
Genetic Analysis of Choroideremia-Related Rab Escort Proteins

 , , ,
, , ,  , , ,
, , ,  , and
, and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
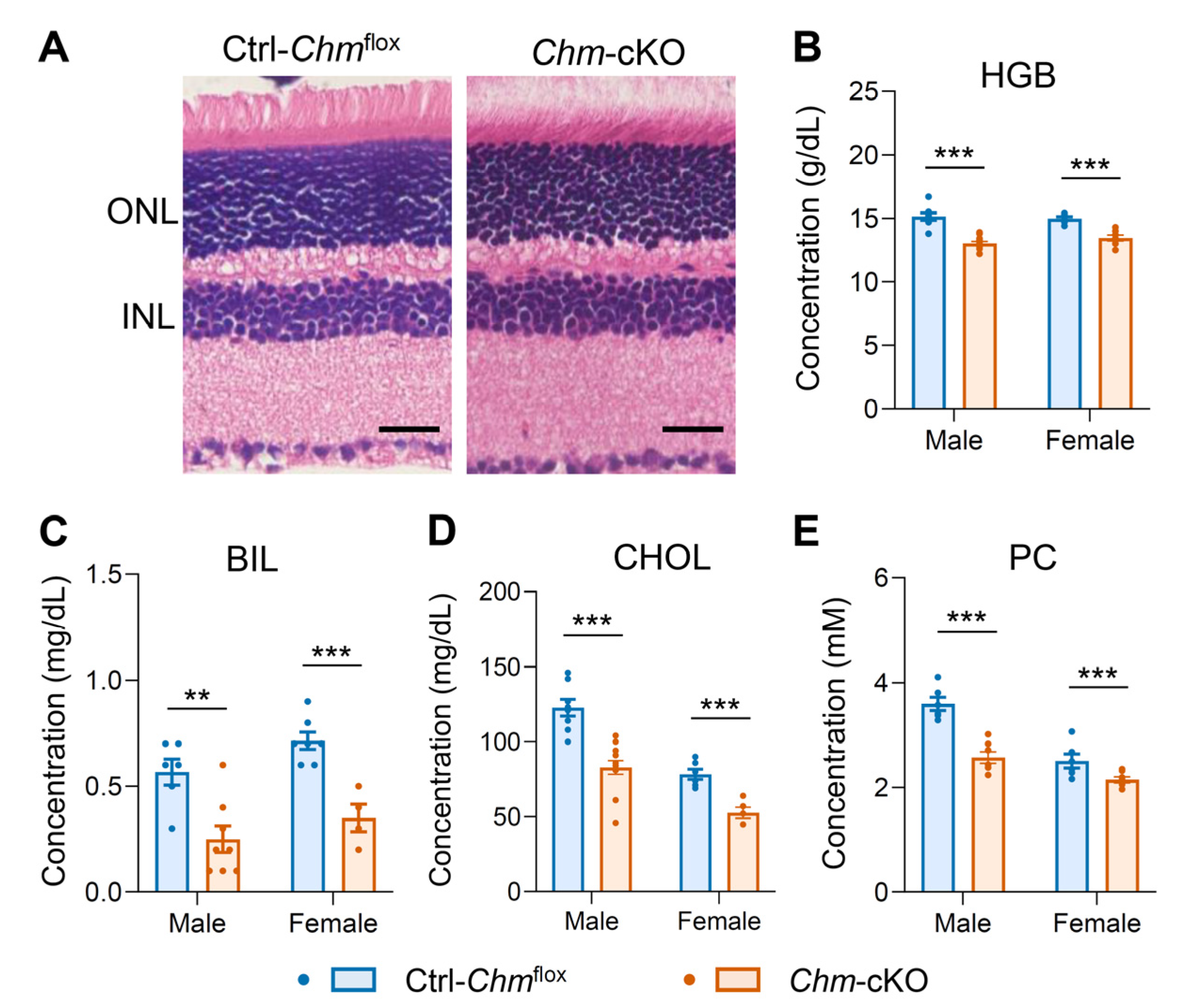
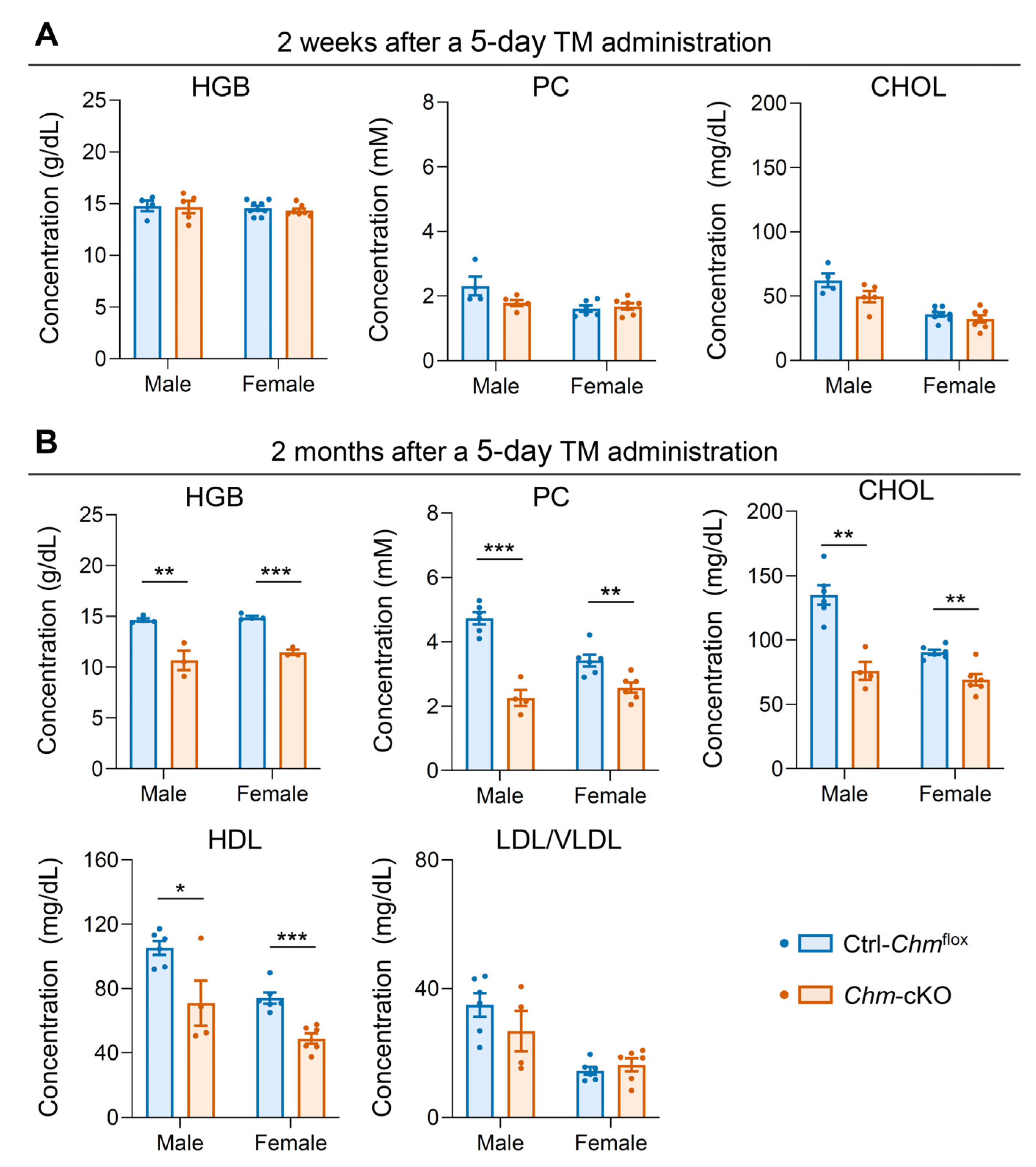
2.1. Mouse Model of Choroideremia Exhibits Metabolic Abnormalities That Mimic the Human Phenotype
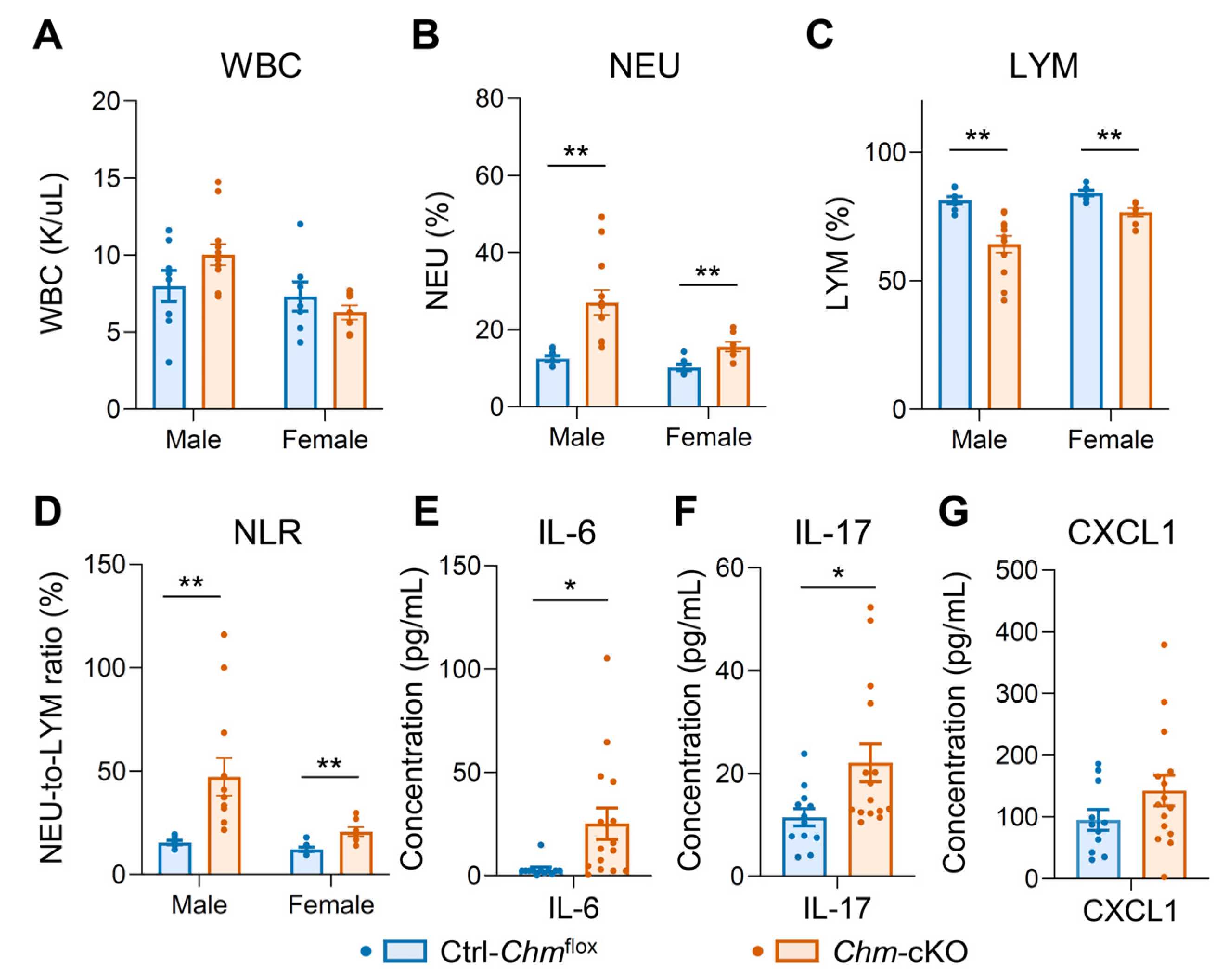
2.2. Mouse Model of Choroideremia Exhibits Elevation of Systemic Inflammatory Biomarkers
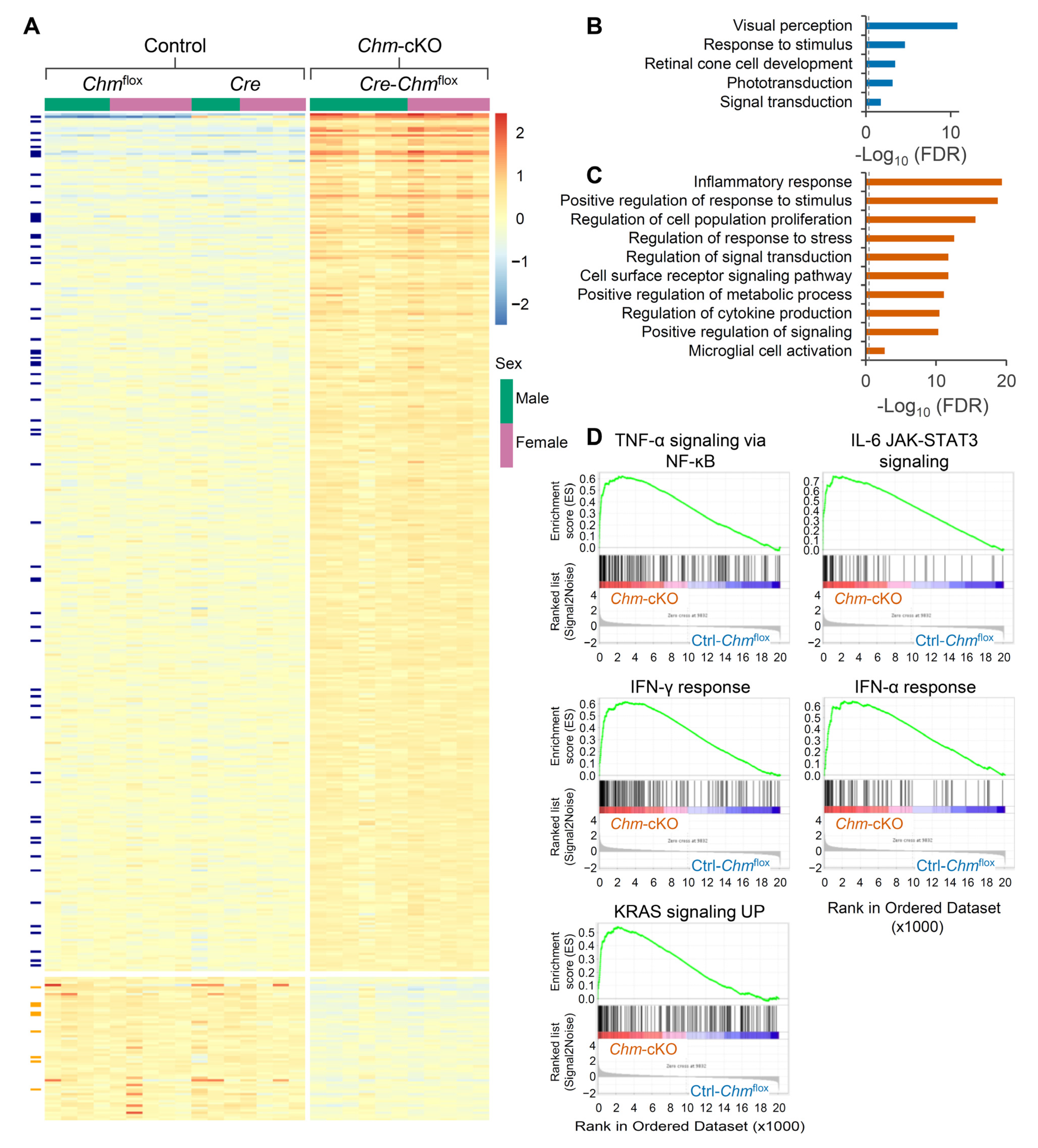
2.3. Retinas of Mouse Model of Choroideremia Exhibit Elevated Inflammatory Transcriptomic Biomarkers
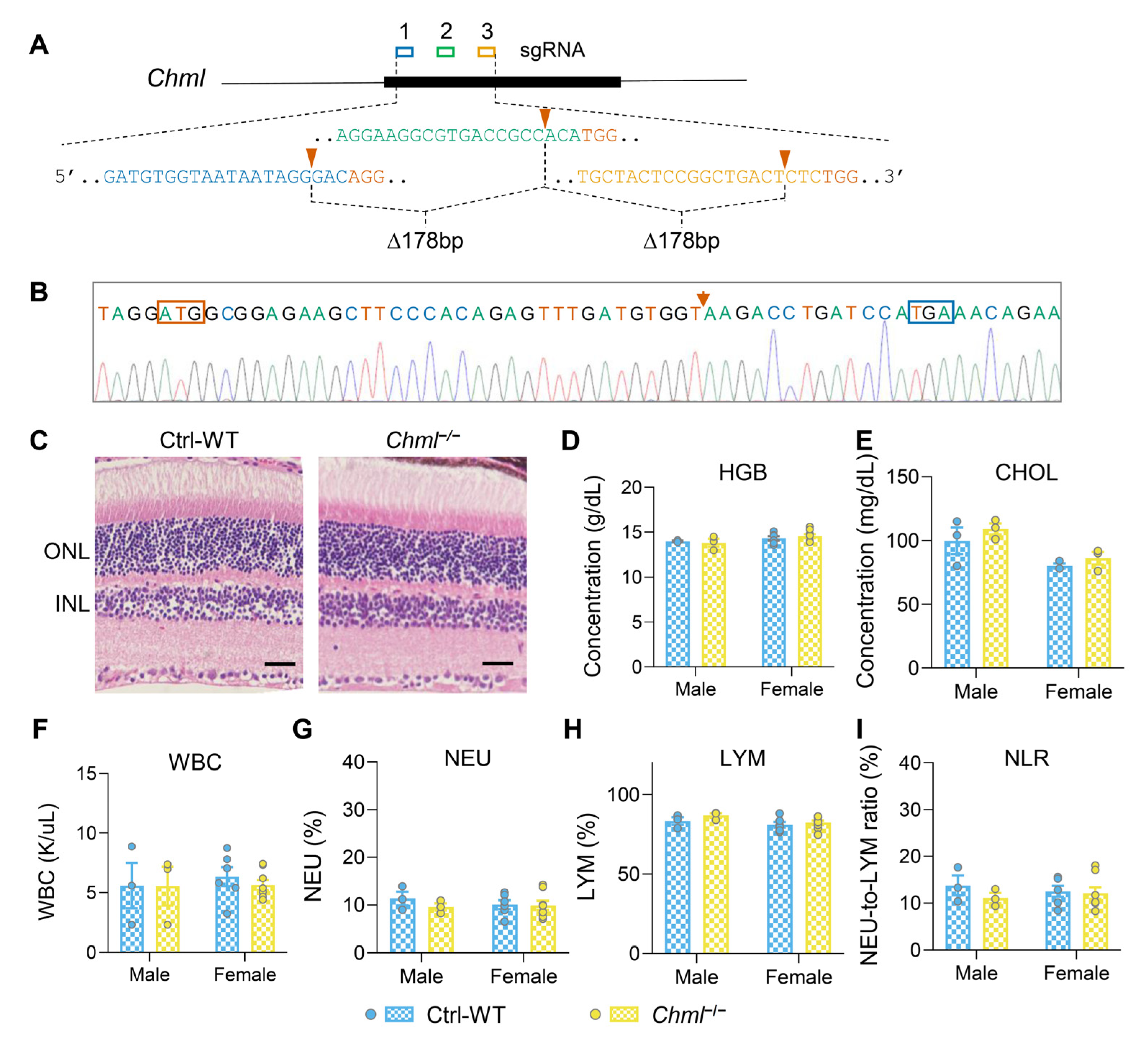
2.4. REP-2 Deficiency Leads to No Detectable Phenotypic Alterations
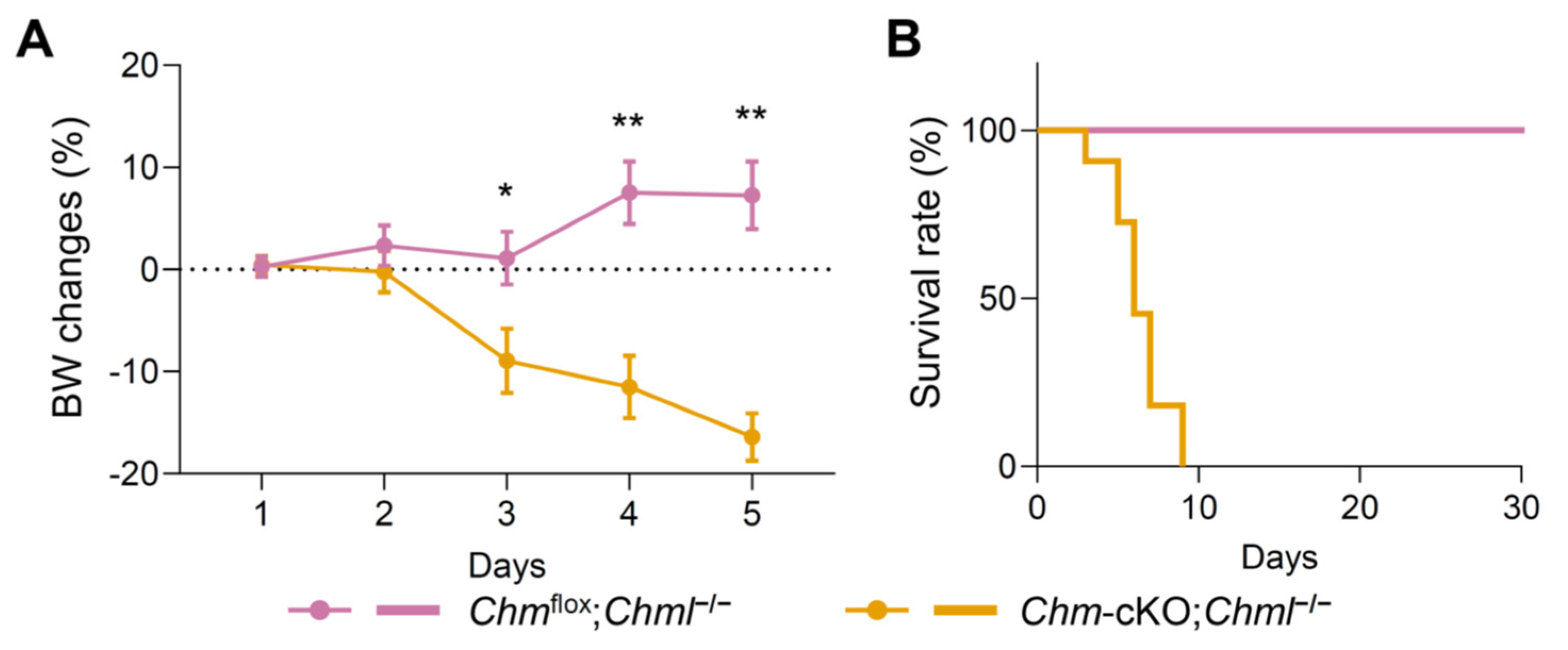
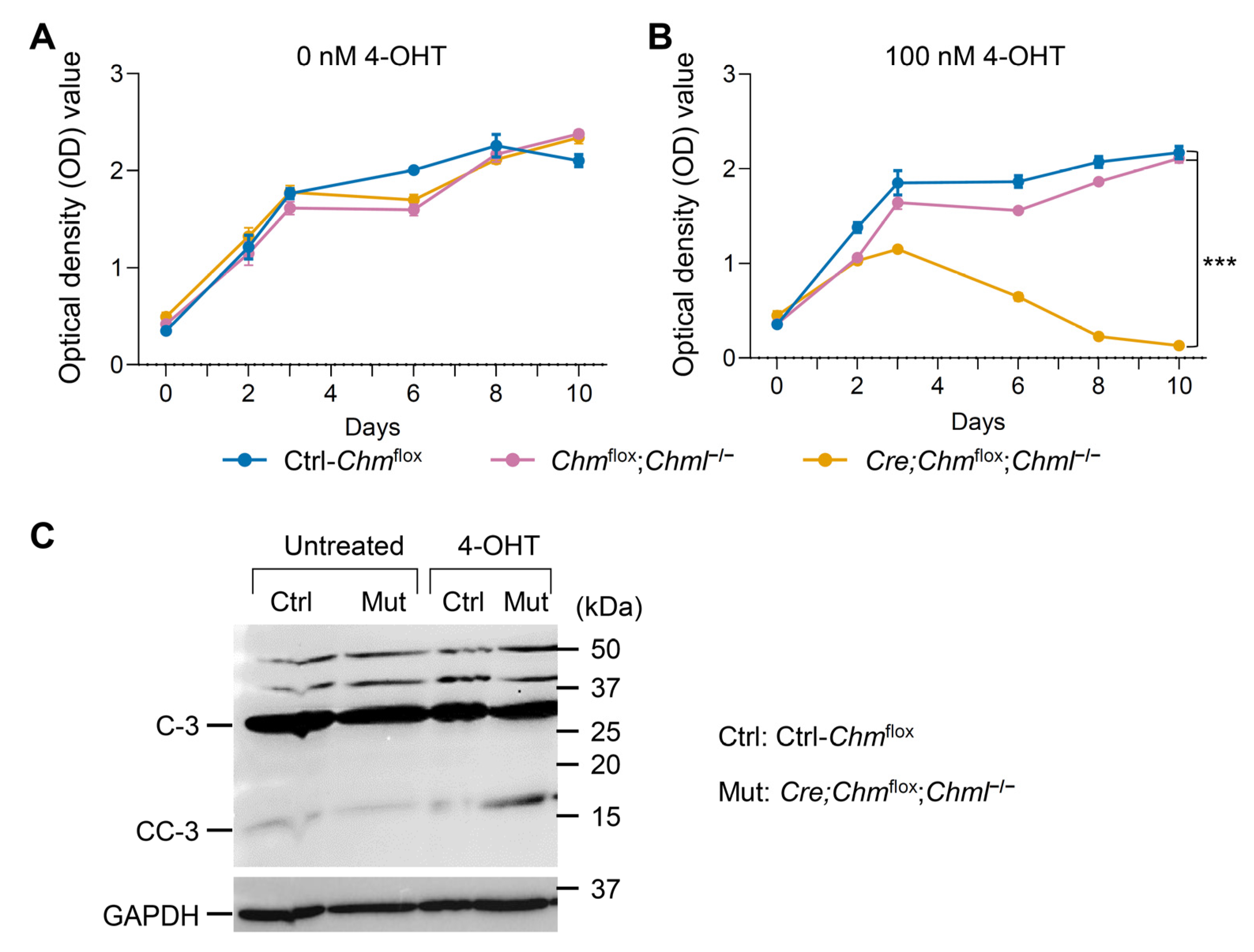
2.5. Dual Deficiency in REP-1 and REP-2 Results in Lethality In Vivo and In Vitro
3. Discussion
Future Direction
4. Materials and Methods
4.1. Generation of Desired Mouse Mutants
4.2. Animals
4.3. Histology Analysis
4.4. Analysis of Mouse Blood
4.5. Cytokine/Chemokine Assay
4.6. RNA Sequencing
4.7. Analysis of RNA-Seq Data
4.8. qPCR Analysis
4.9. Cell Culture and MTT Assay
4.10. Western Blots
4.11. Statistics and Reproducibility
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- MacDonald, I.M.; Hume, S.; Zhai, Y.; Xu, M. Choroideremia. In GeneReviews((R)); Adam, M.P., Feldman, J., Mirzaa, G.M., Pagon, R.A., Wallace, S.E., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 1993. [Google Scholar]
- Dimopoulos, I.S.; Radziwon, A.; St Laurent, C.D.; MacDonald, I.M. Choroideremia. Curr. Opin. Ophthalmol. 2017, 28, 410–415. [Google Scholar] [CrossRef] [PubMed]
- Stenmark, H. Rab GTPases as coordinators of vesicle traffic. Nat. Rev. Mol. Cell Biol. 2009, 10, 513–525. [Google Scholar] [CrossRef] [PubMed]
- Pennesi, M.E.; Birch, D.G.; Duncan, J.L.; Bennett, J.; Girach, A. CHOROIDEREMIA: Retinal Degeneration with an Unmet Need. Retina 2019, 39, 2059–2069. [Google Scholar] [CrossRef] [PubMed]
- MacDonald, I.M.; Russell, L.; Chan, C.C. Choroideremia: New findings from ocular pathology and review of recent literature. Surv. Ophthalmol. 2009, 54, 401–407. [Google Scholar] [CrossRef]
- Tolmachova, T.; Anders, R.; Abrink, M.; Bugeon, L.; Dallman, M.J.; Futter, C.E.; Ramalho, J.S.; Tonagel, F.; Tanimoto, N.; Seeliger, M.W.; et al. Independent degeneration of photoreceptors and retinal pigment epithelium in conditional knockout mouse models of choroideremia. J. Clin. Investig. 2006, 116, 386–394. [Google Scholar] [CrossRef]
- Sarkar, H.; Tracey-White, D.; Hagag, A.M.; Burgoyne, T.; Nair, N.; Jensen, L.D.; Edwards, M.M.; Moosajee, M. Loss of REP1 impacts choroidal melanogenesis and vasculogenesis in choroideremia. Biochim. Biophys. Acta Mol. Basis Dis. 2024, 1870, 166963. [Google Scholar] [CrossRef]
- Moosajee, M.; Tulloch, M.; Baron, R.A.; Gregory-Evans, C.Y.; Pereira-Leal, J.B.; Seabra, M.C. Single choroideremia gene in nonmammalian vertebrates explains early embryonic lethality of the zebrafish model of choroideremia. Investig. Ophthalmol. Vis. Sci. 2009, 50, 3009–3016. [Google Scholar] [CrossRef]
- MacDonald, I.M.; Moen, C.; Duncan, J.L.; Tsang, S.H.; Cehajic-Kapetanovic, J.; Aleman, T.S. Perspectives on Gene Therapy: Choroideremia Represents a Challenging Model for the Treatment of Other Inherited Retinal Degenerations. Transl. Vis. Sci. Technol. 2020, 9, 17. [Google Scholar] [CrossRef]
- Cunha, D.L.; Richardson, R.; Tracey-White, D.; Abbouda, A.; Mitsios, A.; Horneffer-van der Sluis, V.; Takis, P.; Owen, N.; Skinner, J.; Welch, A.A.; et al. REP1 deficiency causes systemic dysfunction of lipid metabolism and oxidative stress in choroideremia. J. Clin. Investig. 2021, 6, e146934. [Google Scholar] [CrossRef]
- Sarkar, H.; Moosajee, M. Choroideremia: Molecular mechanisms and therapies. Trends Mol. Med. 2022, 28, 378–387. [Google Scholar] [CrossRef]
- van den Hurk, J.A.; Hendriks, W.; van de Pol, D.J.; Oerlemans, F.; Jaissle, G.; Ruther, K.; Kohler, K.; Hartmann, J.; Zrenner, E.; van Bokhoven, H.; et al. Mouse choroideremia gene mutation causes photoreceptor cell degeneration and is not transmitted through the female germline. Hum. Mol. Genet. 1997, 6, 851–858. [Google Scholar] [CrossRef] [PubMed]
- Shi, W.; van den Hurk, J.A.; Alamo-Bethencourt, V.; Mayer, W.; Winkens, H.J.; Ropers, H.H.; Cremers, F.P.; Fundele, R. Choroideremia gene product affects trophoblast development and vascularization in mouse extra-embryonic tissues. Dev. Biol. 2004, 272, 53–65. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, S.; McMahon, A.P. Efficient recombination in diverse tissues by a tamoxifen-inducible form of Cre: A tool for temporally regulated gene activation/inactivation in the mouse. Dev. Biol. 2002, 244, 305–318. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.C.; Huang, C.H.; Chung, H.H.; Chen, P.L.; Hu, F.R.; Yang, C.H.; Yang, C.M.; Lin, C.W.; Hsu, C.C.; Chen, T.C. Metabolomics facilitates differential diagnosis in common inherited retinal degenerations by exploring their profiles of serum metabolites. Nat. Commun. 2024, 15, 3562. [Google Scholar] [CrossRef]
- Zahorec, R. Ratio of neutrophil to lymphocyte counts--rapid and simple parameter of systemic inflammation and stress in critically ill. Bratisl. Lek. Listy 2001, 102, 5–14. [Google Scholar]
- Song, M.; Graubard, B.I.; Rabkin, C.S.; Engels, E.A. Neutrophil-to-lymphocyte ratio and mortality in the United States general population. Sci. Rep. 2021, 11, 464. [Google Scholar] [CrossRef]
- Buonacera, A.; Stancanelli, B.; Colaci, M.; Malatino, L. Neutrophil to Lymphocyte Ratio: An Emerging Marker of the Relationships between the Immune System and Diseases. Int. J. Mol. Sci. 2022, 23, 3636. [Google Scholar] [CrossRef]
- Sapkota, D.; Chintala, H.; Wu, F.; Fliesler, S.J.; Hu, Z.; Mu, X. Onecut1 and Onecut2 redundantly regulate early retinal cell fates during development. Proc. Natl. Acad. Sci. USA 2014, 111, E4086–E4095. [Google Scholar] [CrossRef]
- Loza, M.J.; McCall, C.E.; Li, L.; Isaacs, W.B.; Xu, J.; Chang, B.L. Assembly of inflammation-related genes for pathway-focused genetic analysis. PLoS ONE 2007, 2, e1035. [Google Scholar] [CrossRef]
- Lewis, G.P.; Fisher, S.K. Up-regulation of glial fibrillary acidic protein in response to retinal injury: Its potential role in glial remodeling and a comparison to vimentin expression. Int. Rev. Cytol. 2003, 230, 263–290. [Google Scholar] [CrossRef]
- Tang, Y.; Xiao, Z.; Pan, L.; Zhuang, D.; Cho, K.S.; Robert, K.; Chen, X.; Shu, L.; Tang, G.; Wu, J.; et al. Therapeutic Targeting of Retinal Immune Microenvironment With CSF-1 Receptor Antibody Promotes Visual Function Recovery After Ischemic Optic Neuropathy. Front. Immunol. 2020, 11, 585918. [Google Scholar] [CrossRef] [PubMed]
- Mercau, M.E.; Akalu, Y.T.; Mazzoni, F.; Gyimesi, G.; Alberto, E.J.; Kong, Y.; Hafler, B.P.; Finnemann, S.C.; Rothlin, C.V.; Ghosh, S. Inflammation of the retinal pigment epithelium drives early-onset photoreceptor degeneration in Mertk-associated retinitis pigmentosa. Sci. Adv. 2023, 9, eade9459. [Google Scholar] [CrossRef] [PubMed]
- Sherry, D.M.; Mitchell, R.; Li, H.; Graham, D.R.; Ash, J.D. Leukemia inhibitory factor inhibits neuronal development and disrupts synaptic organization in the mouse retina. J. Neurosci. Res. 2005, 82, 316–332. [Google Scholar] [CrossRef] [PubMed]
- Noda, K.; Nakao, S.; Ishida, S.; Ishibashi, T. Leukocyte adhesion molecules in diabetic retinopathy. J. Ophthalmol. 2012, 2012, 279037. [Google Scholar] [CrossRef]
- Deng, W.; Yi, C.; Pan, W.; Liu, J.; Qi, J.; Chen, J.; Zhou, Z.; Duan, Y.; Ning, X.; Li, J.; et al. Vascular Cell Adhesion Molecule-1 (VCAM-1) contributes to macular fibrosis in neovascular age-related macular degeneration through modulating macrophage functions. Immun. Ageing 2023, 20, 65. [Google Scholar] [CrossRef]
- Wavre-Shapton, S.T.; Tolmachova, T.; Lopes da Silva, M.; Futter, C.E.; Seabra, M.C. Conditional ablation of the choroideremia gene causes age-related changes in mouse retinal pigment epithelium. PLoS ONE 2013, 8, e57769. [Google Scholar] [CrossRef]
- Moosajee, M.; Tracey-White, D.; Smart, M.; Weetall, M.; Torriano, S.; Kalatzis, V.; da Cruz, L.; Coffey, P.; Webster, A.R.; Welch, E. Functional rescue of REP1 following treatment with PTC124 and novel derivative PTC-414 in human choroideremia fibroblasts and the nonsense-mediated zebrafish model. Hum. Mol. Genet. 2016, 25, 3416–3431. [Google Scholar] [CrossRef]
- Cremers, F.P.; Armstrong, S.A.; Seabra, M.C.; Brown, M.S.; Goldstein, J.L. REP-2, a Rab escort protein encoded by the choroideremia-like gene. J. Biol. Chem. 1994, 269, 2111–2117. [Google Scholar] [CrossRef]
- Meschede, I.P.; Burgoyne, T.; Tolmachova, T.; Seabra, M.C.; Futter, C.E. Chronically shortened rod outer segments accompany photoreceptor cell death in Choroideremia. PLoS ONE 2020, 15, e0242284. [Google Scholar] [CrossRef]
- Okita, A.; Murakami, Y.; Shimokawa, S.; Funatsu, J.; Fujiwara, K.; Nakatake, S.; Koyanagi, Y.; Akiyama, M.; Takeda, A.; Hisatomi, T.; et al. Changes of Serum Inflammatory Molecules and Their Relationships with Visual Function in Retinitis Pigmentosa. Investig. Ophthalmol. Vis. Sci. 2020, 61, 30. [Google Scholar] [CrossRef]
- Murakami, Y.; Ikeda, Y.; Nakatake, S.; Fujiwara, K.; Tachibana, T.; Yoshida, N.; Notomi, S.; Hisatomi, T.; Yoshida, S.; Ishibashi, T.; et al. C-Reactive protein and progression of vision loss in retinitis pigmentosa. Acta Ophthalmol. 2018, 96, e174–e179. [Google Scholar] [CrossRef] [PubMed]
- He, M.; Wu, T.; Zhang, L.; Ye, W.; Ma, J.; Zhao, C.; Liu, J.; Zhou, J. Correlation between neutrophil-to-lymphocyte ratio and clinical manifestations and complications of retinitis pigmentosa. Acta Ophthalmol. 2022, 100, e278–e287. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Liu, G.; Liu, X.; Wang, Y.; He, F.; Nie, D.; Liu, X.; Liu, X. RNA-seq analysis reveals differentially expressed inflammatory chemokines in a rat retinal degeneration model induced by sodium iodate. J. Int. Med. Res. 2022, 50, 3000605221119376. [Google Scholar] [CrossRef]
- Niazi, S.; Krogh Nielsen, M.; Sorensen, T.L.; Subhi, Y. Neutrophil-to-lymphocyte ratio in age-related macular degeneration: A systematic review and meta-analysis. Acta Ophthalmol. 2019, 97, 558–566. [Google Scholar] [CrossRef]
- Rozing, M.P.; Durhuus, J.A.; Krogh Nielsen, M.; Subhi, Y.; Kirkwood, T.B.; Westendorp, R.G.; Sorensen, T.L. Age-related macular degeneration: A two-level model hypothesis. Prog. Retin. Eye Res. 2020, 76, 100825. [Google Scholar] [CrossRef]
- Nahavandipour, A.; Krogh Nielsen, M.; Sorensen, T.L.; Subhi, Y. Systemic levels of interleukin-6 in patients with age-related macular degeneration: A systematic review and meta-analysis. Acta Ophthalmol. 2020, 98, 434–444. [Google Scholar] [CrossRef]
- Valle, M.L.; Dworshak, J.; Sharma, A.; Ibrahim, A.S.; Al-Shabrawey, M.; Sharma, S. Inhibition of interleukin-6 trans-signaling prevents inflammation and endothelial barrier disruption in retinal endothelial cells. Exp. Eye Res. 2019, 178, 27–36. [Google Scholar] [CrossRef]
- Jo, D.H.; Yun, J.H.; Cho, C.S.; Kim, J.H.; Kim, J.H.; Cho, C.H. Interaction between microglia and retinal pigment epithelial cells determines the integrity of outer blood-retinal barrier in diabetic retinopathy. Glia 2019, 67, 321–331. [Google Scholar] [CrossRef]
- Cameron, J.D.; Fine, B.S.; Shapiro, I. Histopathologic observations in choroideremia with emphasis on vascular changes of the uveal tract. Ophthalmology 1987, 94, 187–196. [Google Scholar] [CrossRef]
- Bonilha, V.L.; Trzupek, K.M.; Li, Y.; Francis, P.J.; Hollyfield, J.G.; Rayborn, M.E.; Smaoui, N.; Weleber, R.G. Choroideremia: Analysis of the retina from a female symptomatic carrier. Ophthalmic Genet. 2008, 29, 99–110. [Google Scholar] [CrossRef]
- Dimopoulos, I.S.; Chan, S.; MacLaren, R.E.; MacDonald, I.M. Pathogenic mechanisms and the prospect of gene therapy for choroideremia. Expert. Opin. Orphan Drugs 2015, 3, 787–798. [Google Scholar] [CrossRef] [PubMed]
- Drag, S.; Dotiwala, F.; Upadhyay, A.K. Gene Therapy for Retinal Degenerative Diseases: Progress, Challenges, and Future Directions. Investig. Ophthalmol. Vis. Sci. 2023, 64, 39. [Google Scholar] [CrossRef] [PubMed]
- Jalil, A.; Ivanova, T.; Moussa, G.; Parry, N.R.A.; Black, G.C.M. Retinal gene therapy in RPE-65 gene mediated inherited retinal dystrophy. Eye 2023, 37, 1874–1877. [Google Scholar] [CrossRef] [PubMed]
- Dimopoulos, I.S.; Hoang, S.C.; Radziwon, A.; Binczyk, N.M.; Seabra, M.C.; MacLaren, R.E.; Somani, R.; Tennant, M.T.S.; MacDonald, I.M. Two-Year Results After AAV2-Mediated Gene Therapy for Choroideremia: The Alberta Experience. Am. J. Ophthalmol. 2018, 193, 130–142. [Google Scholar] [CrossRef]
- Zhai, Y.; Xu, M.; Radziwon, A.; Dimopoulos, I.S.; Crichton, P.; Mah, R.; MacLaren, R.E.; Somani, R.; Tennant, M.T.; MacDonald, I.M. AAV2-Mediated Gene Therapy for Choroideremia: 5-Year Results and Alternate Anti-sense Oligonucleotide Therapy. Am. J. Ophthalmol. 2023, 248, 145–156. [Google Scholar] [CrossRef]
- Chan, Y.K.; Dick, A.D.; Hall, S.M.; Langmann, T.; Scribner, C.L.; Mansfield, B.C.; Ocular Gene Therapy Inflammation Working Group. Inflammation in Viral Vector-Mediated Ocular Gene Therapy: A Review and Report from a Workshop Hosted by the Foundation Fighting Blindness, 9/2020. Transl. Vis. Sci. Technol. 2021, 10, 3. [Google Scholar] [CrossRef]
- Lane, K.T.; Beese, L.S. Thematic review series: Lipid posttranslational modifications. Structural biology of protein farnesyltransferase and geranylgeranyltransferase type I. J. Lipid Res. 2006, 47, 681–699. [Google Scholar] [CrossRef]
- Storck, E.M.; Morales-Sanfrutos, J.; Serwa, R.A.; Panyain, N.; Lanyon-Hogg, T.; Tolmachova, T.; Ventimiglia, L.N.; Martin-Serrano, J.; Seabra, M.C.; Wojciak-Stothard, B.; et al. Dual chemical probes enable quantitative system-wide analysis of protein prenylation and prenylation dynamics. Nat. Chem. 2019, 11, 552–561. [Google Scholar] [CrossRef]
- Shirakawa, R.; Goto-Ito, S.; Goto, K.; Wakayama, S.; Kubo, H.; Sakata, N.; Trinh, D.A.; Yamagata, A.; Sato, Y.; Masumoto, H.; et al. A SNARE geranylgeranyltransferase essential for the organization of the Golgi apparatus. EMBO J. 2020, 39, e104120. [Google Scholar] [CrossRef]
- Kuchay, S.; Wang, H.; Marzio, A.; Jain, K.; Homer, H.; Fehrenbacher, N.; Philips, M.R.; Zheng, N.; Pagano, M. GGTase3 is a newly identified geranylgeranyltransferase targeting a ubiquitin ligase. Nat. Struct. Mol. Biol. 2019, 26, 628–636. [Google Scholar] [CrossRef]
- Rowell, C.A.; Kowalczyk, J.J.; Lewis, M.D.; Garcia, A.M. Direct demonstration of geranylgeranylation and farnesylation of Ki-Ras in vivo. J. Biol. Chem. 1997, 272, 14093–14097. [Google Scholar] [CrossRef] [PubMed]
- Whyte, D.B.; Kirschmeier, P.; Hockenberry, T.N.; Nunez-Oliva, I.; James, L.; Catino, J.J.; Bishop, W.R.; Pai, J.K. K- and N-Ras are geranylgeranylated in cells treated with farnesyl protein transferase inhibitors. J. Biol. Chem. 1997, 272, 14459–14464. [Google Scholar] [CrossRef] [PubMed]
- Kohnke, M.; Delon, C.; Hastie, M.L.; Nguyen, U.T.; Wu, Y.W.; Waldmann, H.; Goody, R.S.; Gorman, J.J.; Alexandrov, K. Rab GTPase prenylation hierarchy and its potential role in choroideremia disease. PLoS ONE 2013, 8, e81758. [Google Scholar] [CrossRef]
- Seabra, M.C.; Ho, Y.K.; Anant, J.S. Deficient geranylgeranylation of Ram/Rab27 in choroideremia. J. Biol. Chem. 1995, 270, 24420–24427. [Google Scholar] [CrossRef]
- Cong, L.; Ran, F.A.; Cox, D.; Lin, S.; Barretto, R.; Habib, N.; Hsu, P.D.; Wu, X.; Jiang, W.; Marraffini, L.A.; et al. Multiplex genome engineering using CRISPR/Cas systems. Science 2013, 339, 819–823. [Google Scholar] [CrossRef]
- Kent, W.J.; Sugnet, C.W.; Furey, T.S.; Roskin, K.M.; Pringle, T.H.; Zahler, A.M.; Haussler, D. The human genome browser at UCSC. Genome Res. 2002, 12, 996–1006. [Google Scholar] [CrossRef]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R.; Genome Project Data Processing, S. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef]
- Wang, L.; Wang, S.; Li, W. RSeQC: Quality control of RNA-seq experiments. Bioinformatics 2012, 28, 2184–2185. [Google Scholar] [CrossRef]
- Liao, Y.; Smyth, G.K.; Shi, W. featureCounts: An efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 2014, 30, 923–930. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef] [PubMed]
- Dennis, G., Jr.; Sherman, B.T.; Hosack, D.A.; Yang, J.; Gao, W.; Lane, H.C.; Lempicki, R.A. DAVID: Database for Annotation, Visualization, and Integrated Discovery. Genome Biol. 2003, 4, P3. [Google Scholar] [CrossRef]
- Liberzon, A.; Birger, C.; Thorvaldsdottir, H.; Ghandi, M.; Mesirov, J.P.; Tamayo, P. The Molecular Signatures Database (MSigDB) hallmark gene set collection. Cell Syst. 2015, 1, 417–425. [Google Scholar] [CrossRef]
- Durkin, M.E.; Qian, X.; Popescu, N.C.; Lowy, D.R. Isolation of Mouse Embryo Fibroblasts. Bio Protoc. 2013, 3, e908. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xing, Z.; Wu, F.; Cortes-Gomez, E.; Pao, A.; Gao, L.; Douglas, A.; Li, Y.; Spernyak, J.A.; Wong, G.W.; Singh, P.K.; et al. Genetic Analysis of Choroideremia-Related Rab Escort Proteins. Int. J. Mol. Sci. 2025, 26, 3636. https://doi.org/10.3390/ijms26083636
Xing Z, Wu F, Cortes-Gomez E, Pao A, Gao L, Douglas A, Li Y, Spernyak JA, Wong GW, Singh PK, et al. Genetic Analysis of Choroideremia-Related Rab Escort Proteins. International Journal of Molecular Sciences. 2025; 26(8):3636. https://doi.org/10.3390/ijms26083636
Chicago/Turabian StyleXing, Zhuo, Fuguo Wu, Eduardo Cortes-Gomez, Annie Pao, Lingqiu Gao, Avrium Douglas, Yichen Li, Joseph A. Spernyak, G. William Wong, Prashant K. Singh, and et al. 2025. "Genetic Analysis of Choroideremia-Related Rab Escort Proteins" International Journal of Molecular Sciences 26, no. 8: 3636. https://doi.org/10.3390/ijms26083636
APA StyleXing, Z., Wu, F., Cortes-Gomez, E., Pao, A., Gao, L., Douglas, A., Li, Y., Spernyak, J. A., Wong, G. W., Singh, P. K., Wang, J., Liu, S., Thanavala, Y., MacDonald, I. M., Mu, X., & Yu, Y. E. (2025). Genetic Analysis of Choroideremia-Related Rab Escort Proteins. International Journal of Molecular Sciences, 26(8), 3636. https://doi.org/10.3390/ijms26083636