
Transcription-Coupled Repair and R-Loop Crosstalk in Genome Stability



{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
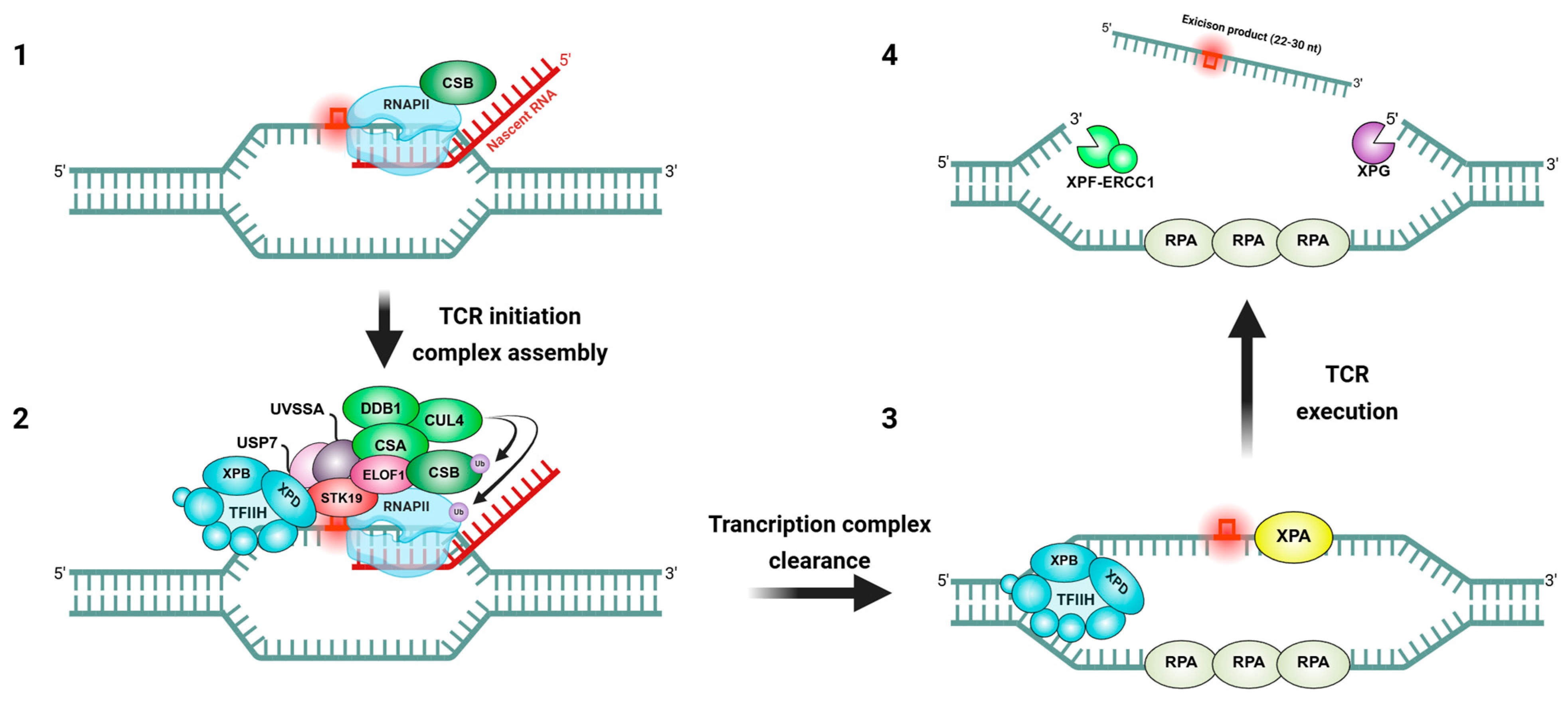
2. Molecular Mechanisms of Transcription-Coupled Repair
3. Biogenesis and Regulation of R-Loops
4. Interplay Between TCR and R-Loops
4.1. TCR Factors as a Source of Genome Instability via R-Loop Processing
4.2. Protective Roles of R-Loops as Cooperative Intermediates in TCR
4.3. TCR-Mediated Suppression of R-Loop Accumulation
5. Pathological Consequences of Dysregulated TCR and R-Loops
5.1. Cockayne Syndrome: A Prototypical Disorder Caused by Loss of TCR
5.2. R-Loop Dysregulation in Neurodegenerative Disorders
5.3. Cancer Predisposition Linked to TCR or R-Loop Deficiencies
6. Therapeutic Implications
6.1. Gene Therapy and Enzyme Replacement
6.2. Small-Molecule Modulators
6.3. Exploiting R-Loop Vulnerabilities with Synthetic Lethality
6.4. Therapeutic Targeting of TCR in Progeria and Neurodegeneration
7. Future Directions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| AAV | Adeno-associated virus |
| ALS4 | Amyotrophic lateral sclerosis type 4 |
| AOA2 | Ataxia with oculomotor apraxia type 2 |
| ATR | Ataxia telangiectasia and rad3-related |
| ATRX | ATRX chromatin remodeler |
| ATP | Adenosine triphosphate |
| AQR | Aquarius |
| BIR | Break-induced replication |
| BRCA1/2 | Breast cancer susceptibility gene 1/2 |
| BLM | Blood syndrome protein |
| cGAS | Cyclic GMP–AMP synthase |
| STING | Stimulator of interferon genes |
| Cas3 | CRISPR-associated protein 3 |
| CDK9 | Cycle-dependent kinase 9 |
| CS | Cockayne syndrome |
| CSA | Cockayne syndrome A |
| CSB | Cockayne syndrome B |
| CUL4 | Cullin 4 |
| DNA | Deoxyribonucleic acid |
| DDX1 | DEAD-box RNA helicase 1 |
| DDX17 | DEAD-box RNA helicase 17 |
| DDX21 | DEAD-box RNA helicase 21 |
| DDB1 | DNA damage binding protein 1 |
| DHX9 | DExH-box helicase 9 |
| DSB | Double-strand break |
| DNAP δ/ε | DNA polymerase δ/ε |
| ELOF1 | Elongation factor 1 |
| ERCC1 | Excision repair cross-complementation group 1 |
| ERCC6 | Excision repair cross-complementation group 6 |
| ERCC8 | Excision repair cross-complementation group 8 |
| ETAA1 | Ewing’s tumor-associated antigen 1 |
| FANCD2 | Fanconi anemia complementation group D2 |
| FANCI | Fanconi anemia complementation group I |
| FANCM | Fanconi anemia complementation group M |
| FMR1 | Fragile X messenger ribonucleoprotein 1 |
| GGR | Global-genome NER |
| mRNA | Messenger RNA |
| mRNP | Messenger ribonucleoprotein |
| m6A | N6-methyladenosine |
| METTL3 | Methyltransferase 3 |
| NAD+ | Nicotinamide adenine dinucleotide |
| NER | Nucleotide excision repair |
| PARP1 | Poly(ADP-ribose) polymerase 1 |
| PIF1 | PIF1 helicase |
| Pol δ/ε | DNA polymerase delta/epsilon |
| POLD3 | DNA polymerase delta subunit 3 |
| RNA | Ribonucleic acid |
| RAD52 | DNA repair protein RAD52 |
| RNAPII | RNA polymerase II |
| RNase H | Ribonuclease H |
| RPA | Replication protein A |
| RTEL1 | Regulator of telomere elongation helicase 1 |
| SETX | Senataxin |
| SF3B1 | Splicing factor 3B subunit 1 |
| SRSF1 | Serine/arginine-rich splicing factor 1 |
| SRSF2 | Serine/arginine-rich splicing factor 2 |
| STK19 | Serine/threonine kinase 19 |
| ssDNA | Single-stranded DNA |
| TCR | Transcription-coupled repair |
| TC-NER | Transcription-coupled nucleotide excision repair |
| TERRA | Telomeric repeat-containing RNA |
| THO | Subcomplex of transcription-export complex |
| TREX | Transcription-export complex |
| TFIIH | Transcription factor IIH |
| TopI | Topoisomerase I |
| TopII | Topoisomerase II |
| UV | Ultraviolet |
| UVSSA | UV-stimulated scaffold protein A |
| U2AF1 | U2 small nuclear RNA auxiliary factor 1 |
| UAP56 | U2AF65-associated protein 56 |
| USP7 | Ubiquitin-specific protease 7 |
| WRN | Werner syndrome protein |
| XP | Xeroderma pigmentosum |
| XPA | Xeroderma pigmentosum group A protein |
| XPB | Xeroderma pigmentosum group B protein |
| XPC | Xeroderma pigmentosum group C protein |
| XPD | Xeroderma pigmentosum group D protein |
| XPF | Xeroderma pigmentosum group F protein |
| XPG | Xeroderma pigmentosum group G protein |
References
- Marteijn, J.A.; Lans, H.; Vermeulen, W.; Hoeijmakers, J.H. Understanding nucleotide excision repair and its roles in cancer and ageing. Nat. Rev. Mol. Cell Biol. 2014, 15, 465–481. [Google Scholar] [CrossRef] [PubMed]
- Selby, C.P.; Lindsey-Boltz, L.A.; Li, W.; Sancar, A. Molecular mechanisms of transcription-coupled repair. Annu. Rev. Biochem. 2023, 92, 115–144. [Google Scholar] [CrossRef]
- Park, J.M.; Kang, T.H. Transcriptional and posttranslational regulation of nucleotide excision repair: The guardian of the genome against ultraviolet radiation. Int. J. Mol. Sci. 2016, 17, 1840. [Google Scholar] [CrossRef] [PubMed]
- Spivak, G. Transcription-coupled repair: An update. Arch. Toxicol. 2016, 90, 2583–2594. [Google Scholar] [CrossRef] [PubMed]
- Lee, T.H.; Kang, T.H. DNA oxidation and excision repair pathways. Int. J. Mol. Sci. 2019, 20, 6092. [Google Scholar] [CrossRef]
- Cho, I.; Tsai, P.F.; Lake, R.J.; Basheer, A.; Fan, H.Y. ATP-dependent chromatin remodeling by cockayne syndrome protein B and NAP1-like histone chaperones is required for efficient transcription-coupled DNA repair. PLoS Genet. 2013, 9, e1003407. [Google Scholar] [CrossRef]
- Bohm, K.A.; Hodges, A.J.; Czaja, W.; Selvam, K.; Smerdon, M.J.; Mao, P.; Wyrick, J.J. Distinct roles for RSC and SWI/SNF chromatin remodelers in genomic excision repair. Genome Res. 2021, 31, 1047–1059. [Google Scholar] [CrossRef]
- Santos-Pereira, J.M.; Aguilera, A. R loops: New modulators of genome dynamics and function. Nat. Rev. Genet. 2015, 16, 583–597. [Google Scholar] [CrossRef]
- Brickner, J.R.; Garzon, J.L.; Cimprich, K.A. Walking a tightrope: The complex balancing act of R-loops in genome stability. Mol. Cell 2022, 82, 2267–2297. [Google Scholar] [CrossRef]
- Garcia-Muse, T.; Aguilera, A. R loops: From physiological to pathological roles. Cell 2019, 179, 604–618. [Google Scholar] [CrossRef]
- Hamperl, S.; Bocek, M.J.; Saldivar, J.C.; Swigut, T.; Cimprich, K.A. Transcription-replication conflict orientation modulates r-loop levels and activates distinct DNA damage responses. Cell 2017, 170, 774–786.E19. [Google Scholar] [CrossRef] [PubMed]
- Goulielmaki, E.; Tsekrekou, M.; Batsiotos, N.; Ascensao-Ferreira, M.; Ledaki, E.; Stratigi, K.; Chatzinikolaou, G.; Topalis, P.; Kosteas, T.; Altmuller, J.; et al. The splicing factor XAB2 interacts with ERCC1-XPF and XPG for R-loop processing. Nat. Commun. 2021, 12, 3153. [Google Scholar] [CrossRef]
- Richard, P.; Manley, J.L. R loops and links to human disease. J. Mol. Biol. 2017, 429, 3168–3180. [Google Scholar] [CrossRef]
- Perego, M.G.L.; Taiana, M.; Bresolin, N.; Comi, G.P.; Corti, S. R-loops in motor neuron diseases. Mol. Neurobiol. 2019, 56, 2579–2589. [Google Scholar] [CrossRef]
- van den Heuvel, D.; van der Weegen, Y.; Boer, D.E.C.; Ogi, T.; Luijsterburg, M.S. Transcription-coupled DNA repair: From mechanism to human disorder. Trends Cell Biol. 2021, 31, 359–371. [Google Scholar] [CrossRef]
- Mackay, R.P.; Xu, Q.; Weinberger, P.M. R-loop physiology and pathology: A brief review. DNA Cell Biol. 2020, 39, 1914–1925. [Google Scholar] [CrossRef]
- Qiu, Y.; Man, C.; Zhu, L.; Zhang, S.; Wang, X.; Gong, D.; Fan, Y. R-loops’ m6A modification and its roles in cancers. Mol. Cancer 2024, 23, 232. [Google Scholar] [CrossRef]
- van der Weegen, Y.; Golan-Berman, H.; Mevissen, T.E.T.; Apelt, K.; Gonzalez-Prieto, R.; Goedhart, J.; Heilbrun, E.E.; Vertegaal, A.C.O.; van den Heuvel, D.; Walter, J.C.; et al. The cooperative action of CSB, CSA, and UVSSA target TFIIH to DNA damage-stalled RNA polymerase II. Nat. Commun. 2020, 11, 2104. [Google Scholar] [CrossRef]
- Xu, J.; Lahiri, I.; Wang, W.; Wier, A.; Cianfrocco, M.A.; Chong, J.; Hare, A.A.; Dervan, P.B.; DiMaio, F.; Leschziner, A.E.; et al. Structural basis for the initiation of eukaryotic transcription-coupled DNA repair. Nature 2017, 551, 653–657. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Xu, J.; Chong, J.; Wang, D. Structural basis of DNA lesion recognition for eukaryotic transcription-coupled nucleotide excision repair. DNA Repair 2018, 71, 43–55. [Google Scholar] [CrossRef] [PubMed]
- Duan, M.; Speer, R.M.; Ulibarri, J.; Liu, K.J.; Mao, P. Transcription-coupled nucleotide excision repair: New insights revealed by genomic approaches. DNA Repair 2021, 103, 103126. [Google Scholar] [CrossRef] [PubMed]
- Schwertman, P.; Lagarou, A.; Dekkers, D.H.; Raams, A.; van der Hoek, A.C.; Laffeber, C.; Hoeijmakers, J.H.; Demmers, J.A.; Fousteri, M.; Vermeulen, W.; et al. UV-sensitive syndrome protein UVSSA recruits USP7 to regulate transcription-coupled repair. Nat. Genet. 2012, 44, 598–602. [Google Scholar] [CrossRef]
- Oksenych, V.; Bernardes de Jesus, B.; Zhovmer, A.; Egly, J.M.; Coin, F. Molecular insights into the recruitment of TFIIH to sites of DNA damage. EMBO J. 2009, 28, 2971–2980. [Google Scholar] [CrossRef] [PubMed]
- Kang, T.H. Circadian rhythm of NER and ATR pathways. Biomolecules 2021, 11, 715. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.H.; Kim, G.H.; Kemp, M.G.; Choi, J.H. TREX1 degrades the 3′ end of the small DNA oligonucleotide products of nucleotide excision repair in human cells. Nucleic Acids Res. 2022, 50, 3974–3984. [Google Scholar] [CrossRef]
- Scharer, O.D. Nucleotide excision repair in eukaryotes. Cold Spring Harb. Perspect. Biol. 2013, 5, a012609. [Google Scholar] [CrossRef]
- Donnio, L.M.; Giglia-Mari, G. Keep calm and reboot - how cells restart transcription after DNA damage and DNA repair. FEBS Lett. 2025, 599, 275–294. [Google Scholar] [CrossRef]
- Reese, J.C. New roles for elongation factors in RNA polymerase II ubiquitylation and degradation. Biochim. Biophys. Acta Gene Regul. Mech. 2023, 1866, 194956. [Google Scholar] [CrossRef]
- Spyropoulou, Z.; Papaspyropoulos, A.; Lagopati, N.; Myrianthopoulos, V.; Georgakilas, A.G.; Fousteri, M.; Kotsinas, A.; Gorgoulis, V.G. Cockayne syndrome group B (CSB): The regulatory framework governing the multifunctional protein and its plausible role in cancer. Cells 2021, 10, 866. [Google Scholar] [CrossRef]
- Schwertman, P.; Vermeulen, W.; Marteijn, J.A. UVSSA and USP7, a new couple in transcription-coupled DNA repair. Chromosoma 2013, 122, 275–284. [Google Scholar] [CrossRef]
- Mevissen, T.E.T.; Kummecke, M.; Schmid, E.W.; Farnung, L.; Walter, J.C. STK19 positions TFIIH for cell-free transcription-coupled DNA repair. Cell 2024, 187, 7091–7106.E24. [Google Scholar] [CrossRef] [PubMed]
- Geijer, M.E.; Zhou, D.; Selvam, K.; Steurer, B.; Mukherjee, C.; Evers, B.; Cugusi, S.; van Toorn, M.; van der Woude, M.; Janssens, R.C.; et al. Elongation factor ELOF1 drives transcription-coupled repair and prevents genome instability. Nat. Cell Biol. 2021, 23, 608–619. [Google Scholar] [CrossRef]
- van der Weegen, Y.; de Lint, K.; van den Heuvel, D.; Nakazawa, Y.; Mevissen, T.E.T.; van Schie, J.J.M.; San Martin Alonso, M.; Boer, D.E.C.; Gonzalez-Prieto, R.; Narayanan, I.V.; et al. ELOF1 is a transcription-coupled DNA repair factor that directs RNA polymerase II ubiquitylation. Nat. Cell Biol. 2021, 23, 595–607. [Google Scholar] [CrossRef]
- Tan, Y.; Gao, M.; Huang, Y.; Zhan, D.; Wu, S.; An, J.; Zhang, X.; Hu, J. STK19 is a transcription-coupled repair factor that participates in UVSSA ubiquitination and TFIIH loading. Nucleic. Acids Res. 2024, 52, 12767–12783. [Google Scholar] [CrossRef] [PubMed]
- van den Heuvel, D.; Rodriguez-Martinez, M.; van der Meer, P.J.; Nieto Moreno, N.; Park, J.; Kim, H.S.; van Schie, J.J.M.; Wondergem, A.P.; D’Souza, A.; Yakoub, G.; et al. STK19 facilitates the clearance of lesion-stalled RNAPII during transcription-coupled DNA repair. Cell 2024, 187, 7107–7125.E25. [Google Scholar] [CrossRef]
- Ramadhin, A.R.; Lee, S.H.; Zhou, D.; Salmazo, A.; Gonzalo-Hansen, C.; van Sluis, M.; Blom, C.M.A.; Janssens, R.C.; Raams, A.; Dekkers, D.; et al. STK19 drives transcription-coupled repair by stimulating repair complex stability, RNA Pol II ubiquitylation, and TFIIH recruitment. Mol. Cell 2024, 84, 4740–4757.E12. [Google Scholar] [CrossRef]
- Wu, T.; Nance, J.; Chu, F.; Fazzio, T.G. Characterization of R-loop-interacting proteins in embryonic stem cells reveals roles in rRNA processing and gene expression. Mol. Cell Proteom. 2021, 20, 100142. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Xu, J.; Hu, J.; Zhang, S.; Hao, Y.; Zhang, D.; Qian, H.; Wang, D.; Fu, X.D. Cockayne syndrome linked to elevated r-loops induced by stalled RNA polymerase II during transcription elongation. Nat. Commun. 2024, 15, 6031. [Google Scholar] [CrossRef]
- Belotserkovskii, B.P.; Tornaletti, S.; D’Souza, A.D.; Hanawalt, P.C. R-loop generation during transcription: Formation, processing and cellular outcomes. DNA Repair 2018, 71, 69–81. [Google Scholar] [CrossRef]
- Luna, R.; Gomez-Gonzalez, B.; Aguilera, A. RNA biogenesis and RNA metabolism factors as R-loop suppressors: A hidden role in genome integrity. Genes. Dev. 2024, 38, 504–527. [Google Scholar] [CrossRef]
- Ginno, P.A.; Lim, Y.W.; Lott, P.L.; Korf, I.; Chedin, F. GC skew at the 5′ and 3′ ends of human genes links R-loop formation to epigenetic regulation and transcription termination. Genome. Res. 2013, 23, 1590–1600. [Google Scholar] [CrossRef]
- Chedin, F. Nascent connections: R-loops and chromatin patterning. Trends Genet. 2016, 32, 828–838. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Z. From origin to the present: Establishment, mechanism, evolutions and biomedical applications of the CRISPR/cas-based macromolecular system in brief. Molecules 2025, 30, 947. [Google Scholar] [CrossRef]
- Hyjek, M.; Figiel, M.; Nowotny, M. RNases H: Structure and mechanism. DNA Repair 2019, 84, 102672. [Google Scholar] [CrossRef]
- Yang, S.; Winstone, L.; Mondal, S.; Wu, Y. Helicases in R-loop formation and resolution. J. Biol. Chem. 2023, 299, 105307. [Google Scholar] [CrossRef] [PubMed]
- Moreira, M.C.; Klur, S.; Watanabe, M.; Nemeth, A.H.; Le Ber, I.; Moniz, J.C.; Tranchant, C.; Aubourg, P.; Tazir, M.; Schols, L.; et al. Senataxin, the ortholog of a yeast RNA helicase, is mutant in ataxia-ocular apraxia 2. Nat. Genet. 2004, 36, 225–227. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.Z.; Bennett, C.L.; Huynh, H.M.; Blair, I.P.; Puls, I.; Irobi, J.; Dierick, I.; Abel, A.; Kennerson, M.L.; Rabin, B.A.; et al. DNA/RNA helicase gene mutations in a form of juvenile amyotrophic lateral sclerosis (ALS4). Am. J. Hum. Genet. 2004, 74, 1128–1135. [Google Scholar] [CrossRef]
- Fernandes, R.V.; Feretzaki, M.; Lingner, J. The makings of TERRA R-loops at chromosome ends. Cell Cycle 2021, 20, 1745–1759. [Google Scholar] [CrossRef]
- Pan, X.; Chen, Y.; Biju, B.; Ahmed, N.; Kong, J.; Goldenberg, M.; Huang, J.; Mohan, N.; Klosek, S.; Parsa, K.; et al. FANCM suppresses DNA replication stress at ALT telomeres by disrupting TERRA R-loops. Sci. Rep. 2019, 9, 19110. [Google Scholar] [CrossRef]
- Hu, C.; Ni, D.; Nam, K.H.; Majumdar, S.; McLean, J.; Stahlberg, H.; Terns, M.P.; Ke, A. Allosteric control of type I-A CRISPR-Cas3 complexes and establishment as effective nucleic acid detection and human genome editing tools. Mol. Cell 2022, 82, 2754–2768.E5. [Google Scholar] [CrossRef]
- Ngo, G.H.P.; Grimstead, J.W.; Baird, D.M. UPF1 promotes the formation of R loops to stimulate DNA double-strand break repair. Nat. Commun. 2021, 12, 3849. [Google Scholar] [CrossRef] [PubMed]
- Hraiky, C.; Raymond, M.A.; Drolet, M. RNase H overproduction corrects a defect at the level of transcription elongation during rRNA synthesis in the absence of DNA topoisomerase I in Escherichia coli. J. Biol. Chem. 2000, 275, 11257–11263. [Google Scholar] [CrossRef]
- Achar, Y.J.; Adhil, M.; Choudhary, R.; Gilbert, N.; Foiani, M. Negative supercoil at gene boundaries modulates gene topology. Nature 2020, 577, 701–705. [Google Scholar] [CrossRef]
- Jimeno, S.; Luna, R.; Garcia-Rubio, M.; Aguilera, A. Tho1, a novel hnRNP, and Sub2 provide alternative pathways for mRNP biogenesis in yeast THO mutants. Mol. Cell Biol. 2006, 26, 4387–4398. [Google Scholar] [CrossRef] [PubMed]
- Bhandari, J.; Guillen-Mendoza, C.; Banks, K.; Eliaz, L.; Southwell, S.; Eyaa, D.; Luna, R.; Aguilera, A.; Xue, X. The molecular chaperone ALYREF promotes R-loop resolution and maintains genome stability. J. Biol. Chem. 2024, 300, 107996. [Google Scholar] [CrossRef] [PubMed]
- Olazabal-Herrero, A.; He, B.; Kwon, Y.; Gupta, A.K.; Dutta, A.; Huang, Y.; Boddu, P.; Liang, Z.; Liang, F.; Teng, Y.; et al. The FANCI/FANCD2 complex links DNA damage response to R-loop regulation through SRSF1-mediated mRNA export. Cell Rep. 2024, 43, 113610. [Google Scholar] [CrossRef]
- Patel, P.S.; Abraham, K.J.; Guturi, K.K.N.; Halaby, M.J.; Khan, Z.; Palomero, L.; Ho, B.; Duan, S.; St-Germain, J.; Algouneh, A.; et al. RNF168 regulates R-loop resolution and genomic stability in BRCA1/2-deficient tumors. J. Clin. Invest. 2021, 131, e140105. [Google Scholar] [CrossRef]
- Okamoto, Y.; Abe, M.; Itaya, A.; Tomida, J.; Ishiai, M.; Takaori-Kondo, A.; Taoka, M.; Isobe, T.; Takata, M. FANCD2 protects genome stability by recruiting RNA processing enzymes to resolve R-loops during mild replication stress. FEBS J. 2019, 286, 139–150. [Google Scholar] [CrossRef]
- Sollier, J.; Stork, C.T.; Garcia-Rubio, M.L.; Paulsen, R.D.; Aguilera, A.; Cimprich, K.A. Transcription-coupled nucleotide excision repair factors promote R-loop-induced genome instability. Mol. Cell 2014, 56, 777–785. [Google Scholar] [CrossRef]
- Tan, J.; Duan, M.; Yadav, T.; Phoon, L.; Wang, X.; Zhang, J.M.; Zou, L.; Lan, L. An R-loop-initiated CSB-RAD52-POLD3 pathway suppresses ROS-induced telomeric DNA breaks. Nucleic Acids Res. 2020, 48, 1285–1300. [Google Scholar] [CrossRef]
- Hao, J.D.; Liu, Q.L.; Liu, M.X.; Yang, X.; Wang, L.M.; Su, S.Y.; Xiao, W.; Zhang, M.Q.; Zhang, Y.C.; Zhang, L.; et al. DDX21 mediates co-transcriptional RNA m 6 A modification to promote transcription termination and genome stability. Mol. Cell 2024, 84, 1711–1726. [Google Scholar] [CrossRef] [PubMed]
- Kajitani, G.S.; Nascimento, L.L.S.; Neves, M.R.C.; Leandro, G.D.S.; Garcia, C.C.M.; Menck, C.F.M. Transcription blockage by DNA damage in nucleotide excision repair-related neurological dysfunctions. Semin. Cell Dev. Biol. 2021, 114, 20–35. [Google Scholar] [CrossRef]
- Karikkineth, A.C.; Scheibye-Knudsen, M.; Fivenson, E.; Croteau, D.L.; Bohr, V.A. Cockayne syndrome: Clinical features, model systems and pathways. Ageing Res. Rev. 2017, 33, 3–17. [Google Scholar] [CrossRef]
- Reid-Bayliss, K.S.; Arron, S.T.; Loeb, L.A.; Bezrookove, V.; Cleaver, J.E. Why cockayne syndrome patients do not get cancer despite their DNA repair deficiency. Proc. Natl. Acad. Sci. USA 2016, 113, 10151–10156. [Google Scholar] [CrossRef]
- Licht, C.L.; Stevnsner, T.; Bohr, V.A. Cockayne syndrome group B cellular and biochemical functions. Am. J. Hum. Genet. 2003, 73, 1217–1239. [Google Scholar] [CrossRef]
- Oh, J.; Xu, J.; Chong, J.; Wang, D. Molecular basis of transcriptional pausing, stalling, and transcription-coupled repair initiation. Biochim. Biophys. Acta. Gene. Regul. Mech. 2021, 1864, 194659. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Chong, J.; Wang, D. Strand-specific effect of Rad26 and TFIIS in rescuing transcriptional arrest by CAG trinucleotide repeat slip-outs. Nucleic Acids Res. 2021, 49, 7618–7627. [Google Scholar] [CrossRef] [PubMed]
- Zuniga, G.; Frost, B. Selective neuronal vulnerability to deficits in RNA processing. Prog. Neurobiol. 2023, 229, 102500. [Google Scholar] [CrossRef]
- Richard, P.; Feng, S.; Tsai, Y.L.; Li, W.; Rinchetti, P.; Muhith, U.; Irizarry-Cole, J.; Stolz, K.; Sanz, L.A.; Hartono, S.; et al. SETX (senataxin), the helicase mutated in AOA2 and ALS4, functions in autophagy regulation. Autophagy 2021, 17, 1889–1906. [Google Scholar] [CrossRef]
- Kumari, D.; Gazy, I.; Usdin, K. Pharmacological reactivation of the silenced FMR1 gene as a targeted therapeutic approach for fragile X syndrome. Brain Sci. 2019, 9, 39. [Google Scholar] [CrossRef]
- Chakraborty, A.; Jenjaroenpun, P.; Li, J.; El Hilali, S.; McCulley, A.; Haarer, B.; Hoffman, E.A.; Belak, A.; Thorland, A.; Hehnly, H.; et al. Replication stress induces global chromosome breakage in the fragile X genome. Cell Rep. 2020, 32, 108179. [Google Scholar] [CrossRef] [PubMed]
- Sato, N.; Amino, T.; Kobayashi, K.; Asakawa, S.; Ishiguro, T.; Tsunemi, T.; Takahashi, M.; Matsuura, T.; Flanigan, K.M.; Iwasaki, S.; et al. Spinocerebellar ataxia type 31 is associated with "inserted" penta-nucleotide repeats containing (TGGAA)n. Am. J. Hum. Genet. 2009, 85, 544–557. [Google Scholar] [CrossRef]
- Rabe, B. Aicardi-goutieres syndrome: Clues from the RNase H2 knock-out mouse. J. Mol. Med. 2013, 91, 1235–1240. [Google Scholar] [CrossRef] [PubMed]
- Madabhushi, R.; Pan, L.; Tsai, L.H. DNA damage and its links to neurodegeneration. Neuron 2014, 83, 266–282. [Google Scholar] [CrossRef] [PubMed]
- Schumacher, B.; Pothof, J.; Vijg, J.; Hoeijmakers, J.H.J. The central role of DNA damage in the ageing process. Nature 2021, 592, 695–703. [Google Scholar] [CrossRef]
- Haradhvala, N.J.; Polak, P.; Stojanov, P.; Covington, K.R.; Shinbrot, E.; Hess, J.M.; Rheinbay, E.; Kim, J.; Maruvka, Y.E.; Braunstein, L.Z.; et al. Mutational strand asymmetries in cancer genomes reveal mechanisms of DNA damage and repair. Cell 2016, 164, 538–549. [Google Scholar] [CrossRef]
- Natale, V.; Raquer, H. Xeroderma pigmentosum-cockayne syndrome complex. Orphanet. J. Rare. Dis. 2017, 12, 65. [Google Scholar] [CrossRef]
- Khan, E.S.; Danckwardt, S. Pathophysiological role and diagnostic potential of R-Loops in cancer and beyond. Genes 2022, 13, 2181. [Google Scholar] [CrossRef]
- Li, F.; Zafar, A.; Luo, L.; Denning, A.M.; Gu, J.; Bennett, A.; Yuan, F.; Zhang, Y. R-loops in genome instability and cancer. Cancers 2023, 15, 4986. [Google Scholar] [CrossRef]
- Vohhodina, J.; Goehring, L.J.; Liu, B.; Kong, Q.; Botchkarev, V.V., Jr.; Huynh, M.; Liu, Z.; Abderazzaq, F.O.; Clark, A.P.; Ficarro, S.B.; et al. BRCA1 binds TERRA RNA and suppresses R-loop-based telomeric DNA damage. Nat. Commun. 2021, 12, 3542. [Google Scholar] [CrossRef]
- Garcia-Rubio, M.L.; Perez-Calero, C.; Barroso, S.I.; Tumini, E.; Herrera-Moyano, E.; Rosado, I.V.; Aguilera, A. The fanconi anemia pathway protects genome integrity from R-loops. PLoS Genet. 2015, 11, e1005674. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, H.D.; Leong, W.Y.; Li, W.; Reddy, P.N.G.; Sullivan, J.D.; Walter, M.J.; Zou, L.; Graubert, T.A. Spliceosome mutations induce R loop-associated sensitivity to ATR inhibition in myelodysplastic syndromes. Cancer Res. 2018, 78, 5363–5374. [Google Scholar] [CrossRef]
- Wang, S.; Min, Z.; Ji, Q.; Geng, L.; Su, Y.; Liu, Z.; Hu, H.; Wang, L.; Zhang, W.; Suzuiki, K.; et al. Rescue of premature aging defects in Cockayne syndrome stem cells by CRISPR/Cas9-mediated gene correction. Protein Cell 2020, 11, 1–22. [Google Scholar] [CrossRef]
- Gatti, V.; De Domenico, S.; Melino, G.; Peschiaroli, A. Senataxin and R-loops homeostasis: Multifaced implications in carcinogenesis. Cell Death Discov. 2023, 9, 145. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Xiao, L. Progress in AAV-mediated in vivo gene therapy and its applications in central nervous system diseases. Int. J. Mol. Sci. 2025, 26, 2213. [Google Scholar] [CrossRef]
- Kotsantis, P.; Silva, L.M.; Irmscher, S.; Jones, R.M.; Folkes, L.; Gromak, N.; Petermann, E. Increased global transcription activity as a mechanism of replication stress in cancer. Nat. Commun. 2016, 7, 13087. [Google Scholar] [CrossRef]
- Stork, C.T.; Bocek, M.; Crossley, M.P.; Sollier, J.; Sanz, L.A.; Chedin, F.; Swigut, T.; Cimprich, K.A. Co-transcriptional R-loops are the main cause of estrogen-induced DNA damage. Elife 2016, 5, e17548. [Google Scholar] [CrossRef]
- Kannan, A.; Cuartas, J.; Gangwani, P.; Branzei, D.; Gangwani, L. Mutation in senataxin alters the mechanism of R-loop resolution in amyotrophic lateral sclerosis 4. Brain 2022, 145, 3072–3094. [Google Scholar] [CrossRef] [PubMed]
- Elsakrmy, N.; Cui, H. R-loops and R-loop-binding proteins in cancer progression and drug resistance. Int. J. Mol. Sci. 2023, 24, 7064. [Google Scholar] [CrossRef]
- Camino, L.P.; Dutta, A.; Barroso, S.; Perez-Calero, C.; Katz, J.N.; Garcia-Rubio, M.; Sung, P.; Gomez-Gonzalez, B.; Aguilera, A. DICER ribonuclease removes harmful R-loops. Mol. Cell 2023, 83, 3707–3719. [Google Scholar] [CrossRef]
- Lam, F.C.; Kong, Y.W.; Yaffe, M.B. Inducing DNA damage through R-loops to kill cancer cells. Mol. Cell Oncol. 2020, 8, 1848233. [Google Scholar] [CrossRef] [PubMed]
- Pommier, Y.; Nussenzweig, A.; Takeda, S.; Austin, C. Human topoisomerases and their roles in genome stability and organization. Nat. Rev. Mol. Cell Biol. 2022, 23, 407–427. [Google Scholar] [CrossRef] [PubMed]
- Topka, S.; Steinsnyder, Z.; Ravichandran, V.; Tkachuk, K.; Kemel, Y.; Bandlamudi, C.; Winkel Madsen, M.; Furberg, H.; Ouerfelli, O.; Rudin, C.M.; et al. Targeting germline- and tumor-associated nucleotide excision repair defects in cancer. Clin. Cancer Res. 2021, 27, 1997–2010. [Google Scholar] [CrossRef] [PubMed]
- Huang, R.; Zhou, P.K. DNA damage repair: Historical perspectives, mechanistic pathways and clinical translation for targeted cancer therapy. Signal Transduct. Target Ther. 2021, 6, 254. [Google Scholar] [CrossRef]
- Mandal, R.; Becker, S.; Strebhardt, K. Targeting CDK9 for anti-cancer therapeutics. Cancers 2021, 13, 2181. [Google Scholar] [CrossRef]
- Huang, Q.; Figueiredo-Pereira, M.E. Ubiquitin/proteasome pathway impairment in neurodegeneration: Therapeutic implications. Apoptosis 2010, 15, 1292–1311. [Google Scholar] [CrossRef]
- Liu, Z.S.; Sinha, S.; Bannister, M.; Song, A.; Arriaga-Gomez, E.; McKeeken, A.J.; Bonner, E.A.; Hanson, B.K.; Sarchi, M.; Takashima, K.; et al. R-loop accumulation in spliceosome mutant leukemias confers sensitivity to PARP1 inhibition by triggering transcription-replication conflicts. Cancer Res. 2024, 84, 577–597. [Google Scholar] [CrossRef]
- Yano, K.; Shiotani, B. Emerging strategies for cancer therapy by ATR inhibitors. Cancer Sci. 2023, 114, 2709–2721. [Google Scholar] [CrossRef]
- Liu, J.; Li, F.; Cao, Y.; Lv, Y.; Lei, K.; Tu, Z.; Gong, C.; Wang, H.; Liu, F.; Huang, K. Mechanisms underlining R-loop biology and implications for human disease. Front. Cell Dev. Biol. 2025, 13, 1537731. [Google Scholar] [CrossRef]
- San Martin Alonso, M.; Noordermeer, S.M. Untangling the crosstalk between BRCA1 and R-loops during DNA repair. Nucleic Acids Res. 2021, 49, 4848–4863. [Google Scholar] [CrossRef]
- Rao, S.; Andrs, M.; Shukla, K.; Isik, E.; Konig, C.; Schneider, S.; Bauer, M.; Rosano, V.; Prokes, J.; Muller, A.; et al. Senataxin RNA/DNA helicase promotes replication restart at co-transcriptional R-loops to prevent MUS81-dependent fork degradation. Nucleic Acids Res. 2024, 52, 10355–10369. [Google Scholar] [CrossRef]
- Shadrick, W.R.; Ndjomou, J.; Kolli, R.; Mukherjee, S.; Hanson, A.M.; Frick, D.N. Discovering new medicines targeting helicases: Challenges and recent progress. J. Biomol. Screen 2013, 18, 761–781. [Google Scholar] [CrossRef] [PubMed]
- Stratigi, K.; Siametis, A.; Garinis, G.A. Looping forward: Exploring R-loop processing and therapeutic potential. FEBS Lett. 2025, 599, 244–266. [Google Scholar] [CrossRef] [PubMed]
- Maffia, A.; Ranise, C.; Sabbioneda, S. From R-loops to G-quadruplexes: Emerging new threats for the replication fork. Int. J. Mol. Sci. 2020, 21, 1506. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Sun, H.S.; Wang, X.; Dumont, A.S.; Liu, Q. Cellular senescence, DNA damage, and neuroinflammation in the aging brain. Trends Neurosci. 2024, 47, 461–474. [Google Scholar] [CrossRef]
- Li, X.; Cao, G.; Liu, X.; Tang, T.S.; Guo, C.; Liu, H. Polymerases and DNA repair in neurons: Implications in neuronal survival and neurodegenerative diseases. Front. Cell Neurosci. 2022, 16, 852002. [Google Scholar] [CrossRef]
- Tsutakawa, S.E.; Tsai, C.L.; Yan, C.; Bralic, A.; Chazin, W.J.; Hamdan, S.M.; Scharer, O.D.; Ivanov, I.; Tainer, J.A. Envisioning how the prototypic molecular machine TFIIH functions in transcription initiation and DNA repair. DNA Repair 2020, 96, 102972. [Google Scholar] [CrossRef]
- Shadfar, S.; Parakh, S.; Jamali, M.S.; Atkin, J.D. Redox dysregulation as a driver for DNA damage and its relationship to neurodegenerative diseases. Transl. Neurodegener. 2023, 12, 18. [Google Scholar] [CrossRef]
- Murata, M.M.; Kong, X.; Moncada, E.; Chen, Y.; Imamura, H.; Wang, P.; Berns, M.W.; Yokomori, K.; Digman, M.A. NAD+ consumption by PARP1 in response to DNA damage triggers metabolic shift critical for damaged cell survival. Mol. Biol. Cell 2019, 30, 2584–2597. [Google Scholar] [CrossRef]
- Cristini, A.; Tellier, M.; Constantinescu, F.; Accalai, C.; Albulescu, L.O.; Heiringhoff, R.; Bery, N.; Sordet, O.; Murphy, S.; Gromak, N. RNase H2, mutated in aicardi-goutieres syndrome, resolves co-transcriptional R-loops to prevent DNA breaks and inflammation. Nat. Commun. 2022, 13, 2961. [Google Scholar] [CrossRef]
- Martin, R.M.; de Almeida, M.R.; Gameiro, E.; de Almeida, S.F. Live-cell imaging unveils distinct R-loop populations with heterogeneous dynamics. Nucleic Acids Res. 2023, 51, 11010–11023. [Google Scholar] [CrossRef] [PubMed]
- Das, S.K.; Menezes, M.E.; Bhatia, S.; Wang, X.Y.; Emdad, L.; Sarkar, D.; Fisher, P.B. Gene therapies for cancer: Strategies, challenges and successes. J. Cell Physiol. 2015, 230, 259–271. [Google Scholar] [CrossRef] [PubMed]
- Parambi, D.G.T.; Alharbi, K.S.; Kumar, R.; Harilal, S.; Batiha, G.E.; Cruz-Martins, N.; Magdy, O.; Musa, A.; Panda, D.S.; Mathew, B. Gene therapy approach with an emphasis on growth factors: Theoretical and clinical outcomes in neurodegenerative diseases. Mol. Neurobiol. 2022, 59, 191–233. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.; Dent, S.Y.; Wilson, J.H.; Wells, R.D.; Napierala, M. R loops stimulate genetic instability of CTG.CAG repeats. Proc. Natl. Acad. Sci. USA 2010, 107, 692–697. [Google Scholar] [CrossRef]
- Crowner, A.; Smith, K.; DeSmet, M. Regulation of R-loops in DNA tumor viruses. Pathogens 2024, 13, 863. [Google Scholar] [CrossRef]
- Yan, P.; Liu, Z.; Song, M.; Wu, Z.; Xu, W.; Li, K.; Ji, Q.; Wang, S.; Liu, X.; Yan, K.; et al. Genome-wide R-loop landscapes during cell differentiation and reprogramming. Cell Rep. 2020, 32, 107870. [Google Scholar] [CrossRef]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jeon, J.; Kang, T.-H. Transcription-Coupled Repair and R-Loop Crosstalk in Genome Stability. Int. J. Mol. Sci. 2025, 26, 3744. https://doi.org/10.3390/ijms26083744
Jeon J, Kang T-H. Transcription-Coupled Repair and R-Loop Crosstalk in Genome Stability. International Journal of Molecular Sciences. 2025; 26(8):3744. https://doi.org/10.3390/ijms26083744
Chicago/Turabian StyleJeon, Jeseok, and Tae-Hong Kang. 2025. "Transcription-Coupled Repair and R-Loop Crosstalk in Genome Stability" International Journal of Molecular Sciences 26, no. 8: 3744. https://doi.org/10.3390/ijms26083744
APA StyleJeon, J., & Kang, T.-H. (2025). Transcription-Coupled Repair and R-Loop Crosstalk in Genome Stability. International Journal of Molecular Sciences, 26(8), 3744. https://doi.org/10.3390/ijms26083744