
How Cells Die in Psoriasis?



Abstract
1. Introduction
2. Methods
2.1. The Approach: Bibliographic Research
2.2. Tools for Manuscript Preparation and Visualization
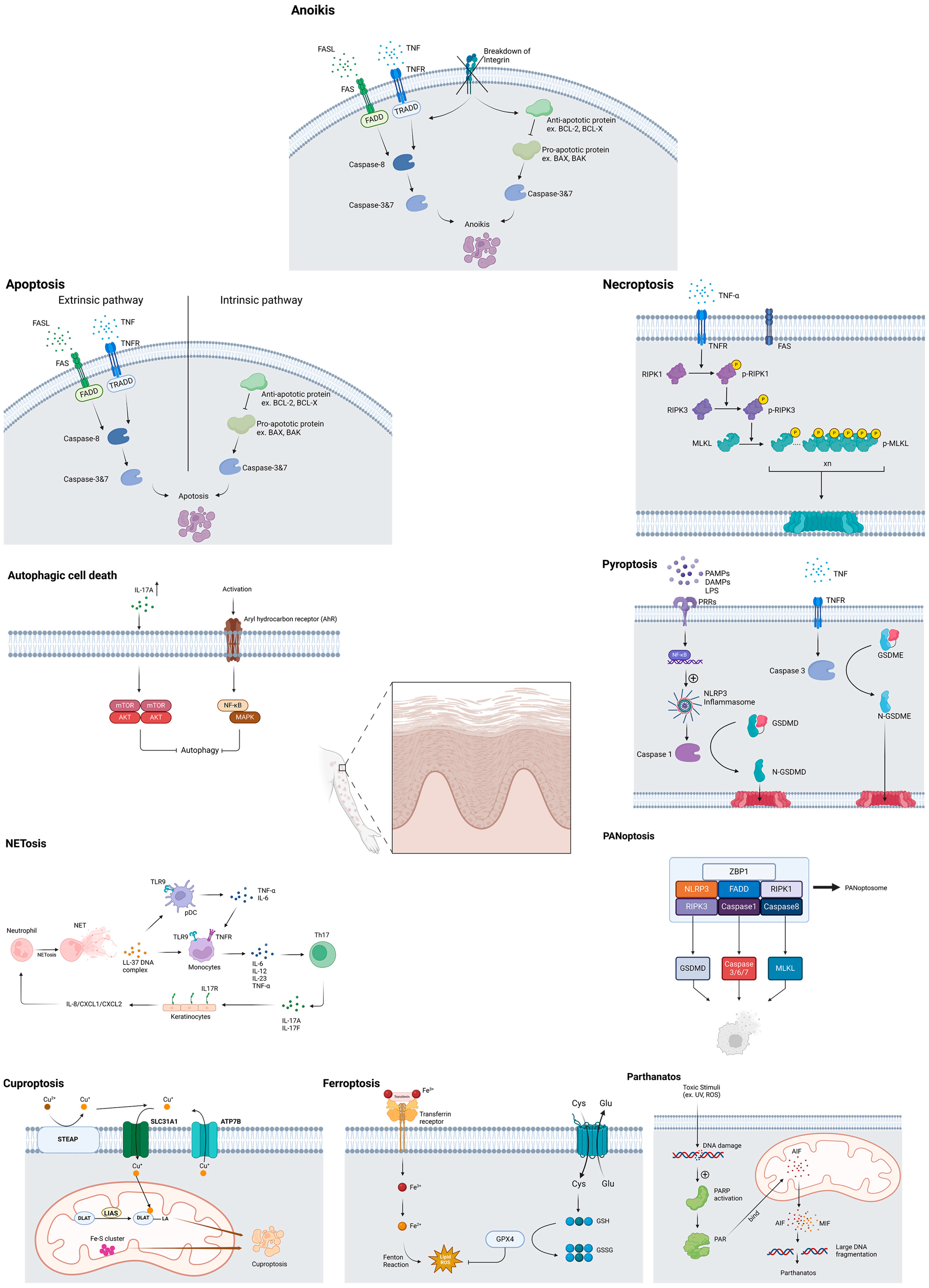
3. Results
3.1. Autophagic Cell DEATH
3.1.1. Autophagic Cell DEATH and Psoriasis
3.1.2. Therapeutic Modulation of Autophagy in Psoriasis
3.2. Apoptosis
3.2.1. Apoptosis and Psoriasis
3.2.2. Therapeutic Modulation of Apoptosis in Psoriasis
3.3. Anoikis
3.3.1. Anoikis and Psoriasis
3.3.2. Therapeutic Modulation of Anoikis in Psoriasis
3.4. Necroptosis
3.4.1. Necroptosis and Psoriasis
3.4.2. Therapeutic Approaches Targeting Necroptosis in Psoriasis
3.5. Pyroptosis
3.5.1. Pyroptosis and Psoriasis
3.5.2. Potential Therapeutic Approaches Targeting Pyroptosis in Psoriasis
3.6. PANoptosis
3.7. Parthanatos
3.7.1. Parthanatos and Psoriasis
3.7.2. Potential Therapeutic Approaches Targeting Parthanatos in Psoriasis
3.8. Ferroptosis
3.8.1. Ferroptosis and Psoriasis
3.8.2. Potential Therapeutic Approaches Targeting Ferroptosis in Psoriasis
3.9. Cuproptosis
3.10. NETosis
3.10.1. NETosis and Psoriasis
3.10.2. Potential Therapeutic Approaches Targeting NETosis in Psoriasis
3.11. Summary of Potential Therapeutic Approaches in Psoriasis
4. Discussion
5. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Greb, J.E.; Goldminz, A.M.; Elder, J.T.; Lebwohl, M.G.; Gladman, D.D.; Wu, J.J.; Mehta, N.N.; Finlay, A.Y.; Gottlieb, A.B. Psoriasis. Nat. Rev. Dis. Primers 2016, 2, 16082. [Google Scholar] [CrossRef] [PubMed]
- Wrone-Smith, T.; Mitra, R.S.; Thompson, C.B.; Jasty, R.; Castle, V.P.; Nickoloff, B.J. Keratinocytes Derived from Psoriatic Plaques Are Resistant to Apoptosis Compared with Normal Skin. Am. J. Pathol. 1997, 151, 1321–1329. [Google Scholar] [PubMed]
- Laporte, M.; Galand, P.; Fokan, D.; de Graef, C.; Heenen, M. Apoptosis in Established and Healing Psoriasis. Dermatology 2000, 200, 314–316. [Google Scholar] [CrossRef]
- Bedoui, S.; Herold, M.J.; Strasser, A. Emerging Connectivity of Programmed Cell Death Pathways and Its Physiological Implications. Nat. Rev. Mol. Cell Biol. 2020, 21, 678–695. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Zhu, Y.; Zhang, L.; Guo, L.; Wang, X.; Pan, Z.; Jiang, X.; Wu, F.; He, G. Mechanisms of Panoptosis and Relevant Small-Molecule Compounds for Fighting Diseases. Cell Death Dis. 2023, 14, 851. [Google Scholar] [CrossRef]
- Li, J.; Cao, F.; Yin, H.-L.; Huang, Z.-J.; Lin, Z.-T.; Mao, N.; Sun, B.; Wang, G. Ferroptosis: Past, Present and Future. Cell Death Dis. 2020, 11, 88. [Google Scholar] [CrossRef]
- Vorobjeva, N.V.; Chernyak, B.V. Netosis: Molecular Mechanisms, Role in Physiology and Pathology. Biochemistry 2020, 85, 1178–1190. [Google Scholar] [CrossRef]
- Han, Y.-H.; Wang, Y.; Lee, S.-J.; Jin, M.-H.; Sun, H.-N.; Kwon, T. Regulation of Anoikis by Extrinsic Death Receptor Pathways. Cell Commun. Signal. 2023, 21, 227. [Google Scholar] [CrossRef]
- Martínez-Morcillo, F.J.; Cantón-Sandoval, J.; Martínez-Navarro, F.J.; Cabas, I.; Martínez-Vicente, I.; Armistead, J.; Hatzold, J.; López-Muñoz, A.; Martínez-Menchón, T.; Corbalán-Vélez, R.; et al. Nampt-Derived Nad+ Fuels Parp1 to Promote Skin Inflammation through Parthanatos Cell Death. PLoS Biol. 2021, 19, e3001455. [Google Scholar] [CrossRef]
- Lin, Q.; Zhu, J.; Chen, J.; Jia, S.; Nie, S. Significance of Cuproptosis- Related Genes in the Diagnosis and Classification of Psoriasis. Front. Mol. Biosci. 2023, 10, 1115091. [Google Scholar] [CrossRef]
- Kroemer, G.; Galluzzi, L.; Vandenabeele, P.; Abrams, J.; Alnemri, E.S.; Baehrecke, E.H.; Blagosklonny, M.V.; El-Deiry, W.S.; Golstein, P.; Green, D.R.; et al. Classification of Cell Death: Recommendations of the Nomenclature Committee on Cell Death 2009. Cell Death Differ. 2009, 16, 3–11. [Google Scholar] [CrossRef]
- Galluzzi, L.; Vitale, I.; Aaronson, S.A.; Abrams, J.M.; Adam, D.; Agostinis, P.; Alnemri, E.S.; Altucci, L.; Amelio, I.; Andrews, D.W.; et al. Molecular Mechanisms of Cell Death: Recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ 2018, 25, 486–541. [Google Scholar] [PubMed]
- Khandia, R.; Dadar, M.; Munjal, A.; Dhama, K.; Karthik, K.; Tiwari, R.; Yatoo, M.I.; Iqbal, H.M.N.; Singh, K.P.; Joshi, S.K.; et al. A Comprehensive Review of Autophagy and Its Various Roles in Infectious, Non-Infectious, and Lifestyle Diseases: Current Knowledge and Prospects for Disease Prevention, Novel Drug Design, and Therapy. Cells 2019, 8, 674. [Google Scholar] [CrossRef] [PubMed]
- Aman, Y.; Schmauck-Medina, T.; Hansen, M.; Morimoto, R.I.; Simon, A.K.; Bjedov, I.; Palikaras, K.; Simonsen, A.; Johansen, T.; Tavernarakis, N.; et al. Autophagy in Healthy Aging and Disease. Nat. Aging 2021, 1, 634–650. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.M.; Shin, D.M.; Yuk, J.M.; Shi, G.; Choi, D.K.; Lee, S.H.; Huang, S.M.; Kim, J.M.; Kim, C.D.; Lee, J.H.; et al. Autophagy Negatively Regulates Keratinocyte Inflammatory Responses Via Scaffolding Protein P62/Sqstm1. J. Immunol. 2011, 186, 1248–1258. [Google Scholar] [CrossRef]
- Varshney, P.; Saini, N. Pi3k/Akt/Mtor Activation and Autophagy Inhibition Plays a Key Role in Increased Cholesterol During Il-17a Mediated Inflammatory Response in Psoriasis. Biochim. Biophys. Acta Mol. Basis Dis. 2018, 1864, 1795–1803. [Google Scholar] [CrossRef]
- Klapan, K.; Simon, D.; Karaulov, A.; Gomzikova, M.; Rizvanov, A.; Yousefi, S.; Simon, H.U. Autophagy and Skin Diseases. Front. Pharmacol. 2022, 13, 844756. [Google Scholar] [CrossRef]
- Wu, X.; Song, J.; Zhang, Y.; Kuai, L.; Liu, C.; Ma, X.; Li, B.; Zhang, Z.; Luo, Y. Exploring the Role of Autophagy in Psoriasis Pathogenesis: Insights into Sustained Inflammation and Dysfunctional Keratinocyte Differentiation. Int. Immunopharmacol. 2024, 135, 112244. [Google Scholar] [CrossRef]
- Duan, Y.; Dong, Y.; Hu, H.; Wang, Q.; Guo, S.; Fu, D.; Song, X.; Kalvakolanu, D.V.; Tian, Z. Il-33 Contributes to Disease Severity in Psoriasis-Like Models of Mouse. Cytokine 2019, 119, 159–167. [Google Scholar] [CrossRef]
- Kim, H.R.; Kang, S.Y.; Kim, H.O.; Park, C.W.; Chung, B.Y. Role of Aryl Hydrocarbon Receptor Activation and Autophagy in Psoriasis-Related Inflammation. Int. J. Mol. Sci. 2020, 21, 2195. [Google Scholar] [CrossRef]
- Wang, Z.; Zhou, H.; Zheng, H.; Zhou, X.; Shen, G.; Teng, X.; Liu, X.; Zhang, J.; Wei, X.; Hu, Z.; et al. Autophagy-Based Unconventional Secretion of Hmgb1 by Keratinocytes Plays a Pivotal Role in Psoriatic Skin Inflammation. Autophagy 2021, 17, 529–552. [Google Scholar] [CrossRef]
- Törmä, H. Regulation of Keratin Expression by Retinoids. Dermato-endocrinology 2011, 3, 136–140. [Google Scholar] [CrossRef]
- Rajawat, Y.; Hilioti, Z.; Bossis, I. Autophagy: A Target for Retinoic Acids. Autophagy 2010, 6, 1224–1226. [Google Scholar] [CrossRef]
- Wong, T.; Hsu, L.; Liao, W. Phototherapy in Psoriasis: A Review of Mechanisms of Action. J. Cutan. Med. Surg. 2013, 17, 6–12. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Wang, H.; Wang, S.; Xu, M.; Liu, M.; Liao, M.; Frank, J.A.; Adhikari, S.; Bower, K.A.; Shi, X.; et al. Gsk3β Signaling Is Involved in Ultraviolet B-Induced Activation of Autophagy in Epidermal Cells. Int. J. Oncol. 2012, 41, 1782–1788. [Google Scholar] [CrossRef] [PubMed]
- Gao, M.; Si, X. Rapamycin Ameliorates Psoriasis by Regulating the Expression and Methylation Levels of Tropomyosin Via Erk1/2 and Mtor Pathways in Vitro and in Vivo. Exp. Dermatol. 2018, 27, 1112–1119. [Google Scholar] [CrossRef] [PubMed]
- Bürger, C.; Shirsath, N.; Lang, V.; Diehl, S.; Kaufmann, R.; Weigert, A.; Han, Y.Y.; Ringel, C.; Wolf, P. Blocking Mtor Signalling with Rapamycin Ameliorates Imiquimod-Induced Psoriasis in Mice. Acta Derm. Venereol. 2017, 97, 1087–1094. [Google Scholar] [CrossRef]
- Wei, K.C.; Lai, P.C. Combination of Everolimus and Tacrolimus: A Potentially Effective Regimen for Recalcitrant Psoriasis. Dermatol. Ther. 2015, 28, 25–27. [Google Scholar] [CrossRef]
- Huang, Z.; Li, J.; Chen, H.; Yu, D.; Sun, S. The Efficacy of Metformin for the Treatment of Psoriasis: A Meta-Analysis Study. Postep. Dermatol. Alergol. 2023, 40, 606–610. [Google Scholar] [CrossRef]
- Park, M.J.; Lee, S.Y.; Moon, S.J.; Son, H.J.; Lee, S.H.; Kim, E.K.; Byun, J.K.; Shin, D.Y.; Park, S.H.; Yang, C.W.; et al. Metformin Attenuates Graft-Versus-Host Disease Via Restricting Mammalian Target of Rapamycin/Signal Transducer and Activator of Transcription 3 and Promoting Adenosine Monophosphate-Activated Protein Kinase-Autophagy for the Balance between T Helper 17 and Tregs. Transl. Res. 2016, 173, 115–130. [Google Scholar]
- Chang, J.E.; Choi, M.S. A Molecular Perspective on the Potential Benefits of Metformin for the Treatment of Inflammatory Skin Disorders. Int. J. Mol. Sci. 2020, 21, 8960. [Google Scholar] [CrossRef] [PubMed]
- Elmore, S. Apoptosis: A Review of Programmed Cell Death. Toxicol. Pathol. 2007, 35, 495–516. [Google Scholar] [CrossRef] [PubMed]
- Vind, A.C.; Wu, Z.; Firdaus, M.J.; Snieckute, G.; Toh, G.A.; Jessen, M.; Martínez, J.F.; Haahr, P.; Andersen, T.L.; Blasius, M.; et al. The Ribotoxic Stress Response Drives Acute Inflammation, Cell Death, and Epidermal Thickening in Uv-Irradiated Skin In vivo. Mol. Cell 2024, 84, 4774–4789.e9. [Google Scholar] [CrossRef] [PubMed]
- Jan, R.; Chaudhry, G.E. Understanding Apoptosis and Apoptotic Pathways Targeted Cancer Therapeutics. Adv. Pharm. Bull. 2019, 9, 205–218. [Google Scholar] [CrossRef]
- Takahashi, H.; Manabe, A.; Ishida-Yamamoto, A.; Hashimoto, Y.; Iizuka, H. Aberrant Expression of Apoptosis-Related Molecules in Psoriatic Epidermis. J. Dermatol. Sci. 2002, 28, 187–197. [Google Scholar] [CrossRef]
- Krawczyk, A.; Miśkiewicz, J.; Strzelec, K.; Wcisło-Dziadecka, D.; Strzalka-Mrozik, B. Apoptosis in Autoimmunological Diseases, with Particular Consideration of Molecular Aspects of Psoriasis. Med. Sci. Monit. 2020, 26, e922035. [Google Scholar] [CrossRef]
- Victor, F.C.; Gottlieb, A.B. Tnf-Alpha and Apoptosis: Implications for the Pathogenesis and Treatment of Psoriasis. J. Drugs Dermatol. 2002, 1, 264–275. [Google Scholar]
- Rückert, R.; Asadullah, K.; Seifert, M.; Budagian, V.M.; Arnold, R.; Trombotto, C.; Paus, R.; Bulfone-Paus, S. Inhibition of Keratinocyte Apoptosis by Il-15: A New Parameter in the Pathogenesis of Psoriasis? J. Immunol. 2000, 165, 2240–2250. [Google Scholar] [CrossRef]
- Stoll, S.W.; Benedict, M.; Mitra, R.; Hiniker, A.; Elder, J.T.; Nuñez, G. Egf Receptor Signaling Inhibits Keratinocyte Apoptosis: Evidence for Mediation by Bcl-Xl. Oncogene 1998, 16, 1493–1499. [Google Scholar] [CrossRef]
- Aufiero, B.M.; Talwar, H.; Young, C.; Krishnan, M.; Hatfield, J.S.; Lee, H.K.; Wong, H.K.; Hamzavi, I.; Murakawa, G.J. Narrow-Band Uvb Induces Apoptosis in Human Keratinocytes. J. Photochem. Photobiol. B Biol. 2006, 82, 132–139. [Google Scholar] [CrossRef]
- Weatherhead, S.C.; Farr, P.M.; Jamieson, D.; Hallinan, J.S.; Lloyd, J.J.; Wipat, A.; Reynolds, N.J. Keratinocyte Apoptosis in Epidermal Remodeling and Clearance of Psoriasis Induced by Uv Radiation. J. Investig. Dermatol. 2011, 131, 1916–1926. [Google Scholar] [CrossRef]
- Ozawa, M.; Ferenczi, K.; Kikuchi, T.; Cardinale, I.; Austin, L.M.; Coven, T.R.; Burack, L.H.; Krueger, J.G. 312-Nanometer Ultraviolet B Light (Narrow-Band Uvb) Induces Apoptosis of T Cells within Psoriatic Lesions. J. Exp. Med. 1999, 189, 711–718. [Google Scholar] [CrossRef] [PubMed]
- Chen, T.-J.; Chung, W.-H.; Chen, C.-B.; Hui, R.C.-Y.; Huang, Y.-H.; Lu, Y.-T.; Wang, C.-W.; Wang, K.-H.; Yang, L.-C.; Hung, S.-I. Methotrexate-Induced Epidermal Necrosis: A Case Series of 24 Patients. J. Am. Acad. Dermatol. 2017, 77, 247–255.e2. [Google Scholar] [CrossRef] [PubMed]
- Ladha, M.A.; Edgerton, B.; Levy, J.; Mahmood, M.N.; Devani, A.R.; Grewal, P.S.; Prajapati, V.H. Methotrexate-Induced Cutaneous Ulceration and Necrosis in Chronic Atopic Dermatitis. JAAD Case Rep. 2020, 6, 864–867. [Google Scholar] [CrossRef] [PubMed]
- Elango, T.; Thirupathi, A.; Subramanian, S.; Ethiraj, P.; Dayalan, H.; Gnanaraj, P. Methotrexate Treatment Provokes Apoptosis of Proliferating Keratinocyte in Psoriasis Patients. Clin. Exp. Med. 2017, 17, 371–381. [Google Scholar] [CrossRef]
- Halprin, K.M. Epidermal "Turnover Time"—A Re-Examination. Br. J. Dermatol. 1972, 86, 14–19. [Google Scholar] [CrossRef]
- Kadry, M.O.; Ammar, N.M.; Hassan, H.A.; Abdel Megeed, R.M. Insights on Attenuating Autophagy Cellular and Molecular Pathways Versus Methotrexate-Induced Toxicity Via Liposomal Turmeric Therapy. J. Genet. Eng. Biotechnol. 2022, 20, 147. [Google Scholar] [CrossRef]
- Elango, T.; Dayalan, H.; Gnanaraj, P.; Malligarjunan, H.; Subramanian, S. Impact of Methotrexate on Oxidative Stress and Apoptosis Markers in Psoriatic Patients. Clin. Exp. Med. 2014, 14, 431–437. [Google Scholar] [CrossRef]
- Soonthornchai, W.; Tangtanatakul, P.; Meephansan, J.; Ruchusatsawat, K.; Reantragoon, R.; Hirankarn, N.; Wongpiyabovorn, J. Down-Regulation of Mir-155 after Treatment with Narrow-Band Uvb and Methotrexate Associates with Apoptosis of Keratinocytes in Psoriasis. Asian Pac. J. Allergy Immunol. 2021, 39, 206–213. [Google Scholar]
- Manggau, M.; Kim, D.S.; Ruwisch, L.; Vogler, R.; Korting, H.C.; Schäfer-Korting, M.; Kleuser, B. 1alpha,25-Dihydroxyvitamin D3 Protects Human Keratinocytes from Apoptosis by the Formation of Sphingosine-1-Phosphate. J. Investig. Dermatol. 2001, 117, 1241–1249. [Google Scholar] [CrossRef]
- De Haes, P.; Garmyn, M.; Carmeliet, G.; Degreef, H.; Vantieghem, K.; Bouillon, R.; Segaert, S. Molecular Pathways Involved in the Anti-Apoptotic Effect of 1,25-Dihydroxyvitamin D3 in Primary Human Keratinocytes. J. Cell Biochem. 2004, 93, 951–967. [Google Scholar] [CrossRef] [PubMed]
- Tiberio, R.; Bozzo, C.; Pertusi, G.; Graziola, F.; Gattoni, M.; Griffanti, P.; Boggio, P.; Colombo, E.; Leigheb, G. Calcipotriol Induces Apoptosis in Psoriatic Keratinocytes. Clin. Exp. Dermatol. 2009, 34, e972–e974. [Google Scholar] [CrossRef] [PubMed]
- Saurat, J.H. Retinoids and Psoriasis: Novel Issues in Retinoid Pharmacology and Implications for Psoriasis Treatment. J. Am. Acad. Dermatol. 1999, 41, S2–S6. [Google Scholar] [CrossRef] [PubMed]
- Kerr, P.E.; DiGiovanna, J.J. From Vitamin to Vesanoid: Systemic Retinoids for the New Millennium. Med. Health 2001, 84, 228–231. [Google Scholar]
- Islam, T.C.; Skarin, T.; Sumitran, S.; Toftgård, R. Retinoids Induce Apoptosis in Cultured Keratinocytes. Br. J. Dermatol. 2000, 143, 709–719. [Google Scholar] [CrossRef]
- Kakavandi, E.; Shahbahrami, R.; Goudarzi, H.; Eslami, G.; Faghihloo, E. Anoikis Resistance and Oncoviruses. J. Cell. Biochem. 2018, 119, 2484–2491. [Google Scholar] [CrossRef]
- Taddei, M.L.; Giannoni, E.; Fiaschi, T.; Chiarugi, P. Anoikis: An Emerging Hallmark in Health and Diseases. J. Pathol. 2012, 226, 380–393. [Google Scholar] [CrossRef]
- De Luca, M.; Pellegrini, G.; Zambruno, G.; Marchisio, P.C. Role of Integrins in Cell Adhesion and Polarity in Normal Keratinocytes and Human Skin Pathologies. J. Dermatol. 1994, 21, 821–828. [Google Scholar] [CrossRef]
- Mei, J.; Jiang, X.Y.; Tian, H.X.; Rong, D.C.; Song, J.N.; Wang, L.; Chen, Y.S.; Wong, R.C.B.; Guo, C.X.; Wang, L.S.; et al. Anoikis in Cell Fate, Physiopathology, and Therapeutic Interventions. MedComm (2020) 2024, 5, e718. [Google Scholar] [CrossRef]
- Kothary, N.; Diak, I.L.; Brinker, A.; Bezabeh, S.; Avigan, M.; Dal Pan, G. Progressive Multifocal Leukoencephalopathy Associated with Efalizumab Use in Psoriasis Patients. J. Am. Acad. Dermatol. 2011, 65, 546–551. [Google Scholar] [CrossRef]
- Sun, L.; Wang, H.; Wang, Z.; He, S.; Chen, S.; Liao, D.; Wang, L.; Yan, J.; Liu, W.; Lei, X.; et al. Mixed Lineage Kinase Domain-Like Protein Mediates Necrosis Signaling Downstream of Rip3 Kinase. Cell 2012, 148, 213–227. [Google Scholar] [CrossRef] [PubMed]
- Estlack, L.E.; Roth, C.C.; Thompson, G.L., 3rd; Lambert, W.A., 3rd; Ibey, B.L. Nanosecond Pulsed Electric Fields Modulate the Expression of Fas/Cd95 Death Receptor Pathway Regulators in U937 and Jurkat Cells. Apoptosis 2014, 19, 1755–1768. [Google Scholar] [CrossRef] [PubMed]
- Feoktistova, M.; Geserick, P.; Kellert, B.; Dimitrova, D.P.; Langlais, C.; Hupe, M.; Cain, K.; MacFarlane, M.; Häcker, G.; Leverkus, M. Ciaps Block Ripoptosome Formation, a Rip1/Caspase-8 Containing Intracellular Cell Death Complex Differentially Regulated by Cflip Isoforms. Mol. Cell 2011, 43, 449–463. [Google Scholar] [CrossRef]
- Murphy, J.M.; Czabotar, P.E.; Hildebrand, J.M.; Lucet, I.S.; Zhang, J.G.; Alvarez-Diaz, S.; Lewis, R.; Lalaoui, N.; Metcalf, D.; Webb, A.I.; et al. The Pseudokinase Mlkl Mediates Necroptosis Via a Molecular Switch Mechanism. Immunity 2013, 39, 443–453. [Google Scholar] [CrossRef] [PubMed]
- Cai, Z.; Jitkaew, S.; Zhao, J.; Chiang, H.-C.; Choksi, S.; Liu, J.; Ward, Y.; Wu, L.-g.; Liu, Z.-G. Plasma Membrane Translocation of Trimerized Mlkl Protein Is Required for Tnf-Induced Necroptosis. Nat. Cell Biol. 2014, 16, 55–65. [Google Scholar] [CrossRef]
- Duan, X.; Liu, X.; Liu, N.; Huang, Y.; Jin, Z.; Zhang, S.; Ming, Z.; Chen, H. Inhibition of Keratinocyte Necroptosis Mediated by Ripk1/Ripk3/Mlkl Provides a Protective Effect against Psoriatic Inflammation. Cell Death Dis. 2020, 11, 134. [Google Scholar] [CrossRef]
- Burlando, M.; Campione, E.; Cuccia, A.; Malara, G.; Naldi, L.; Prignano, F.; Zichichi, L. Real-World Use of Dimethyl Fumarate in Patients with Plaque Psoriasis: A Delphi-Based Expert Consensus. Dermatol. Rep. 2023, 15, 9613. [Google Scholar]
- Corazza, M.; Odorici, G.; Conti, A.; Di Lernia, V.; Motolese, A.; Bardazzi, F.; Di Nuzzo, S.; Monti, A.; Arginelli, F.; Filippi, F.; et al. Dimethyl Fumarate Treatment for Psoriasis in a Real-Life Setting: A Multicentric Retrospective Study. Dermatol. Ther. 2021, 34, e15066. [Google Scholar] [CrossRef]
- Morrison, P.J.; Suhrkamp, I.; Gerdes, S.; Mrowietz, U. Oral Dimethyl Fumarate Induces Changes within the Peripheral Neutrophil Compartment of Patients with Psoriasis That Are Linked with Skin Improvement*. Br. J. Dermatol. 2021, 185, 605–615. [Google Scholar] [CrossRef]
- Shi, F.-L.; Yuan, L.-S.; Wong, T.-S.; Li, Q.; Li, Y.-P.; Xu, R.; You, Y.-P.; Yuan, T.; Zhang, H.-R.; Shi, Z.-J.; et al. Dimethyl Fumarate Inhibits Necroptosis and Alleviates Systemic Inflammatory Response Syndrome by Blocking the Ripk1-Ripk3-Mlkl Axis. Pharmacol. Res. 2023, 189, 106697. [Google Scholar] [CrossRef]
- Li, J.; Liu, X.; Liu, Y.; Huang, F.; Liang, J.; Lin, Y.; Hu, F.; Feng, J.; Han, Z.; Chen, Y.; et al. Saracatinib Inhibits Necroptosis and Ameliorates Psoriatic Inflammation by Targeting Mlkl. Cell Death Dis. 2024, 15, 122. [Google Scholar] [CrossRef] [PubMed]
- Lu, L.; Zhang, Y.; Tan, X.; Merkher, Y.; Leonov, S.; Zhu, L.; Deng, Y.; Zhang, H.; Zhu, D.; Tan, Y.; et al. Emerging Mechanisms of Pyroptosis and Its Therapeutic Strategy in Cancer. Cell Death Discov. 2022, 8, 338. [Google Scholar] [CrossRef] [PubMed]
- Yu, P.; Zhang, X.; Liu, N.; Tang, L.; Peng, C.; Chen, X. Pyroptosis: Mechanisms and Diseases. Signal Transduct. Target. Ther. 2021, 6, 128. [Google Scholar] [CrossRef]
- Liu, X.; Chen, J.; Yue, S.; Zhang, C.; Song, J.; Liang, H.; Liang, C.; Chen, X. Nlrp3-Mediated Il-1β in Regulating the Imbalance between Th17 and Treg in Experimental Autoimmune Prostatitis. Sci. Rep. 2024, 14, 18829. [Google Scholar] [CrossRef]
- Lalor, S.J.; Dungan, L.S.; Sutton, C.E.; Basdeo, S.A.; Fletcher, J.M.; Mills, K.H. Caspase-1-Processed Cytokines Il-1beta and Il-18 Promote Il-17 Production by Gammadelta and Cd4 T Cells That Mediate Autoimmunity. J. Immunol. 2011, 186, 5738–5748. [Google Scholar] [CrossRef]
- Lieberman, J.; Wu, H.; Kagan, J.C. Gasdermin D Activity in Inflammation and Host Defense. Sci. Immunol. 2019, 4, eaav1447. [Google Scholar] [CrossRef] [PubMed]
- Nowowiejska, J.; Baran, A.; Hermanowicz, J.M.; Pryczynicz, A.; Sieklucka, B.; Pawlak, D.; Flisiak, I. Gasdermin D (Gsdmd) Is Upregulated in Psoriatic Skin-a New Potential Link in the Pathogenesis of Psoriasis. Int. J. Mol. Sci. 2023, 24, 13047. [Google Scholar] [CrossRef]
- Lian, N.; Chen, Y.; Chen, S.; Zhang, Y.; Chen, H.; Yang, Y.; Gu, H.; Chen, Q.; Li, M.; Chen, X. Gasdermin D-Mediated Keratinocyte Pyroptosis as a Key Step in Psoriasis Pathogenesis. Cell Death Dis. 2023, 14, 595. [Google Scholar] [CrossRef]
- Wu, J.; Lin, S.; Chen, W.; Lian, G.; Wu, W.; Chen, A.; Sagor, M.I.H.; Luo, L.; Wang, H.; Xie, L. Tnf-A Contributes to Sarcopenia through Caspase-8/Caspase-3/Gsdme-Mediated Pyroptosis. Cell Death Discov. 2023, 9, 76. [Google Scholar] [CrossRef]
- Hu, Y.; Liu, Y.; Zong, L.; Zhang, W.; Liu, R.; Xing, Q.; Liu, Z.; Yan, Q.; Li, W.; Lei, H.; et al. The Multifaceted Roles of Gsdme-Mediated Pyroptosis in Cancer: Therapeutic Strategies and Persisting Obstacles. Cell Death Dis. 2023, 14, 836. [Google Scholar] [CrossRef]
- Li, Y.; He, Y.; Yang, F.; Liang, R.; Xu, W.; Li, Y.; Cheng, J.; Liang, B.; Tang, M.; Shi, X.; et al. Gasdermin E-Mediated Keratinocyte Pyroptosis Participates in the Pathogenesis of Psoriasis by Promoting Skin Inflammation. Br. J. Dermatol. 2024, 191, 385–396. [Google Scholar] [CrossRef] [PubMed]
- Nowowiejska, J.; Baran, A.; Pryczynicz, A.; Hermanowicz, J.M.; Sieklucka, B.; Pawlak, D.; Flisiak, I. Gasdermin E (GSDME)-A New Potential Marker of Psoriasis and Its Metabolic Complications: The First Combined Study on Human Serum, Urine and Tissue. Cells 2023, 12, 2149. [Google Scholar] [CrossRef]
- Lanz, J.; Biniaz-Harris, N.; Kuvaldina, M.; Jain, S.; Lewis, K.; Fallon, B.A. Disulfiram: Mechanisms, Applications, and Challenges. Antibiotics 2023, 12, 524. [Google Scholar] [CrossRef]
- Kaaber, K.; Menné, T.; Veien, N.; Hougaard, P. Treatment of Nickel Dermatitis with Antabuse; a Double Blind Study. Contact Dermat. 1983, 9, 297–299. [Google Scholar] [CrossRef] [PubMed]
- Kaaber, K.; Menne, T.; Tjell, J.C.; Veien, N. Antabuse Treatment of Nickel Dermatitis. Chelation—A New Principle in the Treatment of Nickel Dermatitis. Contact Dermat. 1979, 5, 221–228. [Google Scholar] [CrossRef] [PubMed]
- Jewell, E.W. Does Disulfiram Cure Psoriasis? JAMA 1977, 237, 2189. [Google Scholar] [CrossRef]
- Hu, X.M.; Zheng, S.; Zhang, Q.; Wan, X.; Li, J.; Mao, R.; Yang, R.; Xiong, K. Panoptosis Signaling Enables Broad Immune Response in Psoriasis: From Pathogenesis to New Therapeutic Strategies. Comput. Struct. Biotechnol. J. 2024, 23, 64–76. [Google Scholar] [CrossRef]
- Hu, J.J.; Liu, X.; Xia, S.; Zhang, Z.; Zhang, Y.; Zhao, J.; Ruan, J.; Luo, X.; Lou, X.; Bai, Y.; et al. Fda-Approved Disulfiram Inhibits Pyroptosis by Blocking Gasdermin D Pore Formation. Nat. Immunol. 2020, 21, 736–745. [Google Scholar] [CrossRef]
- Ou, A.-T.; Zhang, J.-X.; Fang, Y.-F.; Wang, R.; Tang, X.-P.; Zhao, P.-F.; Zhao, Y.-G.; Zhang, M.; Huang, Y.-Z. Disulfiram-Loaded Lactoferrin Nanoparticles for Treating Inflammatory Diseases. Acta Pharmacol. Sin. 2021, 42, 1913–1920. [Google Scholar] [CrossRef]
- Samir, P.; Malireddi, R.K.S.; Kanneganti, T.D. The Panoptosome: A Deadly Protein Complex Driving Pyroptosis, Apoptosis, and Necroptosis (Panoptosis). Front. Cell Infect. Microbiol. 2020, 10, 238. [Google Scholar] [CrossRef]
- Banoth, B.; Tuladhar, S.; Karki, R.; Sharma, B.R.; Briard, B.; Kesavardhana, S.; Burton, A.; Kanneganti, T.D. Zbp1 Promotes Fungi-Induced Inflammasome Activation and Pyroptosis, Apoptosis, and Necroptosis (Panoptosis). J. Biol. Chem. 2020, 295, 18276–18283. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Xiong, A.; Liu, J.; Li, X.; Wang, J.; Zhang, L.; Liu, Y.; Xiong, Y.; Li, G.; He, X. Panoptosis: Bridging Apoptosis, Pyroptosis, and Necroptosis in Cancer Progression and Treatment. Cancer Gene Ther. 2024, 31, 970–983. [Google Scholar] [CrossRef] [PubMed]
- Oh, S.; Lee, S. Recent Advances in Zbp1-Derived Panoptosis against Viral Infections. Front. Immunol. 2023, 14, 1148727. [Google Scholar] [CrossRef]
- Yang, Y.; Hounye, A.H.; Chen, Y.; Liu, Z.; Shi, G.; Xiao, Y. Characterization of Panoptosis-Related Genes in Crohn’s Disease by Integrated Bioinformatics, Machine Learning and Experiments. Sci. Rep. 2024, 14, 11731. [Google Scholar] [CrossRef]
- Lu, L.; Zhang, B.; Shi, M.; Liu, A. Identification of Panoptosis-Related Biomarkers and Immune Infiltration Characteristics in Psoriasis. Medicine 2023, 102, e35627. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.; Jiao, X.-L.; Jing, M.; Zhang, S.-X.; Wang, Y.; Li, C.-L.; Shi, G.-X.; Li, Z.-Y.; Liu, G.-L.; Yan, K.; et al. Discovery of Panoptosis-Related Signatures Correlates with Immune Cell Infiltration in Psoriasis. PLoS ONE 2024, 19, e0310362. [Google Scholar] [CrossRef]
- Liu, L.; Li, J.; Ke, Y.; Zeng, X.; Gao, J.; Ba, X.; Wang, R. The Key Players of Parthanatos: Opportunities for Targeting Multiple Levels in the Therapy of Parthanatos-Based Pathogenesis. Cell. Mol. Life Sci. 2022, 79, 60. [Google Scholar] [CrossRef]
- Zhang, Z.; Cheng, B.; Du, W.; Zeng, M.; He, K.; Yin, T.; Shang, S.; Su, T.; Han, D.; Gan, X.; et al. The Role of Nicotinamide Mononucleotide Supplementation in Psoriasis Treatment. Antioxidants 2024, 13, 186. [Google Scholar] [CrossRef]
- El-Khalawany, M.; Nouh, A.H.; Kadah, A.S.; Elsheikh, M.; Said, M. Evaluation of Safety and Efficacy of Topical 4% Nicotinamide in Treatment of Psoriasis; among a Representative Sample of Egyptians (an Analytical Observational Study). Dermatol. Ther. 2022, 35, e15734. [Google Scholar] [CrossRef]
- Jiang, X.; Stockwell, B.R.; Conrad, M. Ferroptosis: Mechanisms, Biology and Role in Disease. Nat. Rev. Mol. Cell Biol. 2021, 22, 266–282. [Google Scholar] [CrossRef]
- Yan, H.-f.; Zou, T.; Tuo, Q.-z.; Xu, S.; Li, H.; Belaidi, A.A.; Lei, P. Ferroptosis: Mechanisms and Links with Diseases. Signal Transduct. Target. Ther. 2021, 6, 49. [Google Scholar] [CrossRef] [PubMed]
- Ingold, I.; Berndt, C.; Schmitt, S.; Doll, S.; Poschmann, G.; Buday, K.; Roveri, A.; Peng, X.; Porto Freitas, F.; Seibt, T.; et al. Selenium Utilization by GPX4 Is Required to Prevent Hydroperoxide-Induced Ferroptosis. Cell 2018, 172, 409–422.e21. [Google Scholar] [CrossRef]
- Li, F.J.; Long, H.Z.; Zhou, Z.W.; Luo, H.Y.; Xu, S.G.; Gao, L.C. System X(C) (-)/Gsh/Gpx4 Axis: An Important Antioxidant System for the Ferroptosis in Drug-Resistant Solid Tumor Therapy. Front. Pharmacol. 2022, 13, 910292. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Green, M.; Choi, J.E.; Gijón, M.; Kennedy, P.D.; Johnson, J.K.; Liao, P.; Lang, X.; Kryczek, I.; Sell, A.; et al. Cd8+ T Cells Regulate Tumour Ferroptosis During Cancer Immunotherapy. Nature 2019, 569, 270–274. [Google Scholar] [CrossRef] [PubMed]
- Shou, Y.; Yang, L.; Yang, Y.; Xu, J. Inhibition of Keratinocyte Ferroptosis Suppresses Psoriatic Inflammation. Cell Death Dis. 2021, 12, 1009. [Google Scholar] [CrossRef]
- Bds, N.K.; Kothari, R.; Sandhu, S. Intravenous Glutathione Can Improve Scalp Psoriasis: A Serendipity. Biomed. J. Sci. Tech. Res. 2022, 43, 34162–34164. [Google Scholar]
- Prussick, R.; Prussick, L.; Gutman, J. Psoriasis Improvement in Patients Using Glutathione-enhancing, Nondenatured Whey Protein Isolate: A Pilot Study. J. Clin. Aesthet. Dermatol. 2013, 6, 23–26. [Google Scholar]
- Nazıroğlu, M.; Yıldız, K.; Tamtürk, B.; Erturan, İ.; Flores-Arce, M. Selenium and Psoriasis. Biol. Trace Elem. Res. 2012, 150, 3–9. [Google Scholar] [CrossRef]
- Chambers, I.G.; Ratan, R.R. Selenium Abandons Selenoproteins to Inhibit Ferroptosis Rapidly. Nat. Metab. 2024, 6, 200–202. [Google Scholar] [CrossRef]
- Al-Oudah, G.A.; Sahib, A.S.; Al-Hattab, M.K.; Al-Ameedee, A.A. Effect of Coq10 Administration to Psoriatic Iraqi Patients on Biological Therapy Upon Severity Index (Pasi) and Quality of Life Index (Dlqi) before and after Therapy. J. Popul. Ther. Clin. Pharmacol. 2022, 29, e52–e60. [Google Scholar]
- Kharaeva, Z.; Gostova, E.; De Luca, C.; Raskovic, D.; Korkina, L. CClinical and Biochemical Effects of Coenzyme Q(10), Vitamin E, and Selenium Supplementation to Psoriasis Patients. Nutrition 2009, 25, 295–302. [Google Scholar] [CrossRef] [PubMed]
- Hadian, K. Ferroptosis Suppressor Protein 1 (Fsp1) and Coenzyme Q(10) Cooperatively Suppress Ferroptosis. Biochemistry 2020, 59, 637–638. [Google Scholar] [CrossRef]
- Li, S.; Luo, X.; Zhang, S.; Su, Y.; Deng, M.; Zhu, Y.; Zhang, P.; Wu, R.; Zhao, M. Ferroptosis Activation Contributes to the Formation of Skin Lesions in Psoriasis Vulgaris. Antioxidants 2023, 12, 310. [Google Scholar] [CrossRef]
- Abboud, E.; Chrayteh, D.; Boussetta, N.; Dalle, H.; Malerba, M.; Wu, T.-D.; Le Gall, M.; Reelfs, O.; Pourzand, C.; Mellett, M.; et al. Skin Hepcidin Initiates Psoriasiform Skin Inflammation Via Fe-Driven Hyperproliferation and Neutrophil Recruitment. Nat. Commun. 2024, 15, 6718. [Google Scholar] [CrossRef] [PubMed]
- Tsvetkov, P.; Coy, S.; Petrova, B.; Dreishpoon, M.; Verma, A.; Abdusamad, M.; Rossen, J.; Joesch-Cohen, L.; Humeidi, R.; Spangler, R.D.; et al. Copper Induces Cell Death by Targeting Lipoylated Tca Cycle Proteins. Science 2022, 375, 1254–1261. [Google Scholar] [CrossRef]
- Tang, D.; Chen, X.; Kroemer, G. Cuproptosis: A Copper-Triggered Modality of Mitochondrial Cell Death. Cell Res. 2022, 32, 417–418. [Google Scholar] [CrossRef] [PubMed]
- Lei, L.; Su, J.; Chen, J.; Chen, W.; Chen, X.; Peng, C. Abnormal Serum Copper and Zinc Levels in Patients with Psoriasis: A Meta-Analysis. Indian. J. Dermatol. 2019, 64, 224–230. [Google Scholar]
- Liao, Y.; Zhao, J.; Bulek, K.; Tang, F.; Chen, X.; Cai, G.; Jia, S.; Fox, P.L.; Huang, E.; Pizarro, T.T.; et al. Inflammation Mobilizes Copper Metabolism to Promote Colon Tumorigenesis Via an Il-17-Steap4-Xiap Axis. Nat. Commun. 2020, 11, 900. [Google Scholar] [CrossRef]
- Liang, Y.; Xing, X.; Beamer, M.A.; Swindell, W.R.; Sarkar, M.K.; Roberts, L.W.; Voorhees, J.J.; Kahlenberg, J.M.; Harms, P.W.; Johnston, A.; et al. Six-Transmembrane Epithelial Antigens of the Prostate Comprise a Novel Inflammatory Nexus in Patients with Pustular Skin Disorders. J. Allergy Clin. Immunol. 2017, 139, 1217–1227. [Google Scholar] [CrossRef]
- Kaya, Ö.; Keskinkaya, Z.; Şehitoğlu, M.H.; Mermutlu, S.I.; Kılıç, S.O. Evaluation of Serum Tumor Necrosis Factor-Alpha-Induced Adipose-Associated Protein (Tiarp/Steap4) Level and Its Association with Disease Activity in Patients with Psoriasis: A Single-Center Prospective Comparative Study. Turk. J. Dermatol. 2024, 18, 119–124. [Google Scholar] [CrossRef]
- Huang, S.U.; O’Sullivan, K.M. The Expanding Role of Extracellular Traps in Inflammation and Autoimmunity: The New Players in Casting Dark Webs. Int. J. Mol. Sci. 2022, 23, 3793. [Google Scholar] [CrossRef]
- Mutua, V.; Gershwin, L.J. A Review of Neutrophil Extracellular Traps (Nets) in Disease: Potential Anti-Nets Therapeutics. Clin. Rev. Allergy Immunol. 2021, 61, 194–211. [Google Scholar] [CrossRef] [PubMed]
- Thiam, H.R.; Wong, S.L.; Wagner, D.D.; Waterman, C.M. Cellular Mechanisms of Netosis. Annu. Rev. Cell Dev. Biol. 2020, 36, 191–218. [Google Scholar] [CrossRef]
- Lambert, S.; Hambro, C.A.; Johnston, A.; Stuart, P.E.; Tsoi, L.C.; Nair, R.P.; Elder, J.T. Neutrophil Extracellular Traps Induce Human th17 Cells: Effect of Psoriasis-Associated Traf3ip2 Genotype. J. Investig. Dermatol. 2019, 139, 1245–1253. [Google Scholar] [CrossRef]
- Lande, R.; Gregorio, J.; Facchinetti, V.; Chatterjee, B.; Wang, Y.H.; Homey, B.; Cao, W.; Wang, Y.H.; Su, B.; Nestle, F.O.; et al. Plasmacytoid Dendritic Cells Sense Self-DNA Coupled with Antimicrobial Peptide. Nature 2007, 449, 564–569. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Feng, Y.; Shi, D. Netosis of Psoriasis: A Critical Step in Amplifying the Inflammatory Response. Front. Immunol. 2024, 15, 1374934. [Google Scholar] [CrossRef] [PubMed]
- Hu, S.C.-S.; Yu, H.-S.; Yen, F.-L.; Lin, C.-L.; Chen, G.-S.; Lan, C.-C.E. Neutrophil Extracellular Trap Formation Is Increased in Psoriasis and Induces Human Β-Defensin-2 Production in Epidermal Keratinocytes. Sci. Rep. 2016, 6, 31119. [Google Scholar] [CrossRef]
- Neu, S.D.; Strzepa, A.; Martin, D.; Sorci-Thomas, M.G.; Pritchard, K.A., Jr.; Dittel, B.N. Myeloperoxidase Inhibition Ameliorates Plaque Psoriasis in Mice. Antioxidants 2021, 10, 1338. [Google Scholar] [CrossRef]
- Gajendran, C.; Fukui, S.; Sadhu, N.M.; Zainuddin, M.; Rajagopal, S.; Gosu, R.; Gutch, S.; Fukui, S.; Sheehy, C.E.; Chu, L.; et al. Alleviation of Arthritis through Prevention of Neutrophil Extracellular Traps by an Orally Available Inhibitor of Protein Arginine Deiminase 4. Sci. Rep. 2023, 13, 3189. [Google Scholar] [CrossRef]
- Czerwińska, J.; Kasprowicz-Furmańczyk, M.; Placek, W.; Owczarczyk-Saczonek, A. Changes in Tumor Necrosis Factor A (Tnfα) and Peptidyl Arginine Deiminase 4 (Pad-4) Levels in Serum of General Treated Psoriatic Patients. Int. J. Environ. Res. Public Health 2022, 19, 8723. [Google Scholar] [CrossRef]

{kind=link}
| Management | Possible Mechanism | References |
|---|---|---|
| Narrowband ultraviolet B (NbUVB) phototherapy | Autophagy, Apoptosis | Y. Yang, et al., 2012 [25] B. M. Aufiero, et al., 2006 [40] S. C. Weatherhead, et al., 2011 [41] M. Ozawa, et al., 1999 [42] |
| Rapamycin | Autophagy | M. Gao and X. Si, 2018 [26] C. Bürger, et al., 2017 [27] |
| Everolimus and tacrolimus | Autophagy | K. C. Wei and P. C. Lai, 2015 [28] |
| Retinoids | Autophagy, Apoptosis | Y. Rajawat, et al., 2010 [23] T. C. Islam, et al., 2000 [55] |
| Metformin | Autophagy | Z. Huang, et al., 2023 [29] |
| Psoralen plus ultraviolet A (PUVA) | Apoptosis | M. Laporte, et al., 2000 [3] |
| Methotrexate (MTX) | Apoptosis | T. Elango, et al., 2017 [45] |
| Vitamin D | Apoptosis | P. De Haes, et al., 2004 [51] |
| Calcipotriol | Apoptosis | R. Tiberio, et al., 2009 [52] |
| Efalizumab and Etaracizumab | Anoikis | J. Mei, et al., 2024 [59] |
| R-7-Cl-O-Necrostatin-1 (Nec-1s) and Necrosulfonamide (NSA) | Necroptosis (Animal model) | X. Duan, et al., 2020 [66] |
| Dimethyl fumarate (DMF) | Necroptosis | M. Burlando, et al., 2023 [67] M. Corazza, et al., 2021 [68] F.-l. Shi, et al., 2023 [70] |
| Saracatinib | Necroptosis | J. Li, et al., 2024 [71] |
| Disulfiram | Pyroptosis | X.-m. Hu, et al., 2024 [87] J. J. Hu, et al., 2020 [88] |
| Nicotinamide and nicotinamide mononucleotide | Parthanatos | Z. Zhang, et al., 2024 [98] M. El-Khalawany, et al., 2022 [99] |
| Ferrostatin-1 (Fer-1) | Ferroptosis (Animal model) | Y. Shou, et al., 2021 [105] |
| Glutathione | Ferroptosis | Nisha Kundu, et al., 2022 [106] R. Prussick, et al., 2013 [107] |
| Selenium | Ferroptosis | M. Nazıroğlu, et al., 2012 [108] I. G. Chambers and R. R. Ratan, 2024 [109] |
| Coenzyme Q10 | Ferroptosis | G. A. Al-Oudah, et al., 2022 [110] Z. Kharaeva, et al., 2009 [111] K. Hadian, 2020 [112] |
| Deferoxamine | Ferroptosis | E. Abboud, et al., 2024 [114] |
| Myeloperoxidase inhibitor | NETosis | S. D. Neu, et al., 2021 [128] |
| Protein arginine deiminase 4 (PAD4) inhibitors | NETosis (Animal model) | C. Gajendran, et al., 2023 [129] J. Czerwińska, et al., 2022 [130] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, C.-H.; Wu, N.-L.; Tsai, T.-F. How Cells Die in Psoriasis? Int. J. Mol. Sci. 2025, 26, 3747. https://doi.org/10.3390/ijms26083747
Chen C-H, Wu N-L, Tsai T-F. How Cells Die in Psoriasis? International Journal of Molecular Sciences. 2025; 26(8):3747. https://doi.org/10.3390/ijms26083747
Chicago/Turabian StyleChen, Chung-Han, Nan-Lin Wu, and Tsen-Fang Tsai. 2025. "How Cells Die in Psoriasis?" International Journal of Molecular Sciences 26, no. 8: 3747. https://doi.org/10.3390/ijms26083747
APA StyleChen, C.-H., Wu, N.-L., & Tsai, T.-F. (2025). How Cells Die in Psoriasis? International Journal of Molecular Sciences, 26(8), 3747. https://doi.org/10.3390/ijms26083747