
Methylation Risk Score Modelling in Endometriosis: Evidence for Non-Genetic DNA Methylation Effects in a Case–Control Study

 , , and
, , and
Abstract
1. Introduction
2. Results
2.1. Factors Contributing to Variation in Endometrial DNAm
2.2. Variation in Endometriosis Status Captured by DNAm in Endometrium Independent of Common Genetic Variants
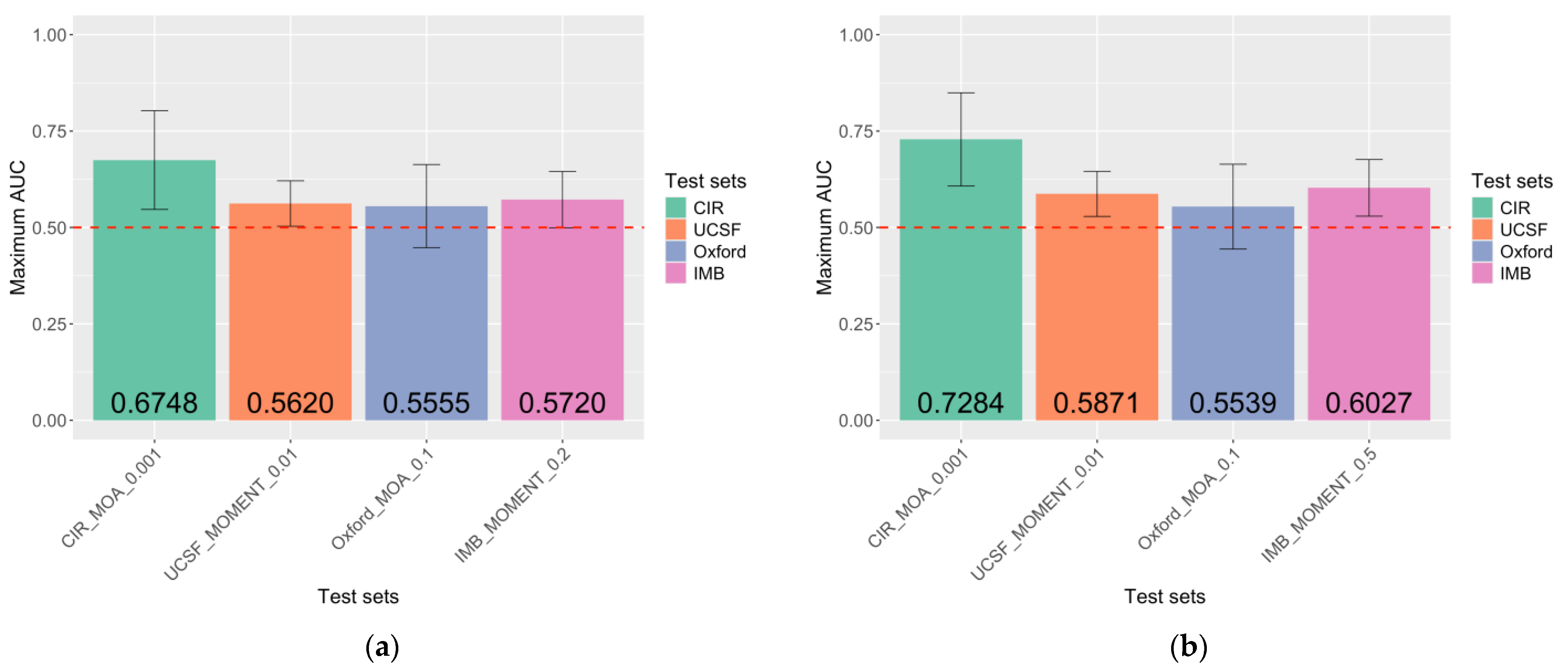
2.3. MRS Captures DNAm Differences Between Endometriosis Cases and Controls
- MRSs derived from p-value thresholds of 1 × 10−4 and 1 × 10−5 were excluded.
- MRSs that yielded the highest AUC within the test set and classification model and demonstrated a significant association with endometriosis were selected.
- If none of the MRSs had a significant association, the MRSs with the highest AUC were chosen.
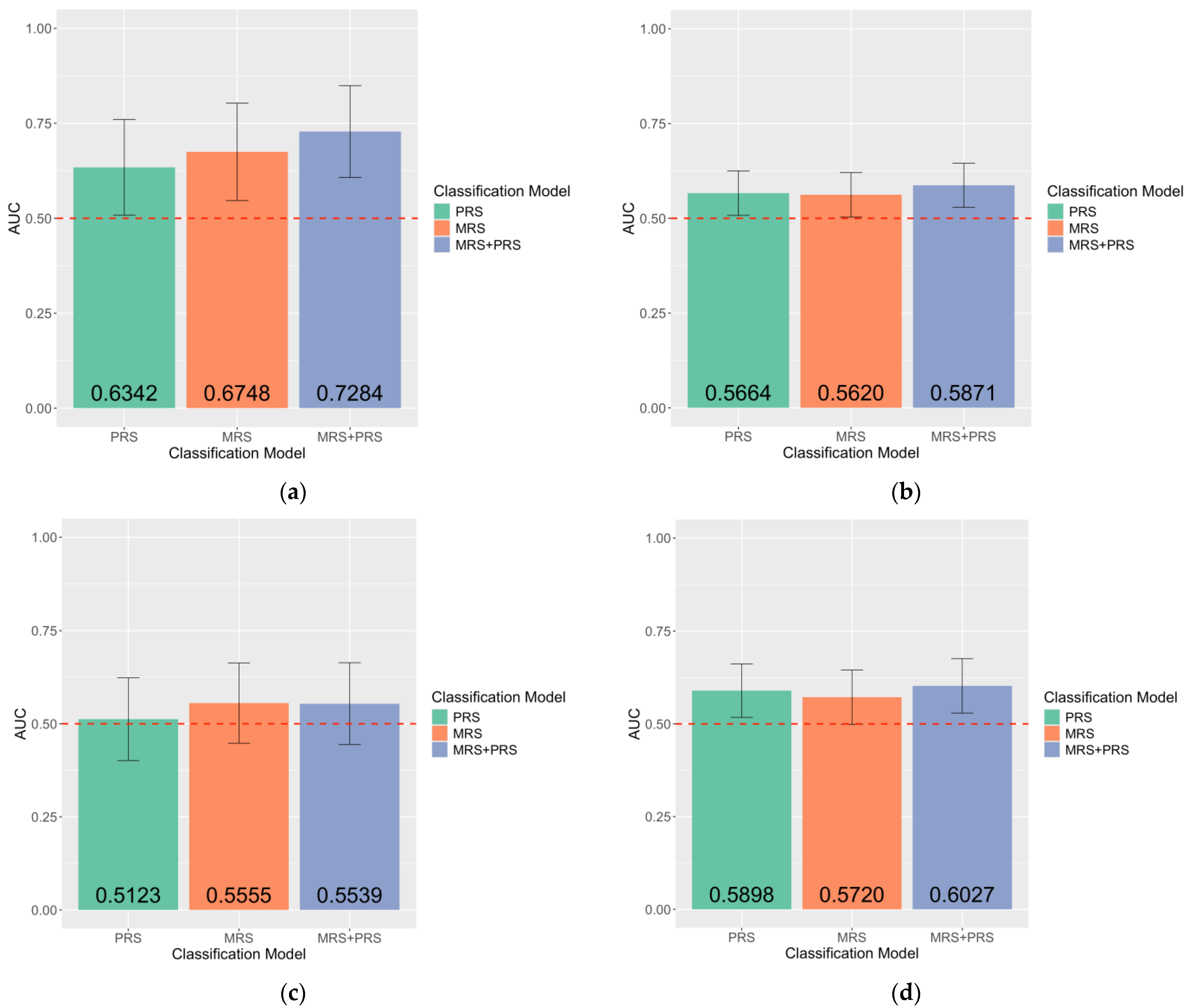
2.4. Unique Contribution of MRS in the Case–Control Classification of Endometriosis
2.5. Sensitivity Analyses: Performance of MRS Within European-Ancestry Samples
3. Discussion
4. Materials and Methods
4.1. Sample Collection and Processing
4.2. DNAm Quality Control
4.3. Genotyping Data Quality Control
4.4. Covariate Selection
4.5. Surrogate Variable Analysis
4.6. Estimation of the Proportion of Variance in Endometriosis Captured by DNAm
- ORM;
- GRM;
- ORM + GRM;
- ORM + GRM + SV;
- ORM + GRM + SV + age + institution + menstrual cycle phase;
4.7. Genetic PC Computation
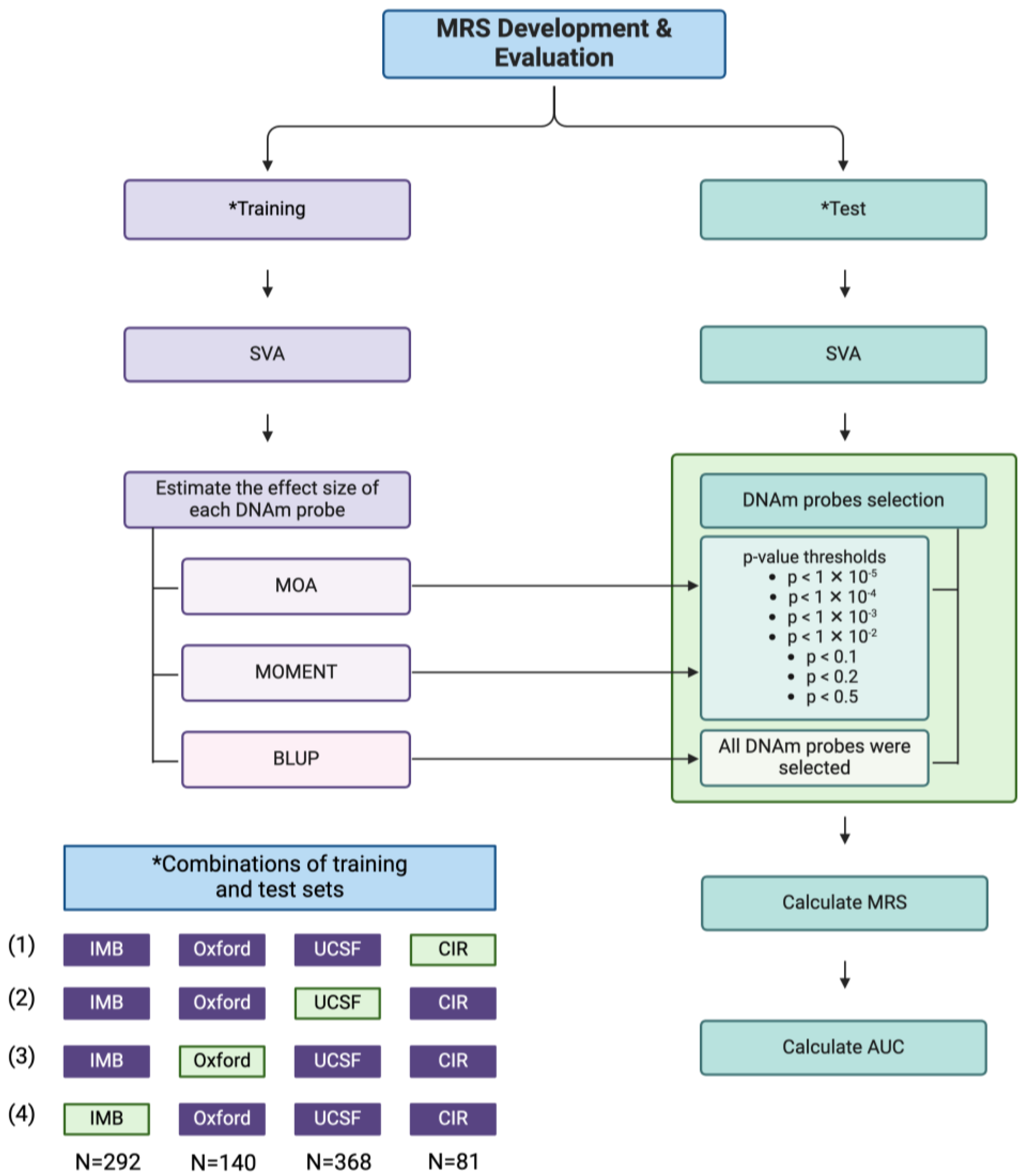
4.8. Methylation Risk Score (MRS) Development
4.9. Correlation Between Effect Sizes Generated from MOA, MOMENT, and BLUP
4.10. MRS Evaluation
4.11. Correlation Between MRSs Generated from Different MRS Models
4.12. PRS Development and Evaluation
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| DNAm | DNA methylation |
| MRS | Methylation risk score |
| AUC | Area under the receiver-operator curve |
| PRS | Polygenic risk score |
| mQTL | DNA methylation quantitative trait loci |
| SV | Surrogate variable |
| ORM | Omics relationship matrix |
| CIR | Centre for Inflammation Research, University of Edinburgh |
| IMB | Institute for Molecular Bioscience, University of Queensland |
| UCSF | University of California San Francisco |
| Oxford | Oxford Endometriosis CaRe Centre |
| OSCA | Omic-data-based Complex Trait Analysis |
| GREML | Genome-based restricted maximum likelihood |
| OREML | Omics residual maximum likelihood |
| GRM | Genomic relationship matrix |
| MOA | MLM-based omic association |
| MOMENT | Multi-component MLM-based association excluding the target |
| BLUP | Best linear unbiased prediction |
References
- Chapron, C.; Marcellin, L.; Borghese, B.; Santulli, P. Rethinking mechanisms, diagnosis and management of endometriosis. Nat. Rev. Endocrinol. 2019, 15, 666–682. [Google Scholar] [CrossRef] [PubMed]
- Rowlands, I.; Abbott, J.; Montgomery, G.; Hockey, R.; Rogers, P.; Mishra, G. Prevalence and incidence of endometriosis in Australian women: A data linkage cohort study. BJOG Int. J. Obstet. Gynaecol. 2021, 128, 657–665. [Google Scholar] [CrossRef] [PubMed]
- Treloar, S.A.; O’connor, D.T.; O’connor, V.M.; Martin, N.G. Genetic influences on endometriosis in an Australian twin sample. Fertil. Steril. 1999, 71, 701–710. [Google Scholar] [CrossRef] [PubMed]
- Saha, R.; Pettersson, H.J.; Svedberg, P.; Olovsson, M.; Bergqvist, A.; Marions, L.; Tornvall, P.; Kuja-Halkola, R. Heritability of endometriosis. Fertil. Steril. 2015, 104, 947–952. [Google Scholar] [CrossRef]
- Rahmioglu, N.; Mortlock, S.; Ghiasi, M.; Møller, P.L.; Stefansdottir, L.; Galarneau, G.; Turman, C.; Danning, R.; Law, M.H.; Sapkota, Y.; et al. The genetic basis of endometriosis and comorbidity with other pain and inflammatory conditions. Nat. Genet. 2023, 55, 423–436. [Google Scholar] [CrossRef]
- McGrath, I.M.; Montgomery, G.W.; Mortlock, S.; International Endometriosis Genetics Consortium. Polygenic risk score phenome-wide association study reveals an association between endometriosis and testosterone. BMC Med. 2023, 21, 482. [Google Scholar] [CrossRef]
- Teschendorff, A.E.; Relton, C.L. Statistical and integrative system-level analysis of DNA methylation data. Nat. Rev. Genet. 2018, 19, 129–147. [Google Scholar] [CrossRef]
- Sun, Y.V. The Influences of Genetic and Environmental Factors on Methylome-Wide Association Studies for Human Diseases. Curr. Genet. Med. Rep. 2014, 2, 261–270. [Google Scholar] [CrossRef]
- Villicaña, S.; Bell, J.T. Genetic impacts on DNA methylation: Research findings and future perspectives. Genome Biol. 2021, 22, 127. [Google Scholar] [CrossRef]
- Fujii, R.; Ando, Y.; Yamada, H.; Tsuboi, Y.; Munetsuna, E.; Yamazaki, M.; Mizuno, G.; Maeda, K.; Ohashi, K.; Ishikawa, H.; et al. Integration of methylation quantitative trait loci (mQTL) on dietary intake on DNA methylation levels: An example of n-3 PUFA and ABCA1 gene. Eur. J. Clin. Nutr. 2023, 77, 881–887. [Google Scholar] [CrossRef]
- Lam, L.L.; Emberly, E.; Fraser, H.B.; Neumann, S.M.; Chen, E.; Miller, G.E.; Kobor, M.S. Factors underlying variable DNA methylation in a human community cohort. Proc. Natl. Acad. Sci. USA 2012, 109, 17253–17260. [Google Scholar] [CrossRef] [PubMed]
- Szyf, M. The early life environment and the epigenome. Biochim. Biophys. Acta (BBA) Gen. Subj. 2009, 1790, 878–885. [Google Scholar] [CrossRef] [PubMed]
- Labonté, B.; Suderman, M.; Maussion, G.; Navaro, L.; Yerko, V.; Mahar, I.; Bureau, A.; Mechawar, N.; Szyf, M.; Meaney, M.J.; et al. Genome-wide Epigenetic Regulation by Early-Life Trauma. Arch. Gen. Psychiatry 2012, 69, 722–731. [Google Scholar] [CrossRef] [PubMed]
- Pogribny, I.P.; Rusyn, I. Environmental Toxicants, Epigenetics, and Cancer. In Epigenetic Alterations in Oncogenesis; Karpf, A.R., Ed.; Springer: New York, NY, USA, 2013; pp. 215–232. [Google Scholar]
- Waterland, R.A.; Jirtle, R.L. Transposable Elements: Targets for Early Nutritional Effects on Epigenetic Gene Regulation. Mol. Cell. Biol. 2003, 23, 5293–5300. [Google Scholar] [CrossRef]
- Rönn, T.; Volkov, P.; Davegårdh, C.; Dayeh, T.; Hall, E.; Olsson, A.H.; Nilsson, E.; Tornberg, Å.; Dekker Nitert, M.; Eriksson, K.-F.; et al. A Six Months Exercise Intervention Influences the Genome-wide DNA Methylation Pattern in Human Adipose Tissue. PLoS Genet. 2013, 9, e1003572. [Google Scholar] [CrossRef]
- Barrès, R.; Yan, J.; Egan, B.; Treebak, J.T.; Rasmussen, M.; Fritz, T.; Caidahl, K.; Krook, A.; O’Gorman, D.J.; Zierath, J.R. Acute Exercise Remodels Promoter Methylation in Human Skeletal Muscle. Cell Metab. 2012, 15, 405–411. [Google Scholar] [CrossRef]
- Fambrini, M.; Sorbi, F.; Bussani, C.; Cioni, R.; Sisti, G.; Andersson, K.L. Hypermethylation of HOXA10 gene in mid-luteal endometrium from women with ovarian endometriomas. Acta Obstet. Gynecol. Scand. 2013, 92, 1331–1334. [Google Scholar] [CrossRef]
- Wu, Y.; Strawn, E.; Basir, Z.; Halverson, G.; Guo, S.-W. Promoter Hypermethylation of Progesterone Receptor Isoform B (PR-B) in Endometriosis. Epigenetics 2006, 1, 106–111. [Google Scholar] [CrossRef]
- Giudice, L.C.; Kao, L.C. Endometriosis. Lancet 2004, 364, 1789–1799. [Google Scholar] [CrossRef]
- Attia, G.R.; Zeitoun, K.; Edwards, D.; Johns, A.; Carr, B.R.; Bulun, S.E. Progesterone receptor isoform A but not B is expressed in endometriosis. J. Clin. Endocrinol. Metab. 2000, 85, 2897–2902. [Google Scholar] [CrossRef]
- Xue, Q.; Lin, Z.; Yin, P.; Milad, M.P.; Cheng, Y.-H.; Confino, E.; Reierstad, S.; Bulun, S.E. Transcriptional Activation of Steroidogenic Factor-1 by Hypomethylation of the 5′ CpG Island in Endometriosis. J. Clin. Endocrinol. Metab. 2007, 92, 3261–3267. [Google Scholar] [CrossRef]
- Xue, Q.; Lin, Z.; Cheng, Y.-H.; Huang, C.-C.; Marsh, E.; Yin, P.; Milad, M.P.; Confino, E.; Reierstad, S.; Innes, J. Promoter Methylation Regulates Estrogen Receptor 2 in Human Endometrium and Endometriosis. Biol. Reprod. 2007, 77, 681–687. [Google Scholar] [CrossRef] [PubMed]
- Naqvi, H.; Ilagan, Y.; Krikun, G.; Taylor, H.S. Altered Genome-Wide Methylation in Endometriosis. Reprod. Sci. 2014, 21, 1237–1243. [Google Scholar] [CrossRef] [PubMed]
- Dyson, M.T.; Roqueiro, D.; Monsivais, D.; Ercan, C.M.; Pavone, M.E.; Brooks, D.C.; Kakinuma, T.; Ono, M.; Jafari, N.; Dai, Y.; et al. Genome-Wide DNA Methylation Analysis Predicts an Epigenetic Switch for GATA Factor Expression in Endometriosis. PLoS Genet. 2014, 10, e1004158. [Google Scholar] [CrossRef]
- Mortlock, S.; Houshdaran, S.; Kosti, I.; Rahmioglu, N.; Nezhat, C.; Vitonis, A.F.; Andrews, S.V.; Grosjean, P.; Paranjpe, M.; Horne, A.W.; et al. Global endometrial DNA methylation analysis reveals insights into mQTL regulation and associated endometriosis disease risk and endometrial function. Commun. Biol. 2023, 6, 780. [Google Scholar] [CrossRef] [PubMed]
- Wahl, S.; Drong, A.; Lehne, B.; Loh, M.; Scott, W.R.; Kunze, S.; Tsai, P.-C.; Ried, J.S.; Zhang, W.; Yang, Y.; et al. Epigenome-wide association study of body mass index, and the adverse outcomes of adiposity. Nature 2017, 541, 81–86. [Google Scholar] [CrossRef]
- Elliott, H.R.; Tillin, T.; McArdle, W.L.; Ho, K.; Duggirala, A.; Frayling, T.M.; Davey Smith, G.; Hughes, A.D.; Chaturvedi, N.; Relton, C.L. Differences in smoking associated DNA methylation patterns in South Asians and Europeans. Clin. Epigenetics 2014, 6, 4. [Google Scholar] [CrossRef]
- Hannon, E.; Dempster, E.; Viana, J.; Burrage, J.; Smith, A.R.; Macdonald, R.; St Clair, D.; Mustard, C.; Breen, G.; Therman, S.; et al. An integrated genetic-epigenetic analysis of schizophrenia: Evidence for co-localization of genetic associations and differential DNA Methylation. Genome Biol. 2016, 17, 176. [Google Scholar] [CrossRef]
- Eze, I.C.; Imboden, M.; Kumar, A.; von Eckardstein, A.; Stolz, D.; Gerbase, M.W.; Künzli, N.; Pons, M.; Kronenberg, F.; Schindler, C.; et al. Air pollution and diabetes association: Modification by type 2 diabetes genetic risk score. Environ. Int. 2016, 94, 263–271. [Google Scholar] [CrossRef]
- Rask-Andersen, M.; Karlsson, T.; Ek, W.E.; Johansson, Å. Gene-environment interaction study for BMI reveals interactions between genetic factors and physical activity, alcohol consumption and socioeconomic Status. PLoS Genet. 2017, 13, e1006977. [Google Scholar] [CrossRef]
- Qi, Q.; Chu, A.Y.; Kang, J.H.; Huang, J.; Rose, L.M.; Jensen, M.K.; Liang, L.; Curhan, G.C.; Pasquale, L.R.; Wiggs, J.L.; et al. Fried food consumption, genetic risk, and body mass index: Gene-diet interaction analysis in three US cohort Studies. BMJ Br. Med. J. 2014, 348, g1610. [Google Scholar] [CrossRef] [PubMed]
- Villanueva, A.; Portela, A.; Sayols, S.; Battiston, C.; Hoshida, Y.; Méndez-González, J.; Imbeaud, S.; Letouzé, E.; Hernandez-Gea, V.; Cornella, H.; et al. DNA methylation-based prognosis and epidrivers in hepatocellular carcinoma. Hepatology 2015, 61, 1945–1956. [Google Scholar] [CrossRef] [PubMed]
- Moreaux, J.; Bruyer, A.; Veyrune, J.-L.; Goldschmidt, H.; Hose, D.; Klein, B. DNA methylation score is predictive of myeloma cell sensitivity to 5-azacitidine. Br. J. Haematol. 2014, 164, 613–616. [Google Scholar] [CrossRef] [PubMed]
- Hüls, A.; Czamara, D. Methodological challenges in constructing DNA methylation risk scores. Epigenetics 2020, 15, 1–11. [Google Scholar] [CrossRef]
- Do, W.L.; Gohar, J.; McCullough, L.E.; Galaviz, K.I.; Conneely, K.N.; Narayan, K.M.V. Examining the association between adiposity and DNA methylation: A systematic review and meta-analysis. Obes. Rev. 2021, 22, e13319. [Google Scholar] [CrossRef]
- Nabais, M.F.; Laws, S.M.; Lin, T.; Vallerga, C.L.; Armstrong, N.J.; Blair, I.P.; Kwok, J.B.; Mather, K.A.; Mellick, G.D.; Sachdev, P.S.; et al. Meta-analysis of genome-wide DNA methylation identifies shared associations across neurodegenerative disorders. Genome Biol. 2021, 22, 90. [Google Scholar] [CrossRef]
- Lee, K.W.; Pausova, Z. Cigarette smoking and DNA methylation. Front. Genet. 2013, 4, 132. [Google Scholar] [CrossRef]
- Grönniger, E.; Weber, B.; Heil, O.; Peters, N.; Stäb, F.; Wenck, H.; Korn, B.; Winnefeld, M.; Lyko, F. Aging and Chronic Sun Exposure Cause Distinct Epigenetic Changes in Human Skin. PLoS Genet. 2010, 6, e1000971. [Google Scholar] [CrossRef]
- Martin, E.M.; Fry, R.C. Environmental Influences on the Epigenome: Exposure- Associated DNA Methylation in Human Populations. Annu. Rev. Public Health 2018, 39, 309–333. [Google Scholar] [CrossRef]
- Volkov, P.; Olsson, A.H.; Gillberg, L.; Jørgensen, S.W.; Brøns, C.; Eriksson, K.-F.; Groop, L.; Jansson, P.-A.; Nilsson, E.; Rönn, T.; et al. A Genome-Wide mQTL Analysis in Human Adipose Tissue Identifies Genetic Variants Associated with DNA Methylation, Gene Expression and Metabolic Traits. PLoS ONE 2016, 11, e0157776. [Google Scholar] [CrossRef]
- Hatton, A.A.; Hillary, R.F.; Bernabeu, E.; McCartney, D.L.; Marioni, R.E.; McRae, A.F. Blood-based genome-wide DNA methylation correlations across body-fat- and adiposity-related biochemical traits. Am. J. Hum. Genet. 2023, 110, 1564–1573. [Google Scholar] [CrossRef] [PubMed]
- Vallerga, C.L.; Zhang, F.; Fowdar, J.; McRae, A.F.; Qi, T.; Nabais, M.F.; Zhang, Q.; Kassam, I.; Henders, A.K.; Wallace, L.; et al. Analysis of DNA methylation associates the cystine–glutamate antiporter SLC7A11 with risk of Parkinson’s disease. Nat. Commun. 2020, 11, 1238. [Google Scholar] [CrossRef]
- Yap, C.X.; Henders, A.K.; Alvares, G.A.; Giles, C.; Huynh, K.; Nguyen, A.; Wallace, L.; McLaren, T.; Yang, Y.; Hernandez, L.M.; et al. Interactions between the lipidome and genetic and environmental factors in autism. Nat. Med. 2023, 29, 936–949. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.H.; Harold, D.; Nyholt, D.R.; Goddard, M.E.; Zondervan, K.T.; Williams, J.; Montgomery, G.W.; Wray, N.R.; Visscher, P.M. Estimation and partitioning of polygenic variation captured by common SNPs for Alzheimer’s disease, multiple sclerosis and endometriosis. Hum. Mol. Genet. 2013, 22, 832–841. [Google Scholar] [CrossRef] [PubMed]
- Barbu, M.C.; Shen, X.; Walker, R.M.; Howard, D.M.; Evans, K.L.; Whalley, H.C.; Porteous, D.J.; Morris, S.W.; Deary, I.J.; Zeng, Y.; et al. Epigenetic prediction of major depressive disorder. Mol. Psychiatry 2021, 26, 5112–5123. [Google Scholar] [CrossRef]
- Nabais, M.F.; Lin, T.; Benyamin, B.; Williams, K.L.; Garton, F.C.; Vinkhuyzen, A.A.E.; Zhang, F.; Vallerga, C.L.; Restuadi, R.; Freydenzon, A.; et al. Significant out-of-sample classification from methylation profile scoring for amyotrophic lateral sclerosis. npj Genom. Med. 2020, 5, 10. [Google Scholar] [CrossRef]
- Kresovich, J.K.; Xu, Z.; O’Brien, K.M.; Shi, M.; Weinberg, C.R.; Sandler, D.P.; Taylor, J.A. Blood DNA methylation profiles improve breast cancer prediction. Mol. Oncol. 2022, 16, 42–53. [Google Scholar] [CrossRef]
- Zhang, F.; Chen, W.; Zhu, Z.; Zhang, Q.; Nabais, M.F.; Qi, T.; Deary, I.J.; Wray, N.R.; Visscher, P.M.; McRae, A.F.; et al. OSCA: A tool for omic-data-based complex trait analysis. Genome Biol. 2019, 20, 107. [Google Scholar] [CrossRef]
- Kloeve-Mogensen, K.; Rohde, P.D.; Twisttmann, S.; Nygaard, M.; Koldby, K.M.; Steffensen, R.; Dahl, C.M.; Rytter, D.; Overgaard, M.T.; Forman, A.; et al. Polygenic Risk Score Prediction for Endometriosis. Front. Reprod. Health 2021, 3, 793226. [Google Scholar] [CrossRef]
- Shah, S.; Bonder, M.J.; Marioni, R.E.; Zhu, Z.; McRae, A.F.; Zhernakova, A.; Harris, S.E.; Liewald, D.; Henders, A.K.; Mendelson, M.M.; et al. Improving Phenotypic Prediction by Combining Genetic and Epigenetic Associations. Am. J. Hum. Genet. 2015, 97, 75–85. [Google Scholar] [CrossRef]
- Mahalingaiah, S.; Hart, J.E.; Laden, F.; Aschengrau, A.; Missmer, S.A. Air Pollution Exposures During Adulthood and Risk of Endometriosis in the Nurses’ Health Study II. Environ. Health Perspect. 2014, 122, 58–64. [Google Scholar] [CrossRef] [PubMed]
- Szczęsna, D.; Wieczorek, K.; Jurewicz, J. An exposure to endocrine active persistent pollutants and endometriosis—A review of current epidemiological studies. Environ. Sci. Pollut. Res. 2023, 30, 13974–13993. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, A.; Harris, H.R.; Vitonis, A.F.; Chavarro, J.E.; Missmer, S.A. A prospective cohort study of meat and fish consumption and endometriosis risk. Am. J. Obstet. Gynecol. 2018, 219, e171–e178. [Google Scholar] [CrossRef] [PubMed]
- Farland, L.V.; Degnan, W.J.; Harris, H.R.; Han, J.; Cho, E.; VoPham, T.; Kvaskoff, M.; Missmer, S.A. Recreational and residential sun exposure and risk of endometriosis: A prospective cohort study. Hum. Reprod. 2020, 36, 199–210. [Google Scholar] [CrossRef]
- Colwell, M.L.; Townsel, C.; Petroff, R.L.; Goodrich, J.M.; Dolinoy, D.C. Epigenetics and the exposome: DNA methylation as a proxy for health impacts of prenatal environmental exposures. Exposome 2023, 3, osad001. [Google Scholar] [CrossRef]
- Ronsini, C.; Fumiento, P.; Iavarone, I.; Greco, P.F.; Cobellis, L.; De Franciscis, P. Liquid Biopsy in Endometriosis: A Systematic Review. Int. J. Mol. Sci. 2023, 24, 6116. [Google Scholar] [CrossRef]
- Gajbhiye, R.; Sonawani, A.; Khan, S.; Suryawanshi, A.; Kadam, S.; Warty, N.; Raut, V.; Khole, V. Identification and validation of novel serum markers for early diagnosis of endometriosis. Hum. Reprod. 2011, 27, 408–417. [Google Scholar] [CrossRef]
- Warren, L.A.; Shih, A.; Renteira, S.M.; Seckin, T.; Blau, B.; Simpfendorfer, K.; Lee, A.; Metz, C.N.; Gregersen, P.K. Analysis of menstrual effluent: Diagnostic potential for endometriosis. Mol. Med. 2018, 24, 1. [Google Scholar] [CrossRef]
- Lokk, K.; Modhukur, V.; Rajashekar, B.; Märtens, K.; Mägi, R.; Kolde, R.; Koltšina, M.; Nilsson, T.K.; Vilo, J.; Salumets, A.; et al. DNA methylome profiling of human tissues identifies global and tissue-specific methylation patterns. Genome Biol. 2014, 15, r54. [Google Scholar] [CrossRef]
- Luo, C.; Hajkova, P.; Ecker, J.R. Dynamic DNA methylation: In the right place at the right time. Science 2018, 361, 1336–1340. [Google Scholar] [CrossRef]
- Rahmioglu, N.; Drong, A.W.; Helen, L.; Thomas, T.; Karin, H.; Merli, S.; Triin, L.-P.; Christine, D.; Emily, T.; George, N.; et al. Variability of genome-wide DNA methylation and mRNA expression profiles in reproductive and endocrine disease related tissues. Epigenetics 2017, 12, 897–908. [Google Scholar] [CrossRef] [PubMed]
- Zhou, W.; Laird, P.W.; Shen, H. Comprehensive characterization, annotation and innovative use of Infinium DNA methylation BeadChip probes. Nucleic Acids Res. 2016, 45, e22. [Google Scholar] [CrossRef] [PubMed]
- Horvath, S.; Zhang, Y.; Langfelder, P.; Kahn, R.S.; Boks, M.P.M.; van Eijk, K.; van den Berg, L.H.; Ophoff, R.A. Aging effects on DNA methylation modules in human brain and blood tissue. Genome Biol. 2012, 13, R97. [Google Scholar] [CrossRef] [PubMed]
- Horvath, S.; Gurven, M.; Levine, M.E.; Trumble, B.C.; Kaplan, H.; Allayee, H.; Ritz, B.R.; Chen, B.; Lu, A.T.; Rickabaugh, T.M.; et al. An epigenetic clock analysis of race/ethnicity, sex, and coronary heart disease. Genome Biol. 2016, 17, 171. [Google Scholar] [CrossRef]
- Ross, J.P.; van Dijk, S.; Phang, M.; Skilton, M.R.; Molloy, P.L.; Oytam, Y. Batch-effect detection, correction and characterisation in Illumina HumanMethylation450 and MethylationEPIC BeadChip array data. Clin. Epigenetics 2022, 14, 58. [Google Scholar] [CrossRef]
- Noyes, R.W.; Hertig, A.T.; Rock, J. Dating the endometrial biopsy. Am. J. Obstet. Gynecol. 1975, 122, 262–263. [Google Scholar] [CrossRef]
- Yang, J.; Lee, S.H.; Goddard, M.E.; Visscher, P.M. GCTA: A Tool for Genome-wide Complex Trait Analysis. Am. J. Hum. Genet. 2011, 88, 76–82. [Google Scholar] [CrossRef]
- Robinson, G.K. That BLUP is a Good Thing: The Estimation of Random Effects. Stat. Sci. 1991, 6, 15–32, 18. [Google Scholar]
- Robin, X.; Turck, N.; Hainard, A.; Tiberti, N.; Lisacek, F.; Sanchez, J.-C.; Müller, M. pROC: An open-source package for R and S+ to analyze and compare ROC curves. BMC Bioinform. 2011, 12, 77. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Characteristics | Endometriosis Status | p-Value | ||
|---|---|---|---|---|
| Controls (N = 318) | Cases (N = 590) | |||
| Age Mean [95% CI] (range) t-test | 37 [36.1–37.9] (18–55) (N = 314) | 34.2 [33.6–34.8] (18–53) (N = 587) | 1.29 × 10−6 | |
| Menstrual cycle phase N (%) Chi-squared | Proliferative | 154 (48.4%) | 285 (48.3%) | 0.75 |
| Secretory (undefined sub-phase) | 7 (2.2%) | 14 (2.4%) | ||
| Early secretory | 41 (12.9%) | 71 (12.0%) | ||
| Mid-secretory | 72 (22.6%) | 121 (20.5%) | ||
| Late secretory | 33 (10.4%) | 66 (11.2%) | ||
| Menstrual | 11 (3.5%) | 33 (5.6%) | ||
| Institutions N (%) Chi-squared | CIR 1 | 31 (9.7%) | 52 (8.8%) | 8.11 × 10−5 |
| IMB 2 | 83 (26.1%) | 213 (36.1%) | ||
| Oxford 3 | 41 (12.9%) | 110 (18.6%) | ||
| UCSF 4 | 163 (51.3%) | 215 (36.4%) | ||
| Genetic ancestry N (%) Chi-squared | ADMIX | 24 (7.5%) | 49 (8.3%) | 1.89 × 10−6 |
| African | 33 (10.4%) | 13 (2.2%) | ||
| American | 21 (6.6%) | 29 (4.9%) | ||
| Eastern Asian | 25 (7.9%) | 47 (8.0%) | ||
| European | 207 (65.1%) | 417 (70.7%) | ||
| Southern Asian | 8 (2.5%) | 35 (5.9%) | ||
| No. | OREML Models | Proportion of Variance Captured 2 (s.e. a) | Phenotypic Variance 1 (s.e. a) | ||
|---|---|---|---|---|---|
| ORM b | GRM c | ORM + GRM e | |||
| 1 | ORM b | 19.58% (0.07) | - | - | 0.2481 (0.02) |
| 2 | GRM c | - | 28.83% (0.17) | - | 0.2251 (0.01) |
| 3 | ORM b + GRM c | 12.35% (0.06) | 22.38% (0.15) | 34.73% | 0.2361 (0.01) |
| 4 | ORM b + GRM c + surrogate variable (SVs) d | 10.70% (0.07) | 27.94% (0.16) | 38.64% | 0.2251 (0.01) |
| 5 | ORM b + GRM c + SVs d + age + institution + menstrual cycle phase | 18.25% (0.08) | 23.78% (0.15) | 42.03% | 0.2187 (0.01) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Thong, L.Y.; McRae, A.F.; Sirota, M.; Giudice, L.; Montgomery, G.W.; Mortlock, S. Methylation Risk Score Modelling in Endometriosis: Evidence for Non-Genetic DNA Methylation Effects in a Case–Control Study. Int. J. Mol. Sci. 2025, 26, 3760. https://doi.org/10.3390/ijms26083760
Thong LY, McRae AF, Sirota M, Giudice L, Montgomery GW, Mortlock S. Methylation Risk Score Modelling in Endometriosis: Evidence for Non-Genetic DNA Methylation Effects in a Case–Control Study. International Journal of Molecular Sciences. 2025; 26(8):3760. https://doi.org/10.3390/ijms26083760
Chicago/Turabian StyleThong, Li Ying, Allan F. McRae, Marina Sirota, Linda Giudice, Grant W. Montgomery, and Sally Mortlock. 2025. "Methylation Risk Score Modelling in Endometriosis: Evidence for Non-Genetic DNA Methylation Effects in a Case–Control Study" International Journal of Molecular Sciences 26, no. 8: 3760. https://doi.org/10.3390/ijms26083760
APA StyleThong, L. Y., McRae, A. F., Sirota, M., Giudice, L., Montgomery, G. W., & Mortlock, S. (2025). Methylation Risk Score Modelling in Endometriosis: Evidence for Non-Genetic DNA Methylation Effects in a Case–Control Study. International Journal of Molecular Sciences, 26(8), 3760. https://doi.org/10.3390/ijms26083760