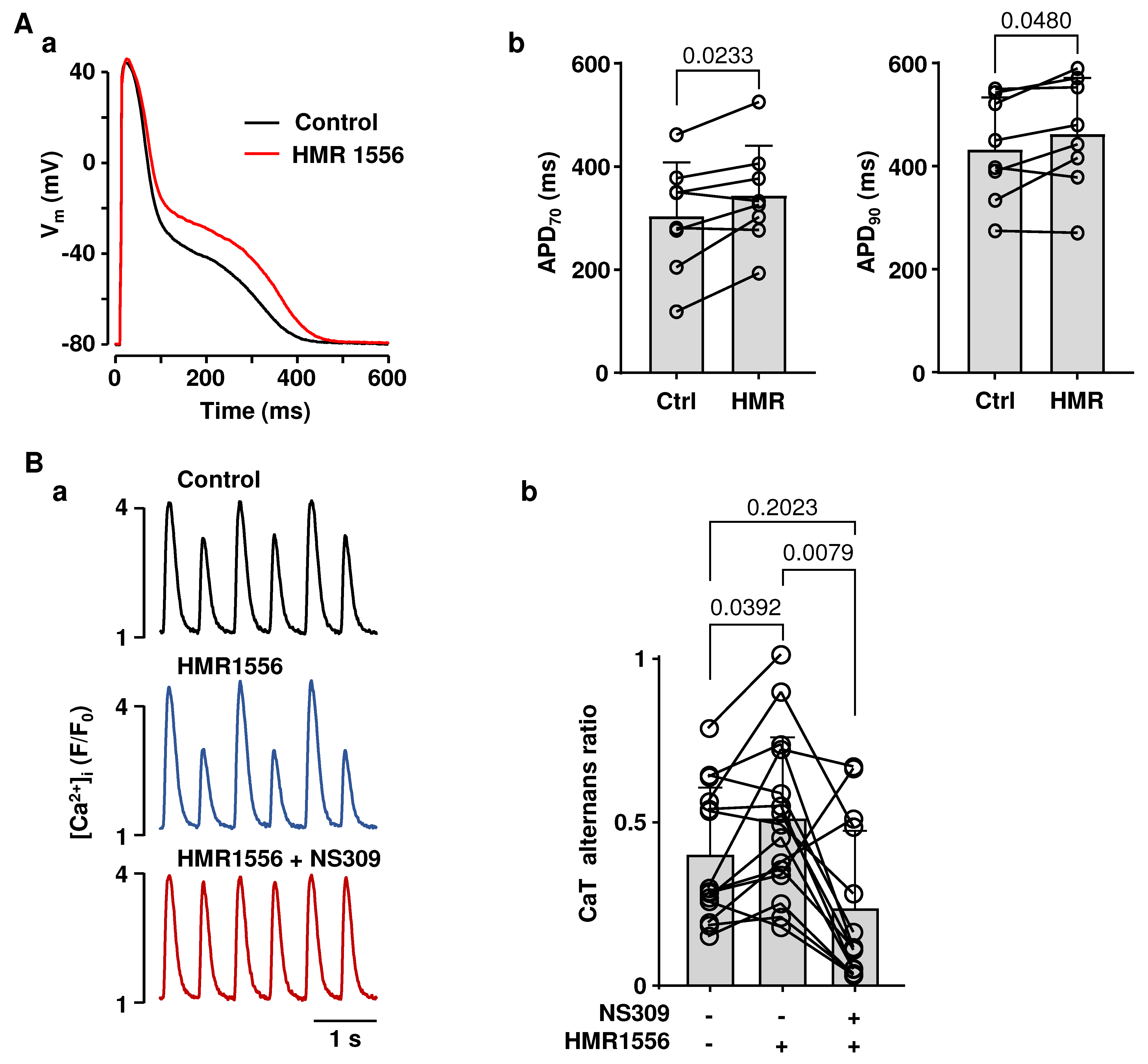
2.1. Effects of SK Channel Inhibition and Activation on Atrial AP Repolarization
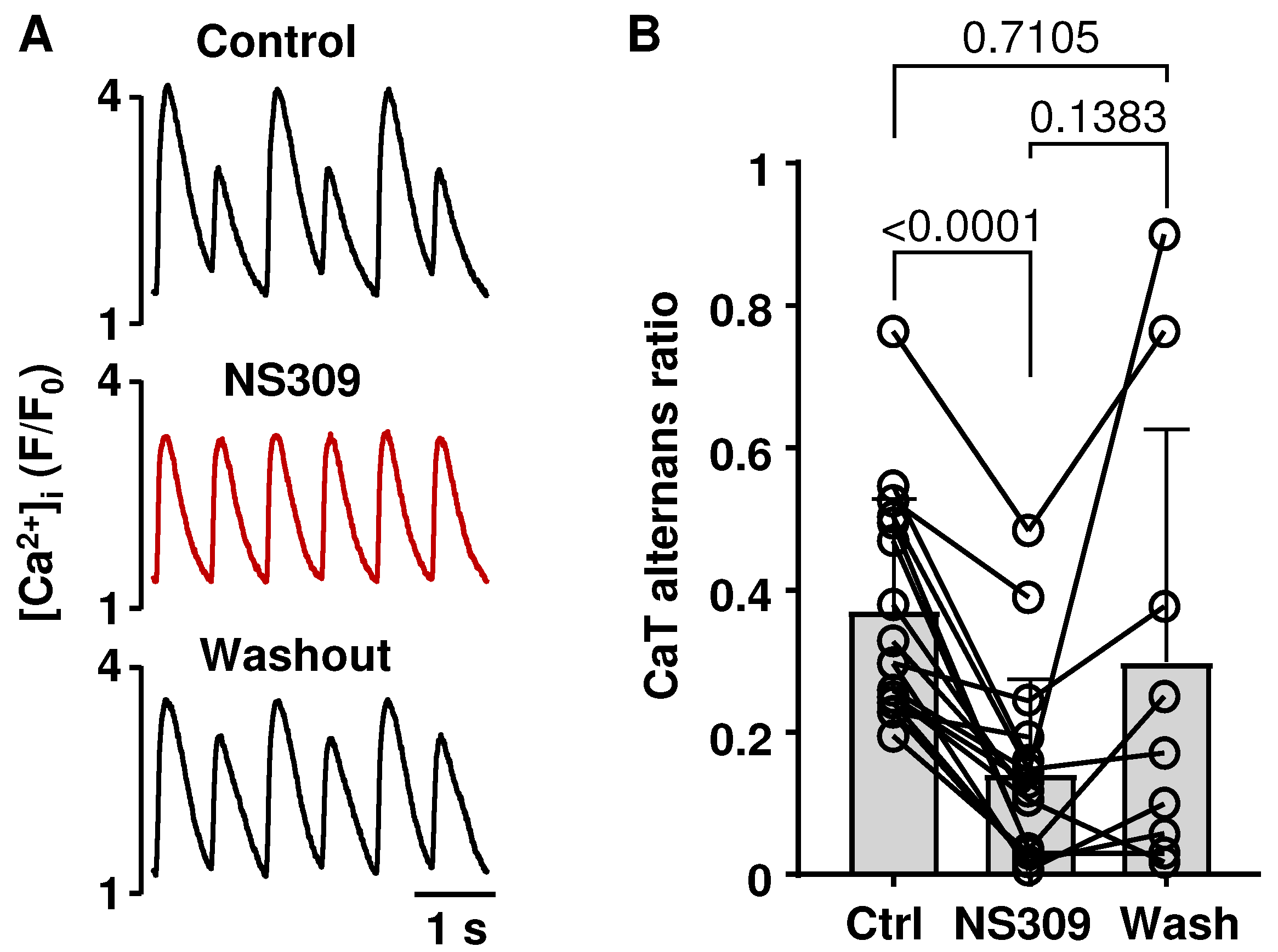
2.2. Activation of SK Channels Suppresses CaT and APD Alternans
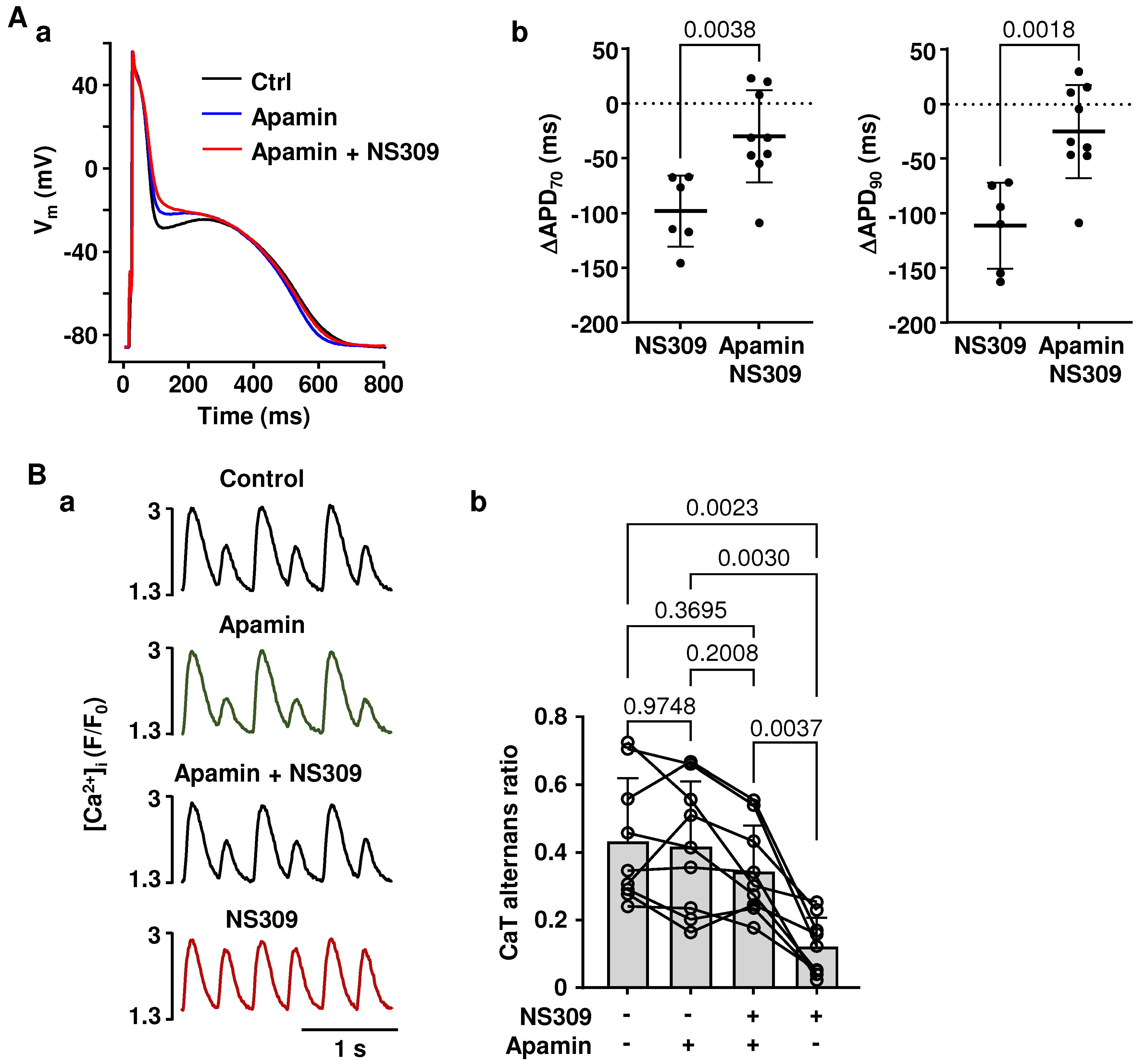
2.3. Effects of NS309 on CaT Alternans and APD Are Induced by SK Channel Activation
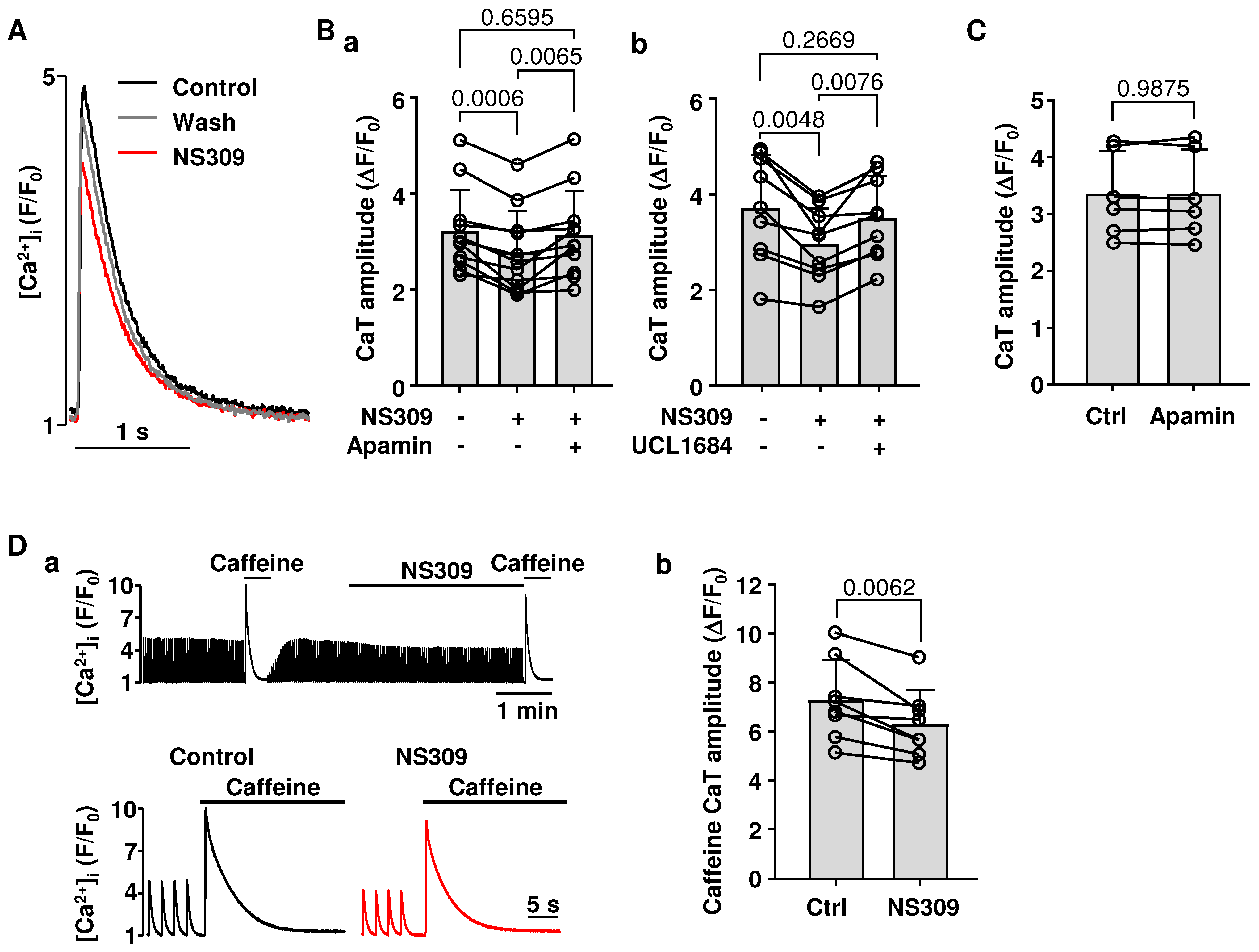
2.4. Effect of SK Channel Activation on CaT Properties
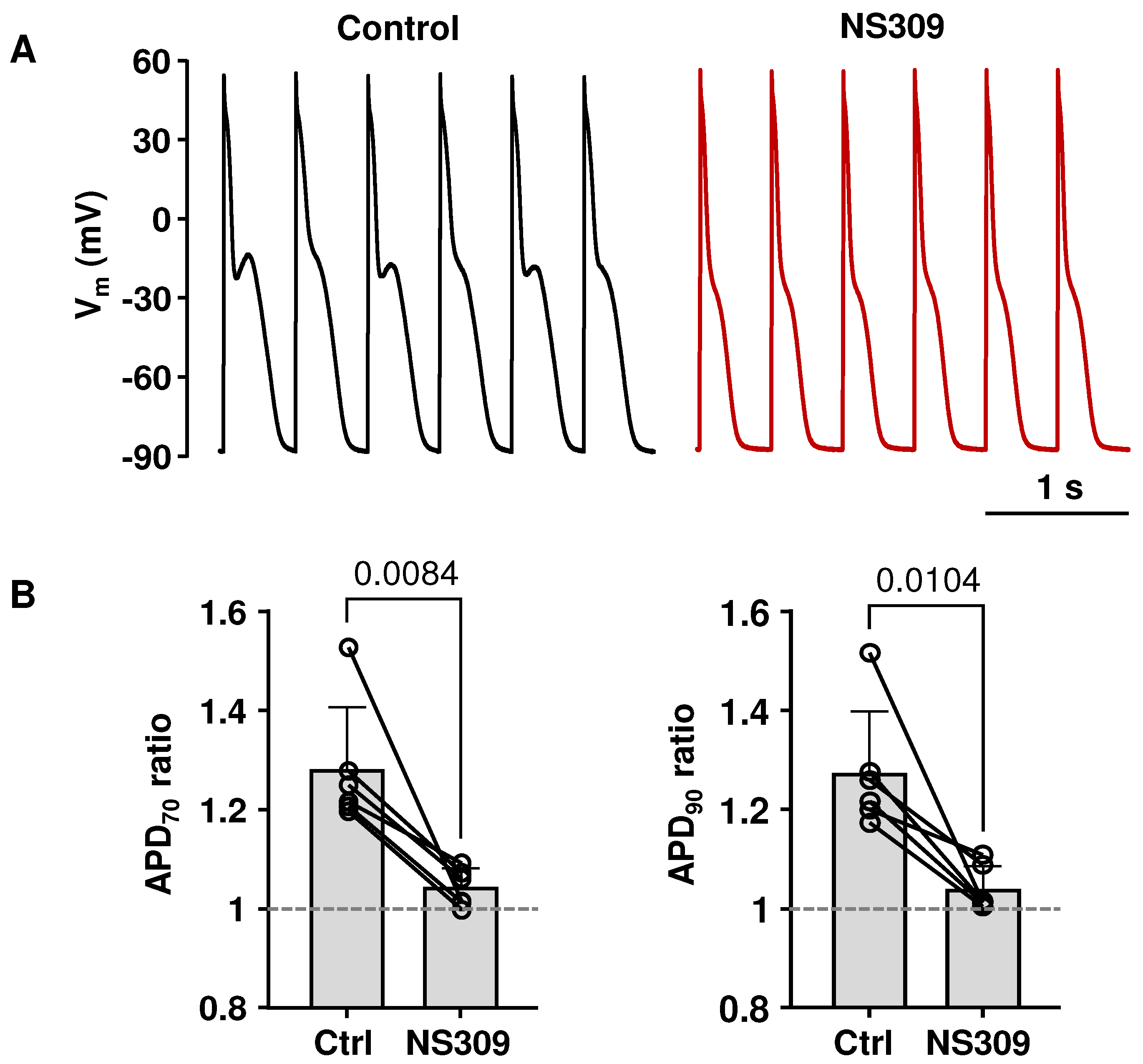
2.5. APD Prolongation Increased the Risk for CaT Alternans, Which Can Be Attenuated by SK Channel Activation
3. Discussion
3.1. SK Channel Activity in Atrial Myocytes
3.2. Cardiac Alternans, SK Channels and Atrial Arrhythmias
3.3. Atrial Alternans in LQT Syndrome
3.4. Limitations and Future Perspectives
4. Materials and Methods
4.1. Ethical Approval
4.2. Myocyte Isolation
4.3. Patch Clamp Experiments
4.4. Ca2+ Measurements
4.5. CaT Alternans
4.6. Drugs
4.7. Data Analysis and Presentation
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations and Acronyms
References
- Xu, Y.; Tuteja, D.; Zhang, Z.; Xu, D.; Zhang, Y.; Rodriguez, J.; Nie, L.; Tuxson, H.R.; Young, J.N.; Glatter, K.A.; et al. Molecular identification and functional roles of a Ca2+-activated K+ channel in human and mouse hearts. J. Biol. Chem. 2003, 278, 49085–49094. [Google Scholar] [CrossRef] [PubMed]
- Tuteja, D.; Xu, D.; Timofeyev, V.; Lu, L.; Sharma, D.; Zhang, Z.; Xu, Y.; Nie, L.; Vazquez, A.E.; Young, J.N.; et al. Differential expression of small-conductance Ca2+-activated K+ channels SK1, SK2, and SK3 in mouse atrial and ventricular myocytes. Am. J. Physiol. Heart Circ. Physiol. 2005, 289, H2714–H2723. [Google Scholar] [CrossRef]
- Skibsbye, L.; Poulet, C.; Diness, J.G.; Bentzen, B.H.; Yuan, L.; Kappert, U.; Matschke, K.; Wettwer, E.; Ravens, U.; Grunnet, M.; et al. Small-conductance calcium-activated potassium (SK) channels contribute to action potential repolarization in human atria. Cardiovasc. Res. 2014, 103, 156–167. [Google Scholar] [CrossRef]
- Zhang, X.D.; Timofeyev, V.; Li, N.; Myers, R.E.; Zhang, D.M.; Singapuri, A.; Lau, V.C.; Bond, C.T.; Adelman, J.; Lieu, D.K.; et al. Critical roles of a small conductance Ca2+-activated K+ channel (SK3) in the repolarization process of atrial myocytes. Cardiovasc. Res. 2014, 101, 317–325. [Google Scholar] [CrossRef] [PubMed]
- Haugaard, M.M.; Hesselkilde, E.Z.; Pehrson, S.; Carstensen, H.; Flethoj, M.; Praestegaard, K.F.; Sorensen, U.S.; Diness, J.G.; Grunnet, M.; Buhl, R.; et al. Pharmacologic inhibition of small-conductance calcium-activated potassium (SK) channels by NS8593 reveals atrial antiarrhythmic potential in horses. Heart Rhythm 2015, 12, 825–835. [Google Scholar] [CrossRef]
- Ozgen, N.; Dun, W.; Sosunov, E.A.; Anyukhovsky, E.P.; Hirose, M.; Duffy, H.S.; Boyden, P.A.; Rosen, M.R. Early electrical remodeling in rabbit pulmonary vein results from trafficking of intracellular SK2 channels to membrane sites. Cardiovasc. Res. 2007, 75, 758–769. [Google Scholar] [CrossRef]
- Chen, M.; Yin, D.; Guo, S.; Xu, D.Z.; Wang, Z.; Chen, Z.; Rubart-von der Lohe, M.; Lin, S.F.; Everett Iv, T.H.; Weiss, J.N.; et al. Sex-specific activation of SK current by isoproterenol facilitates action potential triangulation and arrhythmogenesis in rabbit ventricles. J. Physiol. 2018, 596, 4299–4322. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.D.; Coulibaly, Z.A.; Chen, W.C.; Ledford, H.A.; Lee, J.H.; Sirish, P.; Dai, G.; Jian, Z.; Chuang, F.; Brust-Mascher, I.; et al. Coupling of SK channels, L-type Ca2+ channels, and ryanodine receptors in cardiomyocytes. Sci. Rep. 2018, 8, 4670. [Google Scholar] [CrossRef]
- Hsueh, C.H.; Chang, P.C.; Hsieh, Y.C.; Reher, T.; Chen, P.S.; Lin, S.F. Proarrhythmic effect of blocking the small conductance calcium activated potassium channel in isolated canine left atrium. Heart Rhythm 2013, 10, 891–898. [Google Scholar] [CrossRef]
- Diness, J.G.; Kirchhoff, J.E.; Speerschneider, T.; Abildgaard, L.; Edvardsson, N.; Sorensen, U.S.; Grunnet, M.; Bentzen, B.H. The KCa2 channel inhibitor AP30663 selectively increases atrial refractoriness, converts vernakalant-resistant atrial fibrillation and prevents its reinduction in conscious pigs. Front. Pharmacol. 2020, 11, 159. [Google Scholar] [CrossRef]
- Diness, J.G.; Skibsbye, L.; Simo-Vicens, R.; Santos, J.L.; Lundegaard, P.; Citerni, C.; Sauter, D.R.P.; Bomholtz, S.H.; Svendsen, J.H.; Olesen, S.P.; et al. Termination of vernakalant-resistant atrial fibrillation by inhibition of small-conductance Ca2+-activated K+ channels in pigs. Circ. Arrhythmia Electrophysiol. 2017, 10, e005125. [Google Scholar] [CrossRef] [PubMed]
- Giommi, A.; Gurgel, A.R.B.; Smith, G.L.; Workman, A.J. Does the small conductance Ca2+-activated K+ current ISK flow under physiological conditions in rabbit and human atrial isolated cardiomyocytes? J. Mol. Cell Cardiol. 2023, 183, 70–80. [Google Scholar] [CrossRef]
- Nagy, N.; Szuts, V.; Horvath, Z.; Seprenyi, G.; Farkas, A.S.; Acsai, K.; Prorok, J.; Bitay, M.; Kun, A.; Pataricza, J.; et al. Does small-conductance calcium-activated potassium channel contribute to cardiac repolarization? J. Mol. Cell. Cardiol. 2009, 47, 656–663. [Google Scholar] [CrossRef]
- Qi, X.Y.; Diness, J.G.; Brundel, B.J.; Zhou, X.B.; Naud, P.; Wu, C.T.; Huang, H.; Harada, M.; Aflaki, M.; Dobrev, D.; et al. Role of small-conductance calcium-activated potassium channels in atrial electrophysiology and fibrillation in the dog. Circulation 2014, 129, 430–440. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Li, T.; Xu, D.; Wang, Y.; Zhou, Y.; Wan, J.; Huang, C.L.; Tan, X. Small-conductance calcium-activated potassium channels in the heart: Expression, regulation and pathological implications. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2023, 378, 20220171. [Google Scholar] [CrossRef]
- Zhang, X.D.; Thai, P.N.; Lieu, D.K.; Chiamvimonvat, N. Cardiac small-conductance calcium-activated potassium channels in health and disease. Pflügers Arch.-Eur. J. Physiol. 2021, 473, 477–489. [Google Scholar] [CrossRef]
- Heijman, J.; Zhou, X.; Morotti, S.; Molina, C.E.; Abu-Taha, I.H.; Tekook, M.; Jespersen, T.; Zhang, Y.; Dobrev, S.; Milting, H.; et al. Enhanced Ca2+-dependent SK-channel gating and membrane trafficking in human atrial fibrillation. Circ. Res. 2023, 132, e116–e133. [Google Scholar] [CrossRef] [PubMed]
- Linz, B.; Hesselkilde, E.M.; Skarsfeldt, M.A.; Hertel, J.N.; Sattler, S.M.; Yan, Y.; Tfelt-Hansen, J.; Diness, J.G.; Bentzen, B.H.; Linz, D.; et al. Pharmacological inhibition of SK-channels with AP14145 prevents atrial arrhythmogenic changes in a porcine model for obstructive respiratory events. J. Cardiovasc. Electrophysiol. 2023, 34, 126–134. [Google Scholar] [CrossRef]
- Saljic, A.; Heijman, J.; Dobrev, D. Emerging antiarrhythmic drugs for atrial fibrillation. Int. J. Mol. Sci. 2022, 23, 4096. [Google Scholar] [CrossRef]
- Gatta, G.; Sobota, V.; Citerni, C.; Diness, J.G.; Sorensen, U.S.; Jespersen, T.; Bentzen, B.H.; Zeemering, S.; Kuiper, M.; Verheule, S.; et al. Effective termination of atrial fibrillation by SK channel inhibition is associated with a sudden organization of fibrillatory conduction. Europace 2021, 23, 1847–1859. [Google Scholar] [CrossRef]
- Qi, M.M.; Qian, L.L.; Wang, R.X. Modulation of SK channels: Insight into therapeutics of atrial fibrillation. Heart Lung Circ. 2021, 30, 1130–1139. [Google Scholar] [CrossRef]
- Monigatti-Tenkorang, J.; Jousset, F.; Pascale, P.; Vesin, J.M.; Ruchat, P.; Fromer, M.; Narayan, S.M.; Pruvot, E. Intermittent atrial tachycardia promotes repolarization alternans and conduction slowing during rapid rates, and increases susceptibility to atrial fibrillation in a free-behaving sheep model. J. Cardiovasc. Electrophysiol. 2014, 25, 418–427. [Google Scholar] [CrossRef] [PubMed]
- Verrier, R.L.; Fuller, H.; Justo, F.; Nearing, B.D.; Rajamani, S.; Belardinelli, L. Unmasking atrial repolarization to assess alternans, spatiotemporal heterogeneity, and susceptibility to atrial fibrillation. Heart Rhythm 2016, 13, 953–961. [Google Scholar] [CrossRef]
- Lalani, G.G.; Schricker, A.A.; Clopton, P.; Krummen, D.E.; Narayan, S.M. Frequency analysis of atrial action potential alternans: A sensitive clinical index of individual propensity to atrial fibrillation. Circ. Arrhythmia Electrophysiol. 2013, 6, 859–867. [Google Scholar] [CrossRef]
- Narayan, S.M.; Bode, F.; Karasik, P.L.; Franz, M.R. Alternans of atrial action potentials during atrial flutter as a precursor to atrial fibrillation. Circulation 2002, 106, 1968–1973. [Google Scholar] [CrossRef] [PubMed]
- Narayan, S.M.; Franz, M.R.; Clopton, P.; Pruvot, E.J.; Krummen, D.E. Repolarization alternans reveals vulnerability to human atrial fibrillation. Circulation 2011, 123, 2922–2930. [Google Scholar] [CrossRef] [PubMed]
- Narayan, S.M.; Wright, M.; Derval, N.; Jadidi, A.; Forclaz, A.; Nault, I.; Miyazaki, S.; Sacher, F.; Bordachar, P.; Clementy, J.; et al. Classifying fractionated electrograms in human atrial fibrillation using monophasic action potentials and activation mapping: Evidence for localized drivers, rate acceleration, and nonlocal signal etiologies. Heart Rhythm 2011, 8, 244–253. [Google Scholar] [CrossRef]
- Comtois, P.; Nattel, S. Atrial repolarization alternans as a path to atrial fibrillation. J. Cardiovasc. Electrophysiol. 2012, 23, 1013–1015. [Google Scholar] [CrossRef]
- Berenfeld, O.; Jalife, J. Mechanisms of atrial fibrillation: Rotors, ionic determinants, and excitation frequency. Cardiol. Clin. 2014, 32, 495–506. [Google Scholar] [CrossRef]
- Franz, M.R.; Jamal, S.M.; Narayan, S.M. The role of action potential alternans in the initiation of atrial fibrillation in humans: A review and future directions. Europace 2012, 14 (Suppl. 5), v58–v64. [Google Scholar] [CrossRef]
- Ellinor, P.T.; Lunetta, K.L.; Glazer, N.L.; Pfeufer, A.; Alonso, A.; Chung, M.K.; Sinner, M.F.; de Bakker, P.I.; Mueller, M.; Lubitz, S.A.; et al. Common variants in KCNN3 are associated with lone atrial fibrillation. Nat. Genet. 2010, 42, 240–244. [Google Scholar] [CrossRef]
- Fan, X.; Yu, Y.; Lan, H.; Ou, X.; Yang, L.; Li, T.; Cao, J.; Zeng, X.; Li, M. Ca2+/Calmodulin-dependent protein kinase II (CaMKII) increases small-conductance Ca2+-activated K+ current in patients with chronic atrial fibrillation. Med. Sci. Monit. 2018, 24, 3011–3023. [Google Scholar] [CrossRef] [PubMed]
- Darkow, E.; Nguyen, T.T.; Stolina, M.; Kari, F.A.; Schmidt, C.; Wiedmann, F.; Baczko, I.; Kohl, P.; Rajamani, S.; Ravens, U.; et al. Small conductance Ca2+-activated K+ (SK) channel mRNA expression in human atrial and ventricular tissue: Comparison between donor, atrial fibrillation and heart failure tissue. Front. Physiol. 2021, 12, 650964. [Google Scholar] [CrossRef] [PubMed]
- Yu, T.; Deng, C.; Wu, R.; Guo, H.; Zheng, S.; Yu, X.; Shan, Z.; Kuang, S.; Lin, Q. Decreased expression of small-conductance Ca2+-activated K+ channels SK1 and SK2 in human chronic atrial fibrillation. Life Sci. 2012, 90, 219–227. [Google Scholar] [CrossRef]
- Li, N.; Timofeyev, V.; Tuteja, D.; Xu, D.; Lu, L.; Zhang, Q.; Zhang, Z.; Singapuri, A.; Albert, T.R.; Rajagopal, A.V.; et al. Ablation of a Ca2+-activated K+ channel (SK2 channel) results in action potential prolongation in atrial myocytes and atrial fibrillation. J. Physiol. 2009, 587, 1087–1100. [Google Scholar] [CrossRef]
- Giudicessi, J.R.; Ackerman, M.J. Genotype- and phenotype-guided management of congenital long QT syndrome. Curr. Probl. Cardiol. 2013, 38, 417–455. [Google Scholar] [CrossRef] [PubMed]
- Goldenberg, I.; Zareba, W.; Moss, A.J. Long QT Syndrome. Curr. Probl. Cardiol. 2008, 33, 629–694. [Google Scholar] [CrossRef]
- Zhang, N.; Gong, M.; Tse, G.; Zhang, Z.; Meng, L.; Yan, B.P.; Zhang, L.; Wu, G.; Xia, Y.; Xin-Yan, G.; et al. Prolonged corrected QT interval in predicting atrial fibrillation: A systematic review and meta-analysis. Pacing Clin. Electrophysiol. 2018, 41, 321–327. [Google Scholar] [CrossRef]
- Mandyam, M.C.; Soliman, E.Z.; Alonso, A.; Dewland, T.A.; Heckbert, S.R.; Vittinghoff, E.; Cummings, S.R.; Ellinor, P.T.; Chaitman, B.R.; Stocke, K.; et al. The QT interval and risk of incident atrial fibrillation. Heart Rhythm 2013, 10, 1562–1568. [Google Scholar] [CrossRef]
- Zellerhoff, S.; Pistulli, R.; Monnig, G.; Hinterseer, M.; Beckmann, B.M.; Kobe, J.; Steinbeck, G.; Kaab, S.; Haverkamp, W.; Fabritz, L.; et al. Atrial Arrhythmias in long-QT syndrome under daily life conditions: A nested case control study. J. Cardiovasc. Electrophysiol. 2009, 20, 401–407. [Google Scholar] [CrossRef]
- Kirchhof, P.; Eckardt, L.; Franz, M.R.; Monnig, G.; Loh, P.; Wedekind, H.; Schulze-Bahr, E.; Breithardt, G.; Haverkamp, W. Prolonged atrial action potential durations and polymorphic atrial tachyarrhythmias in patients with long QT syndrome. J. Cardiovasc. Electrophysiol. 2003, 14, 1027–1033. [Google Scholar] [CrossRef] [PubMed]
- Johnson, J.N.; Tester, D.J.; Perry, J.; Salisbury, B.A.; Reed, C.R.; Ackerman, M.J. Prevalence of early-onset atrial fibrillation in congenital long QT syndrome. Heart Rhythm 2008, 5, 704–709. [Google Scholar] [CrossRef] [PubMed]
- Guerrier, K.; Czosek, R.J.; Spar, D.S.; Anderson, J. Long QT genetics manifesting as atrial fibrillation. Heart Rhythm 2013, 10, 1351–1353. [Google Scholar] [CrossRef]
- Calloe, K.; Schmitt, N.; Grubb, S.; Pfeiffer, R.; David, J.P.; Kanter, R.; Cordeiro, J.M.; Antzelevitch, C. Multiple arrhythmic syndromes in a newborn, owing to a novel mutation in SCN5A. Can. J. Physiol. Pharmacol. 2011, 89, 723–736. [Google Scholar] [CrossRef]
- Benito, B.; Brugada, R.; Perich, R.M.; Lizotte, E.; Cinca, J.; Mont, L.; Berruezo, A.; Tolosana, J.M.; Freixa, X.; Brugada, P.; et al. A mutation in the sodium channel is responsible for the association of long QT syndrome and familial atrial fibrillation. Heart Rhythm 2008, 5, 1434–1440. [Google Scholar] [CrossRef]
- Platonov, P.G.; McNitt, S.; Polonsky, B.; Rosero, S.Z.; Zareba, W. Atrial Fibrillation in Long QT Syndrome by Genotype. Circ. Arrhythmia Electrophysiol. 2019, 12, e007213. [Google Scholar] [CrossRef]
- Olesen, M.S.; Yuan, L.; Liang, B.; Holst, A.G.; Nielsen, N.; Nielsen, J.B.; Hedley, P.L.; Christiansen, M.; Olesen, S.P.; Haunso, S.; et al. High prevalence of long QT syndrome-associated SCN5A variants in patients with early-onset lone atrial fibrillation. Circ. Cardiovasc. Genet. 2012, 5, 450–459. [Google Scholar] [CrossRef] [PubMed]
- Bartos, D.C.; Duchatelet, S.; Burgess, D.E.; Klug, D.; Denjoy, I.; Peat, R.; Lupoglazoff, J.M.; Fressart, V.; Berthet, M.; Ackerman, M.J.; et al. R231C mutation in KCNQ1 causes long QT syndrome type 1 and familial atrial fibrillation. Heart Rhythm 2011, 8, 48–55. [Google Scholar] [CrossRef]
- Andreasen, L.; Nielsen, J.B.; Christophersen, I.E.; Holst, A.G.; Sajadieh, A.; Tveit, A.; Haunso, S.; Svendsen, J.H.; Schmitt, N.; Olesen, M.S. Genetic modifier of the QTc interval associated with early-onset atrial fibrillation. Can. J. Cardiol. 2013, 29, 1234–1240. [Google Scholar] [CrossRef]
- Kanaporis, G.; Blatter, L.A. Membrane potential determines calcium alternans through modulation of SR Ca2+ load and L-type Ca2+ current. J. Mol. Cell. Cardiol. 2017, 105, 49–58. [Google Scholar] [CrossRef]
- Kanaporis, G.; Kalik, Z.M.; Blatter, L.A. Action potential shortening rescues atrial calcium alternans. J. Physiol. 2019, 597, 723–740. [Google Scholar] [CrossRef] [PubMed]
- El Yaman, M.; Perry, J.; Makielski, J.C.; Ackerman, M.J. Suppression of atrial fibrillation with mexiletine pharmacotherapy in a young woman with type 1 long QT syndrome. Heart Rhythm 2008, 5, 472–474. [Google Scholar] [CrossRef] [PubMed]
- Mizukami, K.; Yokoshiki, H.; Mitsuyama, H.; Watanabe, M.; Tenma, T.; Takada, S.; Tsutsui, H. Small-conductance Ca2+-activated K+ current is upregulated via the phosphorylation of CaMKII in cardiac hypertrophy from spontaneously hypertensive rats. Am. J. Physiol. Heart Circ. Physiol. 2015, 309, H1066–H1074. [Google Scholar] [CrossRef]
- Kanaporis, G.; Blatter, L.A. Activation of small conductance Ca2+ -activated K+ channels suppresses Ca2+ transient and action potential alternans in ventricular myocytes. J. Physiol. 2023, 601, 51–67. [Google Scholar] [CrossRef]
- Weisbrod, D. Small and Intermediate Calcium Activated Potassium Channels in the Heart: Role and Strategies in the Treatment of Cardiovascular Diseases. Front. Physiol. 2020, 11, 590534. [Google Scholar] [CrossRef]
- Chang, P.C.; Hsieh, Y.C.; Hsueh, C.H.; Weiss, J.N.; Lin, S.F.; Chen, P.S. Apamin induces early afterdepolarizations and torsades de pointes ventricular arrhythmia from failing rabbit ventricles exhibiting secondary rises in intracellular calcium. Heart Rhythm 2013, 10, 1516–1524. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Fei, Y.; Chen, T.Z.; Li, Y.G.; Chen, P.S. The regulation of the small-conductance calcium-activated potassium current and the mechanisms of sex dimorphism in J wave syndrome. Pflügers Arch.-Eur. J. Physiol. 2021, 473, 491–506. [Google Scholar] [CrossRef]
- Bouchard, R.A.; Clark, R.B.; Giles, W.R. Effects of action potential duration on excitation-contraction coupling in rat ventricular myocytes. Action potential voltage-clamp measurements. Circ. Res. 1995, 76, 790–801. [Google Scholar] [CrossRef]
- Schotten, U.; de Haan, S.; Verheule, S.; Harks, E.G.; Frechen, D.; Bodewig, E.; Greiser, M.; Ram, R.; Maessen, J.; Kelm, M.; et al. Blockade of atrial-specific K+-currents increases atrial but not ventricular contractility by enhancing reverse mode Na+/Ca2+-exchange. Cardiovasc. Res. 2007, 73, 37–47. [Google Scholar] [CrossRef]
- Kuzmenkov, A.I.; Peigneur, S.; Nasburg, J.A.; Mineev, K.S.; Nikolaev, M.V.; Pinheiro-Junior, E.L.; Arseniev, A.S.; Wulff, H.; Tytgat, J.; Vassilevski, A.A. Apamin structure and pharmacology revisited. Front. Pharmacol. 2022, 13, 977440. [Google Scholar] [CrossRef]
- Hancock, J.M.; Weatherall, K.L.; Choisy, S.C.; James, A.F.; Hancox, J.C.; Marrion, N.V. Selective activation of heteromeric SK channels contributes to action potential repolarization in mouse atrial myocytes. Heart Rhythm 2015, 12, 1003–1015. [Google Scholar] [CrossRef] [PubMed]
- Kornej, J.; Borschel, C.S.; Benjamin, E.J.; Schnabel, R.B. Epidemiology of Atrial Fibrillation in the 21st Century: Novel Methods and New Insights. Circ. Res. 2020, 127, 4–20. [Google Scholar] [CrossRef]
- Pruvot, E.J.; Katra, R.P.; Rosenbaum, D.S.; Laurita, K.R. Role of calcium cycling versus restitution in the mechanism of repolarization alternans. Circ. Res. 2004, 94, 1083–1090. [Google Scholar] [CrossRef] [PubMed]
- Kanaporis, G.; Blatter, L.A. The mechanisms of calcium cycling and action potential dynamics in cardiac alternans. Circ. Res. 2015, 116, 846–856. [Google Scholar] [CrossRef] [PubMed]
- Kanaporis, G.; Martinez-Hernandez, E.; Blatter, L.A. Calcium- and voltage-driven atrial alternans: Insight from [Ca]i and Vm asynchrony. Physiol. Rep. 2023, 11, e15703. [Google Scholar] [CrossRef]
- Shkryl, V.M.; Maxwell, J.T.; Domeier, T.L.; Blatter, L.A. Refractoriness of sarcoplasmic reticulum Ca2+ release determines Ca2+ alternans in atrial myocytes. Am. J. Physiol. Heart Circ. Physiol. 2012, 302, H2310–H2320. [Google Scholar] [CrossRef]
- Kanaporis, G.; Blatter, L.A. Alternans in atria: Mechanisms and clinical relevance. Medicina 2017, 53, 139–149. [Google Scholar] [CrossRef]
- Diederichsen, S.Z.; Darkner, S.; Chen, X.; Johannessen, A.; Pehrson, S.; Hansen, J.; Svendsen, J.H. QT as a predictor of recurrence after atrial fibrillation ablation and the impact of amiodarone: Results from the placebo-controlled AMIO-CAT trial. Europace 2019, 21, 1055–1062. [Google Scholar] [CrossRef]
- Magnano, M.; Gallo, C.; Bocchino, P.P.; Briguglio, M.; Rivetti, A.; Gaita, F.; Anselmino, M. QT prolongation and variability: New ECG signs of atrial potentials dispersion before atrial fibrillation onset. J. Cardiovasc. Med. 2019, 20, 180–185. [Google Scholar] [CrossRef]
- van Noord, C.; Eijgelsheim, M.; Stricker, B.H. Drug- and non-drug-associated QT interval prolongation. Br. J. Clin. Pharmacol. 2010, 70, 16–23. [Google Scholar] [CrossRef]
- Blana, A.; Kaese, S.; Fortmuller, L.; Laakmann, S.; Damke, D.; van Bragt, K.; Eckstein, J.; Piccini, I.; Kirchhefer, U.; Nattel, S.; et al. Knock-in gain-of-function sodium channel mutation prolongs atrial action potentials and alters atrial vulnerability. Heart Rhythm 2010, 7, 1862–1869. [Google Scholar] [CrossRef] [PubMed]
- Dautova, Y.; Zhang, Y.; Sabir, I.; Grace, A.A.; Huang, C.L. Atrial arrhythmogenesis in wild-type and Scn5a+/delta murine hearts modelling LQT3 syndrome. Pflugers. Arch. 2009, 458, 443–457. [Google Scholar] [CrossRef]
- Shimizu, W.; Horie, M. Phenotypic manifestations of mutations in genes encoding subunits of cardiac potassium channels. Circ. Res. 2011, 109, 97–109. [Google Scholar] [CrossRef] [PubMed]
- Huser, J.; Wang, Y.G.; Sheehan, K.A.; Cifuentes, F.; Lipsius, S.L.; Blatter, L.A. Functional coupling between glycolysis and excitation-contraction coupling underlies alternans in cat heart cells. J. Physiol. 2000, 524 Pt 3, 795–806. [Google Scholar] [CrossRef]
- Myles, R.C.; Burton, F.L.; Cobbe, S.M.; Smith, G.L. Alternans of action potential duration and amplitude in rabbits with left ventricular dysfunction following myocardial infarction. J. Mol. Cell. Cardiol. 2011, 50, 510–521. [Google Scholar] [CrossRef] [PubMed]
- Pastore, J.M.; Girouard, S.D.; Laurita, K.R.; Akar, F.G.; Rosenbaum, D.S. Mechanism linking T-wave alternans to the genesis of cardiac fibrillation. Circulation 1999, 99, 1385–1394. [Google Scholar] [CrossRef]
- Bonilla, I.M.; Long, V.P., 3rd; Vargas-Pinto, P.; Wright, P.; Belevych, A.; Lou, Q.; Mowrey, K.; Yoo, J.; Binkley, P.F.; Fedorov, V.V.; et al. Calcium-activated potassium current modulates ventricular repolarization in chronic heart failure. PLoS ONE 2014, 9, e108824. [Google Scholar] [CrossRef]
- Hamilton, S.; Polina, I.; Terentyeva, R.; Bronk, P.; Kim, T.Y.; Roder, K.; Clements, R.T.; Koren, G.; Choi, B.R.; Terentyev, D. PKA phosphorylation underlies functional recruitment of sarcolemmal SK2 channels in ventricular myocytes from hypertrophic hearts. J. Physiol. 2020, 598, 2847–2873. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kanaporis, G.; Blatter, L.A. Activation of Small Conductance Ca2+-Activated K+ Channels Suppresses Electrical and Calcium Alternans in Atrial Myocytes. Int. J. Mol. Sci. 2025, 26, 3597. https://doi.org/10.3390/ijms26083597
Kanaporis G, Blatter LA. Activation of Small Conductance Ca2+-Activated K+ Channels Suppresses Electrical and Calcium Alternans in Atrial Myocytes. International Journal of Molecular Sciences. 2025; 26(8):3597. https://doi.org/10.3390/ijms26083597
Chicago/Turabian StyleKanaporis, Giedrius, and Lothar A. Blatter. 2025. "Activation of Small Conductance Ca2+-Activated K+ Channels Suppresses Electrical and Calcium Alternans in Atrial Myocytes" International Journal of Molecular Sciences 26, no. 8: 3597. https://doi.org/10.3390/ijms26083597
APA StyleKanaporis, G., & Blatter, L. A. (2025). Activation of Small Conductance Ca2+-Activated K+ Channels Suppresses Electrical and Calcium Alternans in Atrial Myocytes. International Journal of Molecular Sciences, 26(8), 3597. https://doi.org/10.3390/ijms26083597