
p.Phe508del-CFTR Trafficking: A Protein Quality Control Perspective Through UPR, UPS, and Autophagy



{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Biogenesis of CFTR and Differences with the Biogenesis of p.Phe508del-CFTR
3. Unconventional Pathways
4. UPR Regulates the Autophagy-Mediated UPS of p.Phe508del-CFTR
5. Evidence Supporting the Autophagosome-Mediated UPS of p.Phe508del-CFTR
6. Discussion and Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| ATF6 | Activating transcription factor 6 |
| ATGs | Autophagy-related proteins |
| CNX | Calnexin |
| CRT | Calreticulin |
| CFTR | Cystic fibrosis transmembrane conductance regulator |
| CPS | Conventional protein secretion |
| CF | Cystic fibrosis |
| ER | Endoplasmic reticulum |
| ESCRT | Endosomal sorting complex required for transport |
| ERAD | ER-associated degradation |
| EDEM | ER degradation-enhanced mannosidase |
| ERES | ER exit sites |
| ERQC | ER quality control |
| GA | Golgi apparatus |
| GRASP | Golgi reassembly stacking protein |
| IRE1 | Inositol-requiring enzyme 1 |
| LC3 | Light chain 3 |
| LIR | LC3-interacting region |
| NEFs | Nucleotide-exchange factors |
| OST | Oligosaccharyltransferase |
| PI3P | Phosphatidylinositol 3-phosphate |
| PDI | Protein disulfide isomerase |
| PERK | Protein kinase R (PKR)-like endoplasmic reticulum kinase |
| SARs | Secretory autophagy receptors |
| SP | Signal peptide |
| SRP | Signal recognition particle |
| SNAREs | Soluble N-ethylmaleimide-sensitive-factor Attachment protein Receptors |
| TRAP | Translocon-associated protein |
| UPS | Unconventional protein secretion |
| UPR | Unfolded protein response |
| UDP | Uridine diphosphate |
| WIPI | WD-repeat domain phosphoinositide-interacting protein |
References
- Cheng, S.H.; Gregory, R.J.; Marshall, J.; Paul, S.; Souza, D.W.; White, G.A.; O’Riordan, C.R.; Smith, A.E. Defective intracellular transport and processing of CFTR is the molecular basis of most cystic fibrosis. Cell 1990, 63, 827–834. [Google Scholar] [CrossRef] [PubMed]
- Kerem, B.S.; Rommens, J.M.; Buchanan, J.A.; Markiewicz, D.; Cox, T.K.; Chakravarti, A.; Buchwald, M.; Tsui, L.C. Identification of the cystic fibrosis gene: Genetic analysis. Science 1989, 245, 1073–1080. [Google Scholar] [CrossRef] [PubMed]
- Anderson, M.P.; Gregory, R.J.; Thompson, S.; Souza, D.W.; Paul, S.; Mulligan, R.C.; Smith, A.E.; Welsh, M.J. Demonstration That CFTR Is a Chloride Channel by Alteration of Its Anion Selectivity. Science 1991, 253, 202–205. [Google Scholar] [CrossRef]
- Rich, D.P.; Anderson, M.P.; Gregory, R.J.; Cheng, S.H.; Paul, S.; Jefferson, D.M.; McCann, J.D.; Klinger, K.W.; Smith, A.E.; Welsh, M.J. Expression of cystic fibrosis transmembrane conductance regulator corrects defective chloride channel regulation in cystic fibrosis airway epithelial cells. Nature 1990, 347, 358–363. [Google Scholar] [CrossRef] [PubMed]
- Riordan, J.R.; Rommens, J.M.; Kerem, B.S.; Alon, N.O.; Rozmahel, R.; Grzelczak, Z.; Zielenski, J.; Lok, S.I.; Plavsic, N.; Chou, J.L.; et al. Identification of the cystic fibrosis gene: Cloning and characterization of complementary DNA. Science 1989, 245, 1066–1073. [Google Scholar] [CrossRef]
- Sheppard, D.N.; Welsh, M.J. Structure and Function of the CFTR Chloride Channel. Physiol. Rev. 1999, 79, S23–S45. [Google Scholar] [CrossRef]
- Heda, G.D.; Marino, C.R. Surface Expression of the Cystic Fibrosis Transmembrane Conductance Regulator Mutant ΔF508 Is Markedly Upregulated by Combination Treatment with Sodium Butyrate and Low Temperature. Biochem. Biophys. Res. Commun. 2000, 271, 659–664. [Google Scholar] [CrossRef]
- Du, K.; Sharma, M.; Lukacs, G.L. The ΔF508 cystic fibrosis mutation impairs domain-domain interactions and arrests post-translational folding of CFTR. Nat. Struct. Mol. Biol. 2004, 12, 17–25. [Google Scholar] [CrossRef]
- Farinha, C.M.; Amaral, M.D. Most F508del-CFTR Is Targeted to Degradation at an Early Folding Checkpoint and Independently of Calnexin. Mol. Cell. Biol. 2005, 25, 5242–5252. [Google Scholar] [CrossRef]
- Jung, J.; Kim, J.; Roh, S.H.; Jun, I.; Sampson, R.D.; Gee, H.Y.; Choi, J.Y.; Lee, M.G. The HSP70 co-chaperone DNAJC14 targets misfolded pendrin for unconventional protein secretion. Nat. Commun. 2016, 7, 11386. [Google Scholar] [CrossRef]
- Rubenstein, R.C.; Zeitlin, P.L. Sodium 4-phenylbutyrate downregulates Hsc70: Implications for intracellular trafficking of DeltaF508-CFTR. Am. J. Physiol. Cell Physiol. 2000, 278, C259–C267. [Google Scholar] [CrossRef]
- Giuliani, F.; Grieve, A.; Rabouille, C. Unconventional secretion: A stress on GRASP. Curr. Opin. Cell Biol. 2011, 23, 498–504. [Google Scholar] [CrossRef] [PubMed]
- Farinha, C.M.; Nogueira, P.; Mendes, F.; Penque, D.; Amaral, M.D. The human DnaJ homologue (Hdj)-1/heat-shock protein (Hsp) 40 co-chaperone is required for the in vivo stabilization of the cystic fibrosis transmembrane conductance regulator by Hsp70. Biochem. J. 2002, 366, 797–806. [Google Scholar] [CrossRef] [PubMed]
- Ogata, M.; Hino, S.-I.; Saito, A.; Morikawa, K.; Kondo, S.; Kanemoto, S.; Murakami, T.; Taniguchi, M.; Tanii, I.; Yoshinaga, K.; et al. Autophagy Is Activated for Cell Survival after Endoplasmic Reticulum Stress. Mol. Cell. Biol. 2006, 26, 9220–9231. [Google Scholar] [CrossRef]
- Gee, H.Y.; Noh, S.H.; Tang, B.L.; Kim, K.H.; Lee, M.G. Rescue of ΔF508-CFTR Trafficking via a GRASP-Dependent Unconventional Secretion Pathway. Cell 2011, 146, 746–760. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Noh, S.H.; Piao, H.; Kim, D.H.; Kim, K.; Cha, J.S.; Chung, W.Y.; Cho, H.; Kim, J.Y.; Lee, M.G. Monomerization and ER Relocalization of GRASP Is a Requisite for Unconventional Secretion of CFTR. Traffic 2016, 17, 733–753. [Google Scholar] [CrossRef]
- Noh, S.H.; Gee, H.Y.; Kim, Y.; Piao, H.; Kim, J.; Kang, C.M.; Lee, G.; Mook-Jung, I.; Lee, Y.; Cho, J.W.; et al. Specific autophagy and ESCRT components participate in the unconventional secretion of CFTR. Autophagy 2018, 14, 1761–1778. [Google Scholar] [CrossRef]
- Cavalli, G.; Cenci, S. Autophagy and Protein Secretion. J. Mol. Biol. 2020, 432, 2525–2545. [Google Scholar] [CrossRef]
- Trouvé, P.; Férec, C.; Génin, E. The Interplay between the Unfolded Protein Response, Inflammation and Infection in Cystic Fibrosis. Cells 2021, 10, 2980. [Google Scholar] [CrossRef]
- Wek, R.C.; Cavener, D.R. Translational Control and the Unfolded Protein Response. Antioxidants Redox Signal. 2007, 9, 2357–2372. [Google Scholar] [CrossRef]
- Liu, C.Y.; Kaufman, R.J. The unfolded protein response. J. Cell Sci. 2003, 116, 1861–1862. [Google Scholar] [CrossRef] [PubMed]
- Lee, A.S. The ER chaperone and signaling regulator GRP78/BiP as a monitor of endoplasmic reticulum stress. Methods 2005, 35, 373–381. [Google Scholar] [CrossRef] [PubMed]
- Haas, I.G.; Wabl, M. Immunoglobulin heavy chain binding protein. Nature 1983, 306, 387–389. [Google Scholar] [CrossRef]
- Ali, M.M.U.; Bagratuni, T.; Davenport, E.L.; Nowak, P.R.; Silva-Santisteban, M.C.; Hardcastle, A.; McAndrews, C.; Rowlands, M.G.; Morgan, G.J.; Aherne, W.; et al. Structure of the Ire1 autophosphorylation complex and implications for the unfolded protein response. EMBO J. 2011, 30, 894–905. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Brandizzi, F. IRE1: ER stress sensor and cell fate executor. Trends Cell Biol. 2013, 23, 547–555. [Google Scholar] [CrossRef]
- McQuiston, A.; Diehl, J.A. Recent insights into PERK-dependent signaling from the stressed endoplasmic reticulum. F1000Research 2017, 6, 1897. [Google Scholar] [CrossRef]
- Chen, X.; Shen, J.; Prywes, R. The Luminal Domain of ATF6 Senses Endoplasmic Reticulum (ER) Stress and Causes Translocation of ATF6 from the ER to the Golgi. J. Biol. Chem. 2002, 277, 13045–13052. [Google Scholar] [CrossRef]
- Hai, T.W.; Liu, F.; Coukos, W.J.; Green, M.R. Transcription factor ATF cDNA clones: An extensive family of leucine zipper proteins able to selectively form DNA-binding heterodimers. Genes Dev. 1989, 3, 2083–2090. [Google Scholar] [CrossRef]
- Shen, J.; Chen, X.; Hendershot, L.; Prywes, R. ER Stress Regulation of ATF6 Localization by Dissociation of BiP/GRP78 Binding and Unmasking of Golgi Localization Signals. Dev. Cell 2002, 3, 99–111. [Google Scholar] [CrossRef]
- Zhang, K.; Kaufman, R.J. Signaling the Unfolded Protein Response from the Endoplasmic Reticulum. J. Biol. Chem. 2004, 279, 25935–25938. [Google Scholar] [CrossRef]
- Bartoszewski, R.; Rab, A.; Twitty, G.; Stevenson, L.; Fortenberry, J.; Piotrowski, A.; Dumanski, J.P.; Bebök, Z. The Mechanism of Cystic Fibrosis Transmembrane Conductance Regulator Transcriptional Repression during the Unfolded Protein Response. J. Biol. Chem. 2008, 283, 12154–12165. [Google Scholar] [CrossRef]
- Rab, A.; Bartoszewski, R.; Jurkuvenaite, A.; Wakefield, J.; Collawn, J.F.; Bebők, Z. Endoplasmic reticulum stress and the unfolded protein response regulate genomic cystic fibrosis transmembrane conductance regulator expression. Am. J. Physiol. Physiol. 2007, 292, C756–C766. [Google Scholar] [CrossRef]
- Viotti, C. ER to Golgi-Dependent Protein Secretion: The Conventional Pathway. Methods Mol. Biol. 2016, 1459, 3–29. [Google Scholar] [PubMed]
- A Rapoport, T.; Rolls, M.M.; Jungnickel, B. Approaching the mechanism of protein transport across the ER membrane. Curr. Opin. Cell Biol. 1996, 8, 499–504. [Google Scholar] [CrossRef] [PubMed]
- Schlenstedt, G.; Zimmermann, R. Import of frog prepropeptide GLa into microsomes requires ATP but does not involve docking protein or ribosomes. EMBO J. 1987, 6, 699–703. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Xiong, X.; Helm, A.; Kimani, K.; Bragin, A.; Skach, W.R. Co- and Posttranslational Translocation Mechanisms Direct Cystic Fibrosis Transmembrane Conductance Regulator N Terminus Transmembrane Assembly. J. Biol. Chem. 1998, 273, 568–576. [Google Scholar] [CrossRef] [PubMed]
- Farinha, C.M.; Canato, S. From the endoplasmic reticulum to the plasma membrane: Mechanisms of CFTR folding and trafficking. Cell. Mol. Life Sci. 2017, 74, 39–55. [Google Scholar] [CrossRef]
- Farinha, C.M.; Matos, P.; Amaral, M.D. Control of cystic fibrosis transmembrane conductance regulator membrane trafficking: Not just from the endoplasmic reticulum to the Golgi. FEBS J. 2013, 280, 4396–4406. [Google Scholar] [CrossRef]
- Piao, H.; Kim, J.; Noh, S.H.; Kweon, H.-S.; Kim, J.Y.; Lee, M.G. Sec16A is critical for both conventional and unconventional secretion of CFTR. Sci. Rep. 2017, 7, 39887. [Google Scholar] [CrossRef]
- Kleizen, B.; van Vlijmen, T.; de Jonge, H.R.; Braakman, I. Folding of CFTR is predominantly cotranslational. Mol. Cell 2005, 20, 277–287. [Google Scholar] [CrossRef]
- Gemmer, M.; Förster, F. A clearer picture of the ER translocon complex. J. Cell Sci. 2020, 133, jcs231340. [Google Scholar] [CrossRef]
- Pfeffer, S.; Dudek, J.; Schaffer, M.; Ng, B.G.; Albert, S.; Plitzko, J.M.; Baumeister, W.; Zimmermann, R.; Freeze, H.H.; Engel, B.D.; et al. Dissecting the molecular organization of the translocon-associated protein complex. Nat. Commun. 2017, 8, 14516. [Google Scholar] [CrossRef]
- Sanders, S.L.; Whitfield, K.M.; Vogel, J.P.; Rose, M.D.; Schekman, R.W. Sec61p and BiP directly facilitate polypeptide translocation into the ER. Cell 1992, 69, 353–365. [Google Scholar] [CrossRef] [PubMed]
- Daverkausen-Fischer, L.; Draga, M.; Pröls, F. Regulation of Translation, Translocation, and Degradation of Proteins at the Membrane of the Endoplasmic Reticulum. Int. J. Mol. Sci. 2022, 23, 5576. [Google Scholar] [CrossRef] [PubMed]
- Lyman, S.K.; Schekman, R. Binding of Secretory Precursor Polypeptides to a Translocon Subcomplex Is Regulated by BiP. Cell 1997, 88, 85–96. [Google Scholar] [CrossRef] [PubMed]
- Pfeffer, S.; Dudek, J.; Gogala, M.; Schorr, S.; Linxweiler, J.; Lang, S.; Becker, T.; Beckmann, R.; Zimmermann, R.; Förster, F. Structure of the mammalian oligosaccharyl-transferase complex in the native ER protein translocon. Nat. Commun. 2014, 5, 3072. [Google Scholar] [CrossRef]
- Plumb, R.; Zhang, Z.-R.; Appathurai, S.; Mariappan, M. A functional link between the co-translational protein translocation pathway and the UPR. eLife 2015, 4, e07426. [Google Scholar] [CrossRef]
- Hammond, C.; Braakman, I.; Helenius, A. Role of N-linked oligosaccharide recognition, glucose trimming, and calnexin in glycoprotein folding and quality control. Proc. Natl. Acad. Sci. USA 1994, 91, 913–917. [Google Scholar] [CrossRef]
- Okura, G.C.; Bharadwaj, A.G.; Waisman, D.M. Calreticulin-Enigmatic Discovery. Biomolecules 2024, 14, 866. [Google Scholar] [CrossRef]
- Zapun, A.; Darby, N.J.; Tessier, D.C.; Michalak, M.; Bergeron, J.J.M.; Thomas, D.Y. Enhanced Catalysis of Ribonuclease B Folding by the Interaction of Calnexin or Calreticulin with ERp57. J. Biol. Chem. 1998, 273, 6009–6012. [Google Scholar] [CrossRef]
- Braakman, I.; Hebert, D.N. Protein folding in the endoplasmic reticulum. Cold Spring Harb. Perspect. Biol. 2013, 5, a013201. [Google Scholar] [CrossRef] [PubMed]
- Helenius, A.; Aebi, M. Roles of N-Linked Glycans in the Endoplasmic Reticulum. Annu. Rev. Biochem. 2004, 73, 1019–1049. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.-X.; Braakman, I.; Matlack, K.E.; Helenius, A. Quality Control in the Secretory Pathway: The Role of Calreticulin, Calnexin and BiP in the Retention of Glycoproteins with C-Terminal Truncations. Mol. Biol. Cell 1997, 8, 1943–1954. [Google Scholar] [CrossRef]
- Caramelo, J.J.; Parodi, A.J. Getting In and Out from Calnexin/Calreticulin Cycles. J. Biol. Chem. 2008, 283, 10221–10225. [Google Scholar] [CrossRef]
- Ellgaard, L.; McCaul, N.; Chatsisvili, A.; Braakman, I. Co- and Post-Translational Protein Folding in the ER. Traffic 2016, 17, 615–638. [Google Scholar] [CrossRef] [PubMed]
- Ruggiano, A.; Foresti, O.; Carvalho, P. Quality control: ER-associated degradation: Protein quality control and beyond. J. Cell Biol. 2014, 204, 869–879. [Google Scholar] [CrossRef]
- McCracken, A.A.; Brodsky, J.L. Recognition and delivery of ERAD substrates to the proteasome and alternative paths for cell survival. Curr. Top. Microbiol. Immunol. 2005, 300, 17–40. [Google Scholar] [CrossRef]
- Lippincott-Schwartz, J.; Bonifacino, J.S.; Yuan, L.C.; Klausner, R.D. Degradation from the endoplasmic reticulum: Disposing of newly synthesized proteins. Cell 1988, 54, 209–220. [Google Scholar] [CrossRef]
- Bonifacino, J.S.; Cosson, P.; Klausner, R.D. Colocalized transmembrane determinants for ER degradation and subunit assembly explain the intracellular fate of TCR chains. Cell 1990, 63, 503–513. [Google Scholar] [CrossRef]
- Amaral, M.D. CFTR and Chaperones: Processing and Degradation. J. Mol. Neurosci. 2004, 23, 041–048. [Google Scholar] [CrossRef]
- Christianson, J.C.; Shaler, T.A.; Tyler, R.E.; Kopito, R.R. OS-9 and GRP94 deliver mutant α1-antitrypsin to the Hrd1–SEL1L ubiquitin ligase complex for ERAD. Nat. Cell Biol. 2008, 10, 272–282. [Google Scholar] [CrossRef] [PubMed]
- Carvalho, P.; Stanley, A.M.; Rapoport, T.A. Retrotranslocation of a misfolded luminal ER protein by the ubiquitin-ligase Hrd1p. Cell 2010, 143, 579–591. [Google Scholar] [CrossRef] [PubMed]
- Cormier, J.H.; Tamura, T.; Sunryd, J.C.; Hebert, D.N. EDEM1 Recognition and Delivery of Misfolded Proteins to the SEL1L-Containing ERAD Complex. Mol. Cell 2009, 34, 627–633. [Google Scholar] [CrossRef]
- Saeed, M.; Suzuki, R.; Watanabe, N.; Masaki, T.; Tomonaga, M.; Muhammad, A.; Kato, T.; Matsuura, Y.; Watanabe, H.; Wakita, T.; et al. Role of the Endoplasmic Reticulum-associated Degradation (ERAD) Pathway in Degradation of Hepatitis C Virus Envelope Proteins and Production of Virus Particles. J. Biol. Chem. 2011, 286, 37264–37273. [Google Scholar] [CrossRef] [PubMed]
- Riemer, J.; Appenzeller-Herzog, C.; Johansson, L.; Bodenmiller, B.; Hartmann-Petersen, R.; Ellgaard, L. A luminal flavoprotein in endoplasmic reticulum-associated degradation. Proc. Natl. Acad. Sci. USA 2009, 106, 14831–14836. [Google Scholar] [CrossRef]
- Hosokawa, N.; Wada, I.; Nagasawa, K.; Moriyama, T.; Okawa, K.; Nagata, K. Human XTP3-B Forms an Endoplasmic Reticulum Quality Control Scaffold with the HRD1-SEL1L Ubiquitin Ligase Complex and BiP. J. Biol. Chem. 2008, 283, 20914–20924. [Google Scholar] [CrossRef]
- Mueller, B.; Lilley, B.N.; Ploegh, H.L. SEL1L, the homologue of yeast Hrd3p, is involved in protein dislocation from the mammalian ER. J. Cell Biol. 2006, 175, 261–270. [Google Scholar] [CrossRef]
- Mueller, B.; Klemm, E.J.; Spooner, E.; Claessen, J.H.; Ploegh, H.L. SEL1L nucleates a protein complex required for dislocation of misfolded glycoproteins. Proc. Natl. Acad. Sci. USA 2008, 105, 12325–12330. [Google Scholar] [CrossRef]
- Iida, Y.; Fujimori, T.; Okawa, K.; Nagata, K.; Wada, I.; Hosokawa, N. SEL1L Protein Critically Determines the Stability of the HRD1-SEL1L Endoplasmic Reticulum-associated Degradation (ERAD) Complex to Optimize the Degradation Kinetics of ERAD Substrates. J. Biol. Chem. 2011, 286, 16929–16939. [Google Scholar] [CrossRef]
- Ye, Y.; Shibata, Y.; Yun, C.; Ron, D.; Rapoport, T.A. A membrane protein complex mediates retro-translocation from the ER lumen into the cytosol. Nature 2004, 429, 841–847. [Google Scholar] [CrossRef]
- Loureiro, J.; Lilley, B.N.; Spooner, E.; Noriega, V.; Tortorella, D.; Ploegh, H.L. Signal peptide peptidase is required for dislocation from the endoplasmic reticulum. Nature 2006, 441, 894–897. [Google Scholar] [CrossRef] [PubMed]
- Scott, D.C.; Schekman, R. Role of Sec61p in the ER-associated degradation of short-lived transmembrane proteins. J. Cell Biol. 2008, 181, 1095–1105. [Google Scholar] [CrossRef]
- Vasic, V.; Denkert, N.; Schmidt, C.C.; Riedel, D.; Stein, A.; Meinecke, M. Hrd1 forms the retrotranslocation pore regulated by auto-ubiquitination and binding of misfolded proteins. Nat. Cell Biol. 2020, 22, 274–281. [Google Scholar] [CrossRef]
- Ye, Y.; Meyer, H.H.; Rapoport, T.A. The AAA ATPase Cdc48/p97 and its partners transport proteins from the ER into the cytosol. Nature 2001, 414, 652–656. [Google Scholar] [CrossRef]
- Alzayady, K.J.; Panning, M.M.; Kelley, G.G.; Wojcikiewicz, J.R. Involvement of the p97-Ufd1-Npl4 Complex in the Regulated Endoplasmic Reticulum-associated Degradation of Inositol 1,4,5-Trisphosphate Receptors. J. Biol. Chem. 2005, 280, 34530–34537. [Google Scholar] [CrossRef]
- Greenblatt, E.J.; Olzmann, J.A.; Kopito, R.R. Derlin-1 is a rhomboid pseudoprotease required for the dislocation of mutant α-1 antitrypsin from the endoplasmic reticulum. Nat. Struct. Mol. Biol. 2011, 18, 1147–1152. [Google Scholar] [CrossRef]
- Cao, J.; Wang, J.; Qi, W.; Miao, H.-H.; Ge, L.; DeBose-Boyd, R.A.; Tang, J.-J.; Li, B.-L.; Song, B.-L. Ufd1 Is a Cofactor of gp78 and Plays a Key Role in Cholesterol Metabolism by Regulating the Stability of HMG-CoA Reductase. Cell Metab. 2007, 6, 115–128. [Google Scholar] [CrossRef] [PubMed]
- Ballar, P.; Shen, Y.; Yang, H.; Fang, S. The role of a novel p97/valosin-containing protein-interacting motif of gp78 in endoplasmic reticulum-associated degradation. J. Biol. Chem. 2006, 281, 35359–35368. [Google Scholar] [CrossRef] [PubMed]
- Preston, G.M.; Brodsky, J.L. The evolving role of ubiquitin modification in endoplasmic reticulum-associated degradation. Biochem. J. 2017, 474, 445–469. [Google Scholar] [CrossRef]
- Cao, H.; Zhou, X.; Xu, B.; Hu, H.; Guo, J.; Ma, Y.; Wang, M.; Li, N.; Jun, Z. Advances in the study of protein folding and endoplasmic reticulum-associated degradation in mammal cells. J. Zhejiang Univ. Sci. B 2024, 25, 212–232. [Google Scholar] [CrossRef]
- Borgo, C.; D’amore, C.; Capurro, V.; Tomati, V.; Sondo, E.; Cresta, F.; Castellani, C.; Pedemonte, N.; Salvi, M. Targeting the E1 ubiquitin-activating enzyme (UBA1) improves elexacaftor/tezacaftor/ivacaftor efficacy towards F508del and rare misfolded CFTR mutants. Cell. Mol. Life Sci. 2022, 79, 192. [Google Scholar] [CrossRef]
- Eisele, M.R.; Reed, R.G.; Rudack, T.; Schweitzer, A.; Beck, F.; Nagy, I.; Pfeifer, G.; Plitzko, J.M.; Baumeister, W.; Tomko, R.J.; et al. Expanded Coverage of the 26S Proteasome Conformational Landscape Reveals Mechanisms of Peptidase Gating. Cell Rep. 2018, 24, 1301–1315.e5. [Google Scholar] [CrossRef] [PubMed]
- Davis, C.; Spaller, B.L.; Matouschek, A. Mechanisms of substrate recognition by the 26S proteasome. Curr. Opin. Struct. Biol. 2020, 67, 161–169. [Google Scholar] [CrossRef]
- I Bannykh, S.; Rowe, T.; E Balch, W. The organization of endoplasmic reticulum export complexes. J. Cell Biol. 1996, 135, 19–35. [Google Scholar] [CrossRef] [PubMed]
- Barlowe, C.; Orci, L.; Yeung, T.; Hosobuchi, M.; Hamamoto, S.; Salama, N.; Rexach, M.F.; Ravazzola, M.; Amherdt, M.; Schekman, R. COPII: A membrane coat formed by Sec proteins that drive vesicle budding from the endoplasmic reticulum. Cell 1994, 77, 895–907. [Google Scholar] [CrossRef]
- Bielli, A.; Haney, C.J.; Gabreski, G.; Watkins, S.C.; Bannykh, S.I.; Aridor, M. Regulation of Sar1 NH2 terminus by GTP binding and hydrolysis promotes membrane deformation to control COPII vesicle fission. J. Cell Biol. 2005, 171, 919–924. [Google Scholar] [CrossRef] [PubMed]
- Yoshihisa, T.; Barlowe, C.; Schekman, R. Requirement for a GTPase-Activating Protein in Vesicle Budding from the Endoplasmic Reticulum. Science 1993, 259, 1466–1468. [Google Scholar] [CrossRef]
- Long, K.R.; Yamamoto, Y.; Baker, A.L.; Watkins, S.C.; Coyne, C.B.; Conway, J.F.; Aridor, M. Sar1 assembly regulates membrane constriction and ER export. J. Cell Biol. 2010, 190, 115–128. [Google Scholar] [CrossRef]
- Stagg, S.M.; LaPointe, P.; Razvi, A.; Gürkan, C.; Potter, C.S.; Carragher, B.; Balch, W.E. Structural Basis for Cargo Regulation of COPII Coat Assembly. Cell 2008, 134, 474–484. [Google Scholar] [CrossRef]
- Lee, M.C.; Orci, L.; Hamamoto, S.; Futai, E.; Ravazzola, M.; Schekman, R. Sar1p N-terminal helix initiates membrane curvature and completes the fission of a COPII vesicle. Cell 2005, 122, 605–617. [Google Scholar] [CrossRef]
- Lord, C.; Bhandari, D.; Menon, S.; Ghassemian, M.; Nycz, D.; Hay, J.; Ghosh, P.; Ferro-Novick, S. Sequential interactions with Sec23 control the direction of vesicle traffic. Nature 2011, 473, 181–186. [Google Scholar] [CrossRef] [PubMed]
- Amaral, M.D.; Farinha, C.M.; Matos, P.; Botelho, H.M. Investigating Alternative Transport of Integral Plasma Membrane Proteins from the ER to the Golgi: Lessons from the Cystic Fibrosis Transmembrane Conductance Regulator (CFTR). Methods Mol. Biol. 2016, 1459, 105–126. [Google Scholar] [CrossRef]
- Lewis, H.A.; Zhao, X.; Wang, C.; Sauder, J.M.; Rooney, I.; Noland, B.W.; Lorimer, D.; Kearins, M.C.; Conners, K.; Condon, B.; et al. Impact of the ΔF508 Mutation in First Nucleotide-binding Domain of Human Cystic Fibrosis Transmembrane Conductance Regulator on Domain Folding and Structure. J. Biol. Chem. 2005, 280, 1346–1353. [Google Scholar] [CrossRef]
- Scott-Ward, T.S.; Amaral, M.D. Deletion of Phe508 in the first nucleotide-binding domain of the cystic fibrosis transmembrane conductance regulator increases its affinity for the heat shock cognate 70 chaperone. FEBS J. 2009, 276, 7097–7109. [Google Scholar] [CrossRef] [PubMed]
- He, L.; Aleksandrov, A.A.; An, J.; Cui, L.; Yang, Z.; Brouillette, C.G.; Riordan, J.R. Restoration of NBD1 Thermal Stability Is Necessary and Sufficient to Correct ∆F508 CFTR Folding and Assembly. J. Mol. Biol. 2014, 427, 106–120. [Google Scholar] [CrossRef] [PubMed]
- Hoelen, H.; Kleizen, B.; Schmidt, A.; Richardson, J.; Charitou, P.; Thomas, P.J.; Braakman, I. The Primary Folding Defect and Rescue of ΔF508 CFTR Emerge during Translation of the Mutant Domain. PLoS ONE 2010, 5, e15458. [Google Scholar] [CrossRef]
- Yang, Y.; Janich, S.; A Cohn, J.; Wilson, J.M. The common variant of cystic fibrosis transmembrane conductance regulator is recognized by hsp70 and degraded in a pre-Golgi nonlysosomal compartment. Proc. Natl. Acad. Sci. USA 1993, 90, 9480–9484. [Google Scholar] [CrossRef]
- Wang, X.; Venable, J.; LaPointe, P.; Hutt, D.M.; Koulov, A.V.; Coppinger, J.; Gurkan, C.; Kellner, W.; Matteson, J.; Plutner, H.; et al. Hsp90 Cochaperone Aha1 Downregulation Rescues Misfolding of CFTR in Cystic Fibrosis. Cell 2006, 127, 803–815. [Google Scholar] [CrossRef]
- Michelsen, K.; Yuan, H.; Schwappach, B. Hide and run. Arginine-based endoplasmic-reticulum-sorting motifs in the assembly of heteromultimeric membrane proteins. EMBO Rep. 2005, 6, 717–722. [Google Scholar] [CrossRef]
- Chang, X.-B.; Cui, L.; Hou, Y.-X.; Jensen, T.J.; Aleksandrov, A.A.; Mengos, A.; Riordan, J.R. Removal of Multiple Arginine-Framed Trafficking Signals Overcomes Misprocessing of ΔF508 CFTR Present in Most Patients with Cystic Fibrosis. Mol. Cell 1999, 4, 137–142. [Google Scholar] [CrossRef]
- Gentzsch, M.; Chang, X.-B.; Cui, L.; Wu, Y.; Ozols, V.V.; Choudhury, A.; Pagano, R.E.; Riordan, J.R. Endocytic Trafficking Routes of Wild Type and ΔF508 Cystic Fibrosis Transmembrane Conductance Regulator. Mol. Biol. Cell 2004, 15, 2684–2696. [Google Scholar] [CrossRef] [PubMed]
- Sharma, M.; Pampinella, F.; Nemes, C.; Benharouga, M.; So, J.; Du, K.; Bache, K.G.; Papsin, B.; Zerangue, N.; Stenmark, H.; et al. Misfolding diverts CFTR from recycling to degradation: Quality control at early endosomes. J. Cell Biol. 2004, 164, 923–933. [Google Scholar] [CrossRef]
- Loureiro, C.A.; Matos, A.M.; Dias-Alves, Â.; Pereira, J.F.; Uliyakina, I.; Barros, P.; Amaral, M.D.; Matos, P. A molecular switch in the scaffold NHERF1 enables misfolded CFTR to evade the peripheral quality control checkpoint. Sci. Signal. 2015, 8, ra48. [Google Scholar] [CrossRef] [PubMed]
- Cushing, P.R.; Vouilleme, L.; Pellegrini, M.; Boisguerin, P.; Madden, D.R. A Stabilizing Influence: CAL PDZ Inhibition Extends the Half-Life of ΔF508-CFTR. Angew. Chem. 2010, 122, 10103–10107. [Google Scholar] [CrossRef]
- Gee, H.Y.; Kim, J.; Lee, M.G. Unconventional secretion of transmembrane proteins. Semin. Cell Dev. Biol. 2018, 83, 59–66. [Google Scholar] [CrossRef]
- Cleves, A.E. Protein transports: The nonclassical ins and outs. Curr. Biol. 1997, 7, R318–R320. [Google Scholar] [CrossRef]
- Nickel, W. The mystery of nonclassical protein secretion. A current view on cargo proteins and potential export routes. Eur. J. Biochem. 2003, 270, 2109–2119. [Google Scholar] [CrossRef]
- Nickel, W. Unconventional Secretory Routes: Direct Protein Export Across the Plasma Membrane of Mammalian Cells. Traffic 2005, 6, 607–614. [Google Scholar] [CrossRef] [PubMed]
- Yoo, J.-S.; Moyer, B.D.; Bannykh, S.; Yoo, H.-M.; Riordan, J.R.; Balch, W.E. Non-conventional Trafficking of the Cystic Fibrosis Transmembrane Conductance Regulator through the Early Secretory Pathway. J. Biol. Chem. 2002, 277, 11401–11409. [Google Scholar] [CrossRef]
- Wu, H.; Li, T.; Zhao, J. GRASP55: A Multifunctional Protein. Curr. Protein Pept. Sci. 2020, 21, 544–552. [Google Scholar] [CrossRef]
- Mendes, L.F.; Fontana, N.A.; Reddy, S.T.; Uversky, V.N.; Costa-Filho, A.J. The exquisite structural biophysics of the Golgi Reassembly and Stacking Proteins. Int. J. Biol. Macromol. 2020, 164, 3632–3644. [Google Scholar] [CrossRef] [PubMed]
- Rabouille, C.; Linstedt, A.D. GRASP: A Multitasking Tether. Front. Cell Dev. Biol. 2016, 4, 1. [Google Scholar] [CrossRef]
- Grieve, A.G.; Rabouille, C. Golgi Bypass: Skirting Around the Heart of Classical Secretion. Cold Spring Harb. Perspect. Biol. 2011, 3, a005298. [Google Scholar] [CrossRef]
- Ahat, E.; Bui, S.; Zhang, J.; Leprevost, F.d.V.; Sharkey, L.; Reid, W.; Nesvizhskii, A.I.; Paulson, H.L.; Wang, Y. GRASP55 regulates the unconventional secretion and aggregation of mutant huntingtin. J. Biol. Chem. 2022, 298, 102219. [Google Scholar] [CrossRef]
- Ponpuak, M.; Mandell, M.A.; Kimura, T.; Chauhan, S.; Cleyrat, C.; Deretic, V. Secretory autophagy. Curr. Opin. Cell Biol. 2015, 35, 106–116. [Google Scholar] [CrossRef]
- Jiang, S.; Dupont, N.; Castillo, E.F.; Deretic, V. Secretory versus Degradative Autophagy: Unconventional Secretion of Inflammatory Mediators. J. Innate Immun. 2013, 5, 471–479. [Google Scholar] [CrossRef] [PubMed]
- Dupont, N.; Jiang, S.; Pilli, M.; Ornatowski, W.; Bhattacharya, D.; Deretic, V. Autophagy-based unconventional secretory pathway for extracellular delivery of IL-1β. EMBO J. 2011, 30, 4701–4711. [Google Scholar] [CrossRef]
- Duran, J.M.; Anjard, C.; Stefan, C.; Loomis, W.F.; Malhotra, V. Unconventional secretion of Acb1 is mediated by autophagosomes. J. Cell Biol. 2010, 188, 527–536. [Google Scholar] [CrossRef] [PubMed]
- Cuervo, A.M.; Wong, E. Chaperone-mediated autophagy: Roles in disease and aging. Cell Res. 2013, 24, 92–104. [Google Scholar] [CrossRef]
- Johansen, T.; Lamark, T. Selective Autophagy: ATG8 Family Proteins, LIR Motifs and Cargo Receptors. J. Mol. Biol. 2020, 432, 80–103. [Google Scholar] [CrossRef]
- Kirkin, V.; McEwan, D.G.; Novak, I.; Dikic, I. A Role for Ubiquitin in Selective Autophagy. Mol. Cell 2009, 34, 259–269. [Google Scholar] [CrossRef] [PubMed]
- Shorter, J.; Watson, R.; Giannakou, M.; Clarke, M.; Warren, G.; Barr, F.A. GRASP55, a second mammalian GRASP protein involved in the stacking of Golgi cisternae in a cell-free system. EMBO J. 1999, 18, 4949–4960. [Google Scholar] [CrossRef]
- Figueras-Novoa, C.; Timimi, L.; Marcassa, E.; Ulferts, R.; Beale, R. Conjugation of ATG8s to single membranes at a glance. J. Cell Sci. 2024, 137, jcs261031. [Google Scholar] [CrossRef] [PubMed]
- Zhen, Y.; Stenmark, H. Autophagosome Biogenesis. Cells 2023, 12, 668. [Google Scholar] [CrossRef]
- Valverde, D.P.; Yu, S.; Boggavarapu, V.; Kumar, N.; Lees, J.A.; Walz, T.; Reinisch, K.M.; Melia, T.J. ATG2 transports lipids to promote autophagosome biogenesis. J. Cell Biol. 2019, 218, 1787–1798. [Google Scholar] [CrossRef] [PubMed]
- Matoba, K.; Kotani, T.; Tsutsumi, A.; Tsuji, T.; Mori, T.; Noshiro, D.; Sugita, Y.; Nomura, N.; Iwata, S.; Ohsumi, Y.; et al. Atg9 is a lipid scramblase that mediates autophagosomal membrane expansion. Nat. Struct. Mol. Biol. 2020, 27, 1185–1193. [Google Scholar] [CrossRef]
- van Vliet, A.R.; Chiduza, G.N.; Maslen, S.L.; Pye, V.E.; Joshi, D.; De Tito, S.; Jefferies, H.B.; Christodoulou, E.; Roustan, C.; Punch, E.; et al. ATG9A and ATG2A form a heteromeric complex essential for autophagosome formation. Mol. Cell 2022, 82, 4324–4339. [Google Scholar] [CrossRef]
- Cheong, H.; Lindsten, T.; Wu, J.; Lu, C.; Thompson, C.B. Ammonia-induced autophagy is independent of ULK1/ULK2 kinases. Proc. Natl. Acad. Sci. USA 2011, 108, 11121–11126. [Google Scholar] [CrossRef]
- Lin, M.G.; Hurley, J.H. Structure and function of the ULK1 complex in autophagy. Curr. Opin. Cell Biol. 2016, 39, 61–68. [Google Scholar] [CrossRef]
- Russell, R.C.; Tian, Y.; Yuan, H.; Park, H.W.; Chang, Y.-Y.; Kim, J.; Kim, H.; Neufeld, T.P.; Dillin, A.; Guan, K.-L. ULK1 induces autophagy by phosphorylating Beclin-1 and activating VPS34 lipid kinase. Nat. Cell Biol. 2013, 15, 741–750. [Google Scholar] [CrossRef]
- Rogov, V.; Dötsch, V.; Johansen, T.; Kirkin, V. Interactions between Autophagy Receptors and Ubiquitin-like Proteins Form the Molecular Basis for Selective Autophagy. Mol. Cell 2014, 53, 167–178. [Google Scholar] [CrossRef] [PubMed]
- Bjørkøy, G.; Lamark, T.; Brech, A.; Outzen, H.; Perander, M.; Overvatn, A.; Stenmark, H.; Johansen, T. p62/SQSTM1 forms protein aggregates degraded by autophagy and has a protective effect on huntingtin-induced cell death. J. Cell Biol. 2005, 171, 603–614. [Google Scholar] [CrossRef]
- Pankiv, S.; Clausen, T.H.; Lamark, T.; Brech, A.; Bruun, J.-A.; Outzen, H.; Øvervatn, A.; Bjørkøy, G.; Johansen, T. p62/SQSTM1 Binds Directly to Atg8/LC3 to Facilitate Degradation of Ubiquitinated Protein Aggregates by Autophagy. J. Biol. Chem. 2007, 282, 24131–24145. [Google Scholar] [CrossRef] [PubMed]
- Johansen, T.; Lamark, T. Selective autophagy mediated by autophagic adapter proteins. Autophagy 2011, 7, 279–296. [Google Scholar] [CrossRef]
- Brunger, A.T. Structure and function of SNARE and SNARE-interacting proteins. Q. Rev. Biophys. 2005, 38, 1–47. [Google Scholar] [CrossRef] [PubMed]
- Urano, F.; Bertolotti, A.; Ron, D. IRE1 and efferent signaling from the endoplasmic reticulum. J. Cell Sci. 2000, 113, 3697–3702. [Google Scholar] [CrossRef]
- Yoshida, H.; Matsui, T.; Yamamoto, A.; Okada, T.; Mori, K. XBP1 mRNA Is Induced by ATF6 and Spliced by IRE1 in Response to ER Stress to Produce a Highly Active Transcription Factor. Cell 2001, 107, 881–891. [Google Scholar] [CrossRef]
- Calfon, M.; Zeng, H.; Urano, F.; Till, J.H.; Hubbard, S.R.; Harding, H.P.; Clark, S.G.; Ron, D. IRE1 couples endoplasmic reticulum load to secretory capacity by processing the XBP-1 mRNA. Nature 2002, 415, 92–96. [Google Scholar] [CrossRef]
- Ribeiro, C.M.P.; Lubamba, B.A. Role of IRE1α/XBP-1 in Cystic Fibrosis Airway Inflammation. Int. J. Mol. Sci. 2017, 18, 118. [Google Scholar] [CrossRef]
- Sriburi, R.; Jackowski, S.; Mori, K.; Brewer, J.W. XBP1: A link between the unfolded protein response, lipid biosynthesis, and biogenesis of the endoplasmic reticulum. J. Cell Biol. 2004, 167, 35–41. [Google Scholar] [CrossRef]
- Vega-Rubín-De-Celis, S. The Role of Beclin 1-Dependent Autophagy in Cancer. Biology 2019, 9, 4. [Google Scholar] [CrossRef]
- Margariti, A.; Li, H.; Chen, T.; Martin, D.; Vizcay-Barrena, G.; Alam, S.; Karamariti, E.; Xiao, Q.; Zampetaki, A.; Zhang, Z.; et al. XBP1 mRNA Splicing Triggers an Autophagic Response in Endothelial Cells through BECLIN-1 Transcriptional Activation. J. Biol. Chem. 2013, 288, 859–872. [Google Scholar] [CrossRef]
- Yang, Q.; Kim, Y.; Lin, Y.; Lewis, J.; Neckers, L.; Liu, Z. Tumour necrosis factor receptor 1 mediates endoplasmic reticulum stress-induced activation of the MAP kinase JNK. Embo Rep. 2006, 7, 622–627. [Google Scholar] [CrossRef] [PubMed]
- Wei, Y.; Pattingre, S.; Sinha, S.; Bassik, M.; Levine, B. JNK1-Mediated Phosphorylation of Bcl-2 Regulates Starvation-Induced Autophagy. Mol. Cell 2008, 30, 678–688. [Google Scholar] [CrossRef]
- Kuo, C.; You, G.; Jian, Y.; Chen, T.; Siao, Y.; Hsu, A.; Ching, T. AMPK-mediated formation of stress granules is required for dietary restriction-induced longevity in Caenorhabditis elegans. Aging Cell 2020, 19, e13157. [Google Scholar] [CrossRef] [PubMed]
- Kouroku, Y.; Fujita, E.; Tanida, I.; Ueno, T.; Isoai, A.; Kumagai, H.; Ogawa, S.; Kaufman, R.J.; Kominami, E.; Momoi, T. ER stress (PERK/eIF2alpha phosphorylation) mediates the polyglutamine-induced LC3 conversion, an essential step for autophagy formation. Cell Death Differ. 2007, 14, 230–239. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Kang, R.; Huang, H.; Xi, X.; Wang, B.; Wang, J.; Zhao, Z. Hepatitis C virus core protein activates autophagy through EIF2AK3 and ATF6 UPR pathway-mediated MAP1LC3B and ATG12 expression. Autophagy 2014, 10, 766–784. [Google Scholar] [CrossRef]
- Rzymski, T.; Milani, M.; Pike, L.; Buffa, F.; Mellor, H.R.; Winchester, L.; Pires, I.; Hammond, E.; Ragoussis, I.; Harris, A.L. Regulation of autophagy by ATF4 in response to severe hypoxia. Oncogene 2010, 29, 4424–4435. [Google Scholar] [CrossRef]
- B’chir, W.; Maurin, A.-C.; Carraro, V.; Averous, J.; Jousse, C.; Muranishi, Y.; Parry, L.; Stepien, G.; Fafournoux, P.; Bruhat, A. The eIF2α/ATF4 pathway is essential for stress-induced autophagy gene expression. Nucleic Acids Res. 2013, 41, 7683–7699. [Google Scholar] [CrossRef]
- Ye, J.; Rawson, R.B.; Komuro, R.; Chen, X.; Davé, U.P.; Prywes, R.; Brown, M.S.; Goldstein, J.L. ER Stress Induces Cleavage of Membrane-Bound ATF6 by the Same Proteases that Process SREBPs. Mol. Cell 2000, 6, 1355–1364. [Google Scholar] [CrossRef]
- Adachi, Y.; Yamamoto, K.; Okada, T.; Yoshida, H.; Harada, A.; Mori, K. ATF6 Is a Transcription Factor Specializing in the Regulation of Quality Control Proteins in the Endoplasmic Reticulum. Cell Struct. Funct. 2008, 33, 75–89. [Google Scholar] [CrossRef]
- Bommiasamy, H.; Back, S.H.; Fagone, P.; Lee, K.; Meshinchi, S.; Vink, E.; Sriburi, R.; Frank, M.; Jackowski, S.; Kaufman, R.J.; et al. ATF6α induces XBP1-independent expansion of the endoplasmic reticulum. J. Cell Sci. 2009, 122, 1626–1636. [Google Scholar] [CrossRef]
- Nanua, S.; Sajjan, U.; Keshavjee, S.; Hershenson, M.B. Absence of typical unfolded protein response in primary cultured cystic fibrosis airway epithelial cells. Biochem. Biophys. Res. Commun. 2006, 343, 135–143. [Google Scholar] [CrossRef]
- Bartoszewski, R.; Rab, A.; Jurkuvenaite, A.; Mazur, M.; Wakefield, J.; Collawn, J.F.; Bebők, Z. Activation of the Unfolded Protein Response by ΔF508 CFTR. Am. J. Respir. Cell Mol. Biol. 2008, 39, 448–457. [Google Scholar] [CrossRef]
- Kerbiriou, M.; Le Drévo, M.-A.; Férec, C.; Trouvé, P. Coupling cystic fibrosis to endoplasmic reticulum stress: Differential role of Grp78 and ATF6. Biochim. Biophys. Acta (BBA)—Mol. Basis Dis. 2007, 1772, 1236–1249. [Google Scholar] [CrossRef] [PubMed]
- Hull-Ryde, E.A.; Minges, J.T.; Martino, M.E.B.; Kato, T.; Norris-Drouin, J.L.; Ribeiro, C.M.P. IRE1α Is a Therapeutic Target for Cystic Fibrosis Airway Inflammation. Int. J. Mol. Sci. 2021, 22, 3063. [Google Scholar] [CrossRef]
- Ribeiro, C.M.P.; Boucher, R.C. Role of Endoplasmic Reticulum Stress in Cystic Fibrosis-Related Airway Inflammatory Responses. Proc. Am. Thorac. Soc. 2010, 7, 387–394. [Google Scholar] [CrossRef] [PubMed]
- Bakunts, A.; Orsi, A.; Vitale, M.; Cattaneo, A.; Lari, F.; Tadè, L.; Sitia, R.; Raimondi, A.; Bachi, A.; van Anken, E. Ratiometric sensing of BiP-client versus BiP levels by the unfolded protein response determines its signaling amplitude. eLife 2017, 6, e27518. [Google Scholar] [CrossRef] [PubMed]
- Vitale, M.; Bakunts, A.; Orsi, A.; Lari, F.; Tadè, L.; Danieli, A.; Rato, C.; Valetti, C.; Sitia, R.; Raimondi, A.; et al. Author response: Inadequate BiP availability defines endoplasmic reticulum stress. Elife 2019, 8, e41168. [Google Scholar] [CrossRef]
- Zhang, M.; Kenny, S.J.; Ge, L.; Xu, K.; Schekman, R.; Berkeley; States, U. Translocation of interleukin-1β into a vesicle intermediate in autophagy-mediated secretion. eLife 2015, 4, e11205. [Google Scholar] [CrossRef]
- Son, S.M.; Cha, M.-Y.; Choi, H.; Kang, S.; Choi, H.; Lee, M.-S.; Park, S.A.; Mook-Jung, I. Insulin-degrading enzyme secretion from astrocytes is mediated by an autophagy-based unconventional secretory pathway in Alzheimer disease. Autophagy 2016, 12, 784–800. [Google Scholar] [CrossRef] [PubMed]
- Lefebvre, C.; Legouis, R.; Culetto, E. ESCRT and autophagies: Endosomal functions and beyond. Semin. Cell Dev. Biol. 2018, 74, 21–28. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Chen, S.; Ammar, A.-B.; Xu, J.; Wu, Q.; Pan, K.; Zhang, J.; Hong, Y. Crosstalk Between Macroautophagy and Chaperone-Mediated Autophagy: Implications for the Treatment of Neurological Diseases. Mol. Neurobiol. 2014, 52, 1284–1296. [Google Scholar] [CrossRef] [PubMed]
- Tedeschi, V.; Sapienza, S.; Ciancio, R.; Canzoniero, L.M.T.; Pannaccione, A.; Secondo, A. Lysosomal Channels as New Molecular Targets in the Pharmacological Therapy of Neurodegenerative Diseases via Autophagy Regulation. Curr. Neuropharmacol. 2025, 23, 375–383. [Google Scholar] [CrossRef]
- Khan, S.; Upadhyay, S.; Hassan, I. Novel prospects in targeting neurodegenerative disorders via autophagy. Eur. J. Pharmacol. 2024, 984, 177060. [Google Scholar] [CrossRef]
- Hoffmeister, H.; Babinger, K.; Gürster, S.; Cedzich, A.; Meese, C.; Schadendorf, K.; Osten, L.; de Vries, U.; Rascle, A.; Witzgall, R. Polycystin-2 takes different routes to the somatic and ciliary plasma membrane. J. Cell Biol. 2011, 192, 631–645. [Google Scholar] [CrossRef]
- Witzgall, R. Golgi bypass of ciliary proteins. Semin. Cell Dev. Biol. 2018, 83, 51–58. [Google Scholar] [CrossRef]
- Maejima, Y.; Isobe, M.; Sadoshima, J. Regulation of autophagy by Beclin 1 in the heart. J. Mol. Cell. Cardiol. 2016, 95, 19–25. [Google Scholar] [CrossRef]
- Yue, Z.; Jin, S.; Yang, C.; Levine, A.J.; Heintz, N. Beclin 1, an autophagy gene essential for early embryonic development, is a haploinsufficient tumor suppressor. Proc. Natl. Acad. Sci. USA 2003, 100, 15077–15082. [Google Scholar] [CrossRef]
- Liang, X.H.; Jackson, S.; Seaman, M.; Brown, K.; Kempkes, B.; Hibshoosh, H.; Levine, B. nduction of autophagy and inhibition of tumorigenesis by beclin 1. Nature 1999, 402, 672–676. [Google Scholar] [CrossRef]
- Claude-Taupin, A.; Fonderflick, L.; Gauthier, T.; Mansi, L.; Pallandre, J.-R.; Borg, C.; Perez, V.; Monnien, F.; Algros, M.-P.; Vigneron, M.; et al. ATG9A Is Overexpressed in Triple Negative Breast Cancer and Its In Vitro Extinction Leads to the Inhibition of Pro-Cancer Phenotypes. Cells 2018, 7, 248. [Google Scholar] [CrossRef] [PubMed]
- Tang, X.; Walter, E.; Wohleb, E.; Fan, Y.; Wang, C. ATG5 (autophagy related 5) in microglia controls hippocampal neurogenesis in Alzheimer disease. Autophagy 2023, 20, 847–862. [Google Scholar] [CrossRef] [PubMed]
- Barrientos-Riosalido, A.; Real, M.; Bertran, L.; Aguilar, C.; Martínez, S.; Parada, D.; Vives, M.; Sabench, F.; Riesco, D.; Del Castillo, D.; et al. Increased Hepatic ATG7 mRNA and ATG7 Protein Expression in Nonalcoholic Steatohepatitis Associated with Obesity. Int. J. Mol. Sci. 2023, 24, 1324. [Google Scholar] [CrossRef] [PubMed]
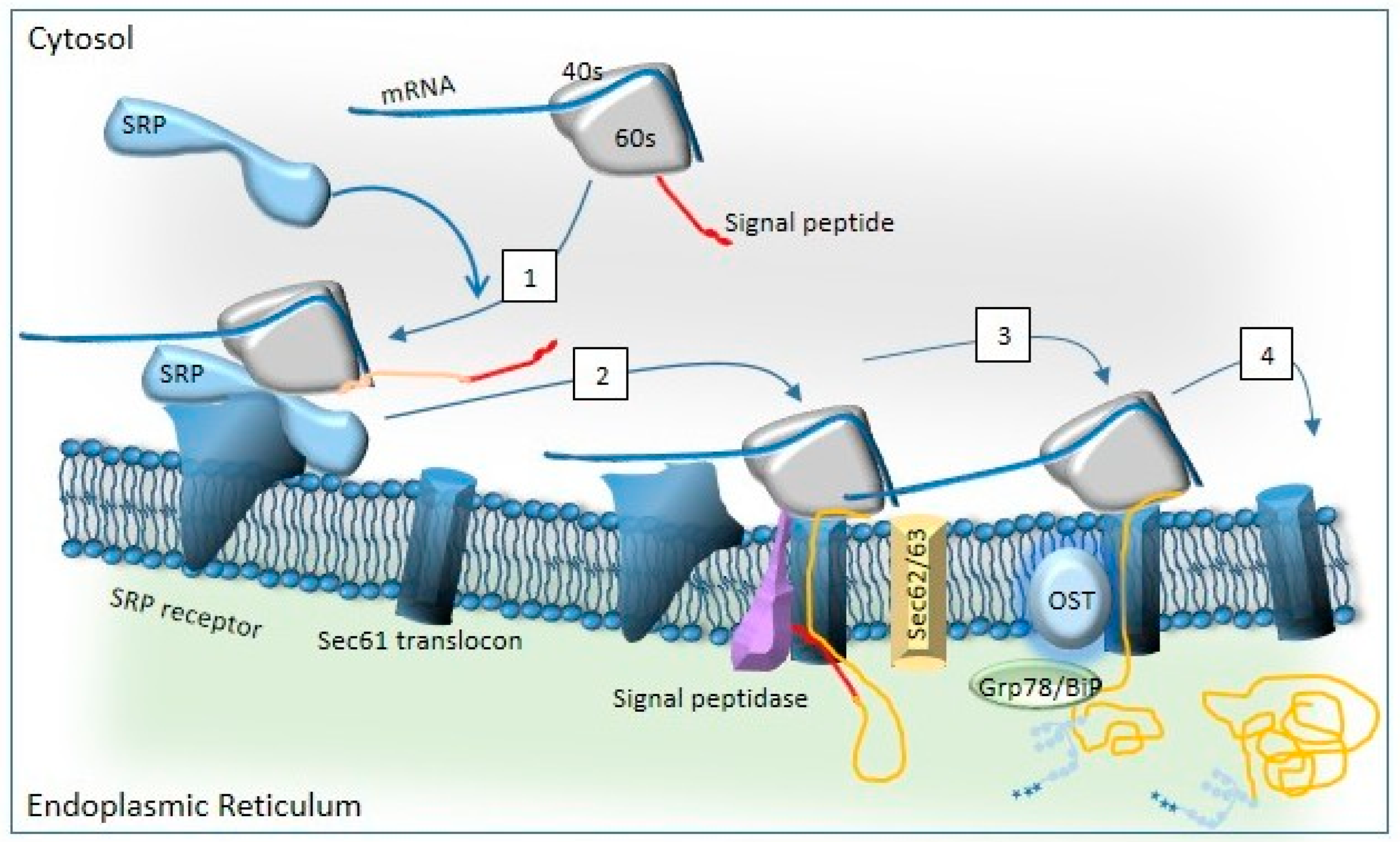
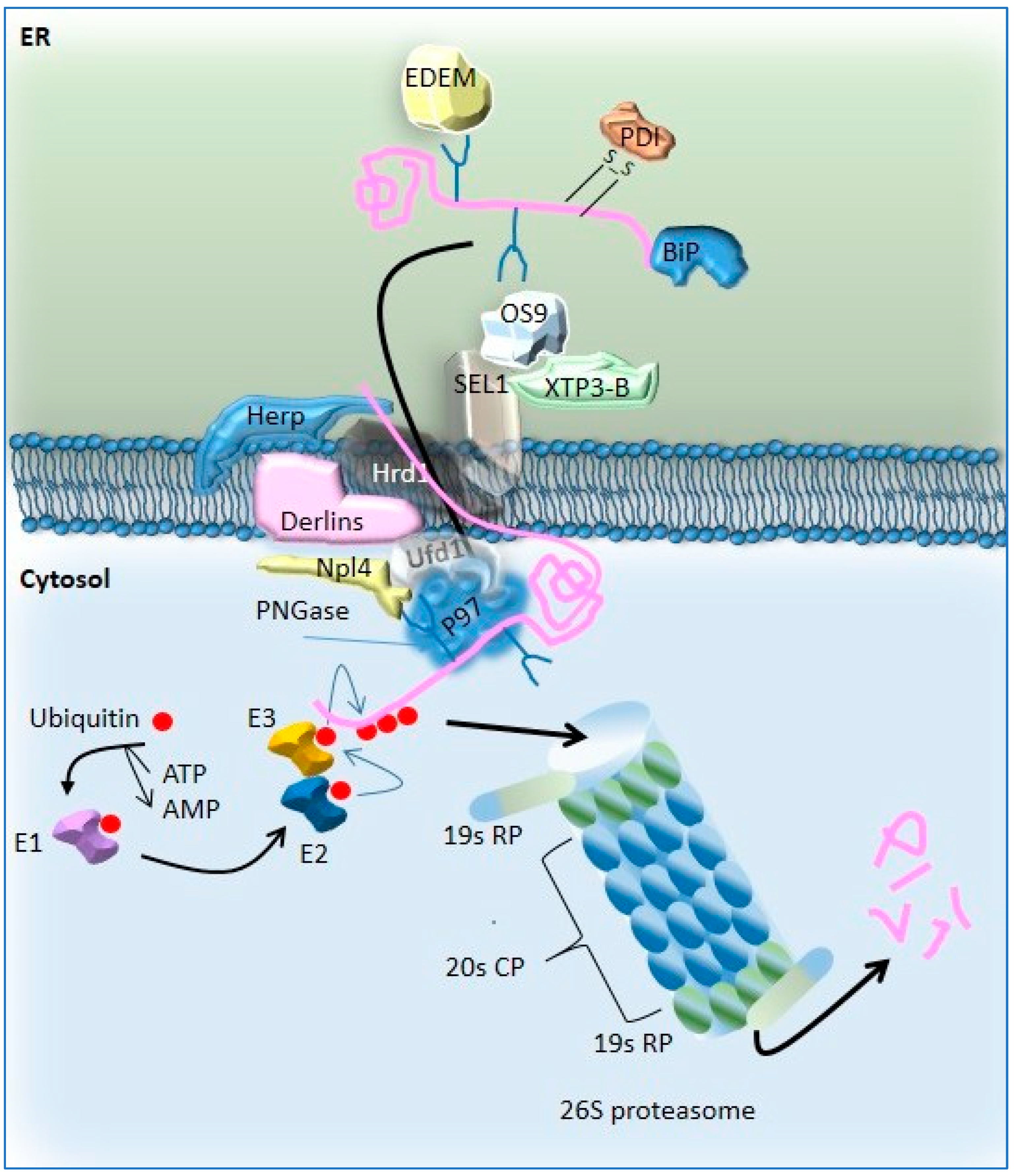

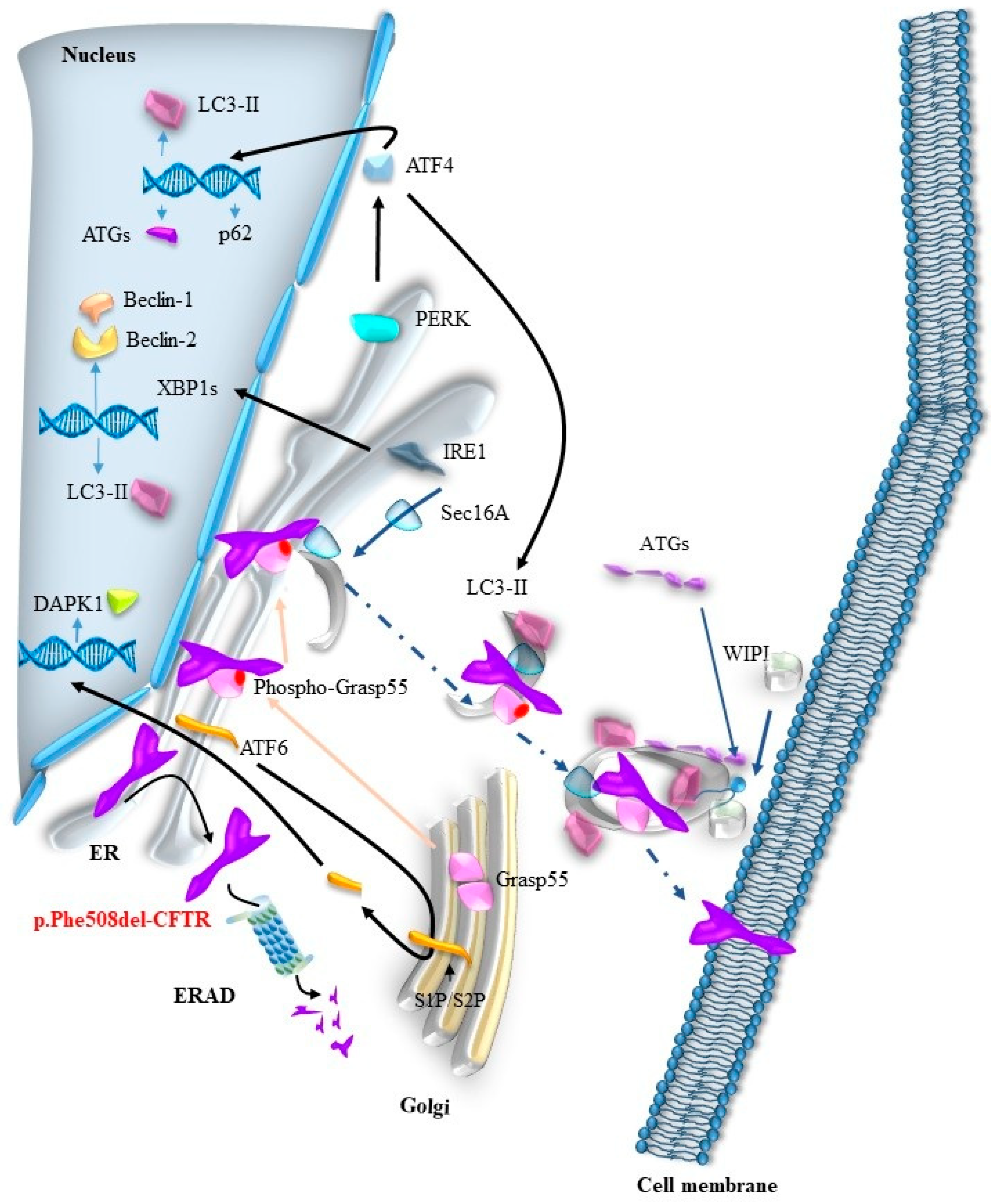
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Trouvé, P.; Férec, C. p.Phe508del-CFTR Trafficking: A Protein Quality Control Perspective Through UPR, UPS, and Autophagy. Int. J. Mol. Sci. 2025, 26, 3623. https://doi.org/10.3390/ijms26083623
Trouvé P, Férec C. p.Phe508del-CFTR Trafficking: A Protein Quality Control Perspective Through UPR, UPS, and Autophagy. International Journal of Molecular Sciences. 2025; 26(8):3623. https://doi.org/10.3390/ijms26083623
Chicago/Turabian StyleTrouvé, Pascal, and Claude Férec. 2025. "p.Phe508del-CFTR Trafficking: A Protein Quality Control Perspective Through UPR, UPS, and Autophagy" International Journal of Molecular Sciences 26, no. 8: 3623. https://doi.org/10.3390/ijms26083623
APA StyleTrouvé, P., & Férec, C. (2025). p.Phe508del-CFTR Trafficking: A Protein Quality Control Perspective Through UPR, UPS, and Autophagy. International Journal of Molecular Sciences, 26(8), 3623. https://doi.org/10.3390/ijms26083623