
Transcriptome Analysis of Cabbage Near-Isogenic Lines Reveals the Involvement of the Plant Defensin Gene PDF1.2 in Fusarium Wilt Resistance


 ,
, 
 , , ,
, , , 
Abstract
1. Introduction
2. Results
2.1. RNA Sequencing of a Near-Isogenic Resistant Line Following Foc Inoculation
2.2. Analysis of DEGs in Cabbage
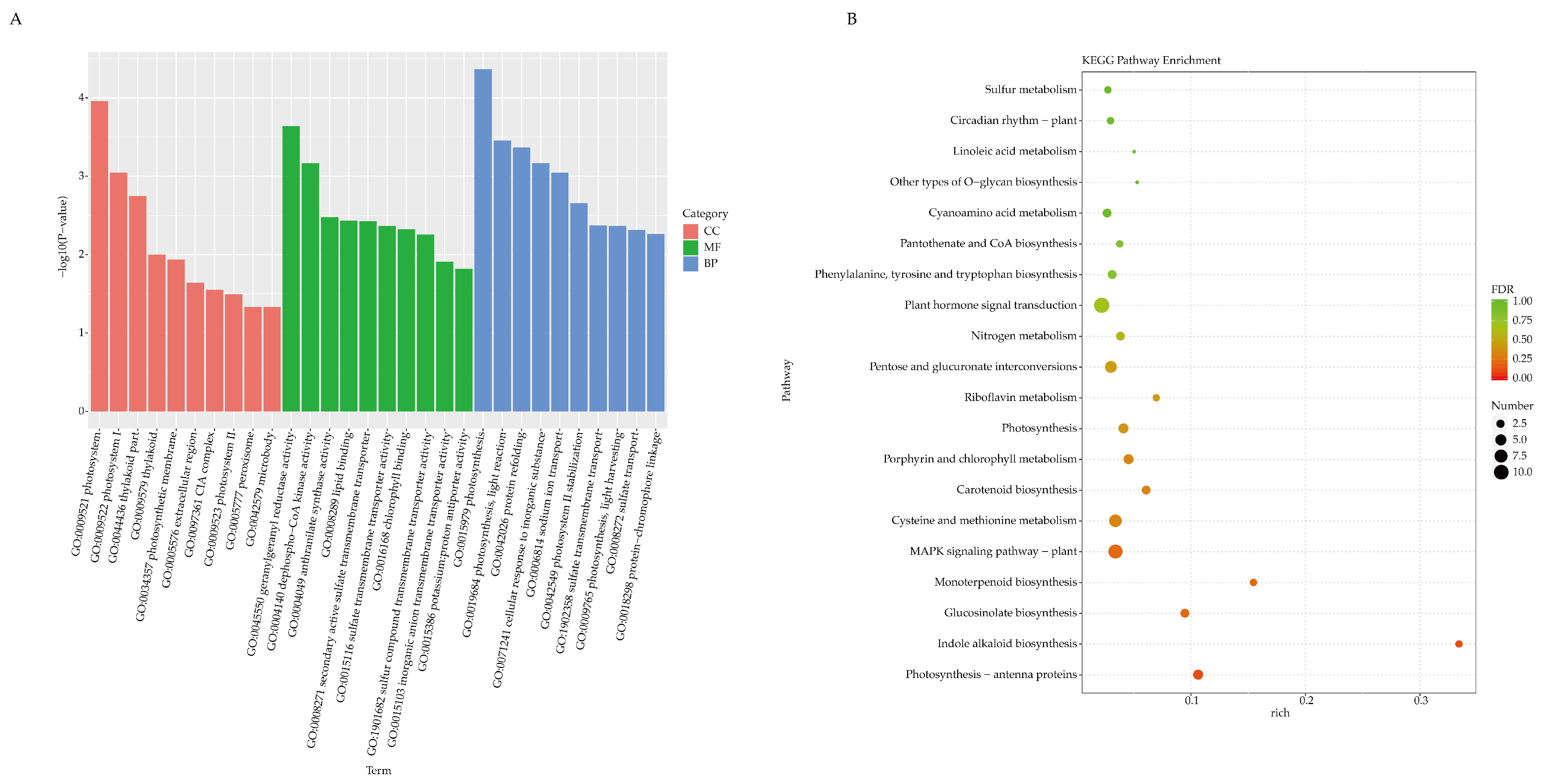
2.3. Functional Enrichment Analysis of DEGs
2.4. Stable Overexpression of PDF1.2 in Cabbage Enhances Resistance Against Foc
3. Discussion
3.1. Cytokinin Signaling in Foc Defense
3.2. ABA Signaling Dynamics
3.3. Growth-Defense Tradeoffs via SAUR/PIFs
3.4. BOP-Mediated Defense Convergence
3.5. CAPE1-like Immune Priming
3.6. MAPK-ET Crosstalk
3.7. The Role of PDF1.2 in Resistance to Foc
4. Materials and Methods
4.1. Fungal Strain and Plant Material
4.2. Pathogen Inoculation and Assessment
4.3. RNA Extraction, cDNA Library Construction, and Sequencing
4.4. Bioinformatics Analysis
4.5. Real-Time Quantitative PCR Analysis
4.6. Plasmid Construction and Agrobacterium-Mediated Plant Genetic Transformation
4.7. Protein Extraction and Western Blotting
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Liu, X.; Zhao, C.; Yang, L.; Zhuang, M.; Zhang, Y.; Wang, Y.; Fang, Z.; Lv, H. A Time-Resolved Dual Transcriptome Analysis Reveals the Molecular Regulating Network Underlying the Compatible/Incompatible Interactions between Cabbage (Brassica Oleracea) and Fusarium Oxysporum f. sp. Conglutinans. Plant Soil 2020, 448, 455–478. [Google Scholar] [CrossRef]
- Farnham, M.W.; Keinath, A.P.; Smith, J.P. Characterization of Fusarium Yellows Resistance in Collard. Plant Disease 2001, 85, 890–894. [Google Scholar] [CrossRef]
- Li, M.; Zhang, T.; Li, X.; Yan, H. Fusarium wilt of Cruciferae and its pathogen identification. Plant Prot. 2003, 29, 44–45. (In Chinese) [Google Scholar]
- Armstrong, G.; Armstrong, J. Formae speciales and races of Fusarium oxysporum causing wilt diseases. In Fusarium: Disease, Biology, and Taxonomy; Pennsylvania State University: University Park, PA, USA, 1981; pp. 391–399. [Google Scholar]
- Lv, H.; Fang, Z.; Yang, L.; Xie, B.; Liu, Y.; Zhuang, M.; Zhang, Y.; Yang, Y. Research on screening of resistant resources to Fusarium wilt and inheritance of the resistant gene in cabbage. Sci. Agric. Sin. 2011, 38, 875–885. (In Chinese) [Google Scholar]
- Lv, H.; Fang, Z.; Yang, L.; Zhang, Y.; Wang, Q.; Liu, Y.; Zhuang, M.; Yang, Y.; Xie, B.; Liu, B.; et al. Mapping and Analysis of a Novel Candidate Fusarium Wilt Resistance Gene FOC1 in Brassica Oleracea. BMC Genom. 2014, 15, 1094. [Google Scholar] [CrossRef]
- Shimizu, M.; Fujimoto, R.; Ying, H.; Pu, Z.; Ebe, Y.; Kawanabe, T.; Saeki, N.; Taylor, J.M.; Kaji, M.; Dennis, E.S.; et al. Identification of Candidate Genes for Fusarium Yellows Resistance in Chinese Cabbage by Differential Expression Analysis. Plant Mol. Biol. 2014, 85, 247–257. [Google Scholar] [CrossRef]
- Shimizu, M.; Pu, Z.; Kawanabe, T.; Kitashiba, H.; Matsumoto, S.; Ebe, Y.; Sano, M.; Funaki, T.; Fukai, E.; Fujimoto, R.; et al. Map-Based Cloning of a Candidate Gene Conferring Fusarium Yellows Resistance in Brassica oleracea. Theor. Appl. Genet. 2015, 128, 119–130. [Google Scholar] [CrossRef]
- Jones, J.D.G.; Dangl, J.L. The Plant Immune System. Nature 2006, 444, 323–329. [Google Scholar] [CrossRef]
- Heath, M.C. Hypersensitive Response-Related Death. In Programmed Cell Death in Higher Plants; Lam, E., Fukuda, H., Greenberg, J., Eds.; Springer: Dordrecht, The Netherlands, 2000; pp. 77–90. ISBN 978-94-010-0934-8. [Google Scholar]
- Lo Presti, L.; Lanver, D.; Schweizer, G.; Tanaka, S.; Liang, L.; Tollot, M.; Zuccaro, A.; Reissmann, S.; Kahmann, R. Fungal Effectors and Plant Susceptibility. Annu. Rev. Plant Biol. 2015, 66, 513–545. [Google Scholar] [CrossRef]
- Shirasu, K.; Schulze-Lefert, P. Regulators of Cell Death in Disease Resistance. Plant Mol. Biol. 2000, 44, 371–385. [Google Scholar] [CrossRef]
- Ngou, B.P.M.; Jones, J.D.G.; Ding, P. Plant Immune Networks. Trends Plant Sci. 2022, 27, 255–273. [Google Scholar] [CrossRef]
- Brotman, Y.; Normantovich, M.; Goldenberg, Z.; Zvirin, Z.; Kovalski, I.; Stovbun, N.; Doniger, T.; Bolger, A.M.; Troadec, C.; Bendahmane, A.; et al. Dual Resistance of Melon to Fusarium Oxysporum Races 0 and 2 and to Papaya Ring-Spot Virus Is Controlled by a Pair of Head-to-Head-Oriented NB-LRR Genes of Unusual Architecture. Mol. Plant 2013, 6, 235–238. [Google Scholar] [CrossRef]
- Pieterse, C.M.J.; Van Der Does, D.; Zamioudis, C.; Leon-Reyes, A.; Van Wees, S.C.M. Hormonal Modulation of Plant Immunity. Annu. Rev. Cell Dev. Biol. 2012, 28, 489–521. [Google Scholar] [CrossRef]
- Ramamoorthy, R.; Jiang, S.-Y.; Kumar, N.; Venkatesh, P.N.; Ramachandran, S. A Comprehensive Transcriptional Profiling of the WRKY Gene Family in Rice Under Various Abiotic and Phytohormone Treatments. Plant Cell Physiol. 2008, 49, 865–879. [Google Scholar] [CrossRef]
- Van Loon, L.C.; Rep, M.; Pieterse, C.M.J. Significance of Inducible Defense-Related Proteins in Infected Plants. Annu. Rev. Phytopathol. 2006, 44, 135–162. [Google Scholar] [CrossRef]
- Zhang, S.; Klessig, D.F. MAPK Cascades in Plant Defense Signaling. Trends Plant Sci. 2001, 6, 520–527. [Google Scholar] [CrossRef]
- Xing, M.; Lv, H.; Ma, J.; Xu, D.; Li, H.; Yang, L.; Kang, J.; Wang, X.; Fang, Z. Transcriptome Profiling of Resistance to Fusarium Oxysporum f. sp. Conglutinans in Cabbage (Brassica Oleracea) Roots. PLoS ONE 2016, 11, e0148048. [Google Scholar] [CrossRef]
- Liu, Z.; Xie, J.; Wang, H.; Zhong, X.; Li, H.; Yu, J.; Kang, J. Identification and Expression Profiling Analysis of NBS–LRR Genes Involved in Fusarium Oxysporum f.sp. Conglutinans Resistance in Cabbage. 3 Biotech 2019, 9, 202. [Google Scholar] [CrossRef]
- Chen, H.; Wang, T.; He, X.; Cai, X.; Lin, R.; Liang, J.; Wu, J.; King, G.; Wang, X. BRAD V3.0: An Upgraded Brassicaceae Database. Nucleic Acids Res. 2022, 50, D1432–D1441. [Google Scholar] [CrossRef]
- Sirén, J.; Välimäki, N.; Mäkinen, V. Indexing Graphs for Path Queries with Applications in Genome Research. IEEE/ACM Trans. Comput. Biol. Bioinform. 2014, 11, 375–388. [Google Scholar] [CrossRef]
- Anders, S.; Pyl, P.T.; Huber, W. HTSeq—A Python Framework to Work with High-Throughput Sequencing Data. Bioinformatics 2015, 31, 166–169. [Google Scholar] [CrossRef] [PubMed]
- Love, M.I.; Huber, W.; Anders, S. Moderated Estimation of Fold Change and Dispersion for RNA-Seq Data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed]
- Kieber, J.J.; Schaller, G.E. Cytokinin Signaling in Plant Development. Development 2018, 145, dev149344. [Google Scholar] [CrossRef] [PubMed]
- Romanov, G.A.; Lomin, S.N.; Schmülling, T. Cytokinin Signaling: From the ER or from the PM? That Is the Question! New Phytol. 2018, 218, 41–53. [Google Scholar] [CrossRef]
- Choi, J.; Huh, S.U.; Kojima, M.; Sakakibara, H.; Paek, K.-H.; Hwang, I. The Cytokinin-Activated Transcription Factor ARR2 Promotes Plant Immunity via TGA3/NPR1-Dependent Salicylic Acid Signaling in Arabidopsis. Dev. Cell 2010, 19, 284–295. [Google Scholar] [CrossRef]
- Takahashi, Y.; Zhang, J.; Hsu, P.-K.; Ceciliato, P.H.O.; Zhang, L.; Dubeaux, G.; Munemasa, S.; Ge, C.; Zhao, Y.; Hauser, F.; et al. MAP3Kinase-Dependent SnRK2-Kinase Activation Is Required for Abscisic Acid Signal Transduction and Rapid Osmotic Stress Response. Nat. Commun. 2020, 11, 12. [Google Scholar] [CrossRef]
- García-Andrade, J.; González, B.; Gonzalez-Guzman, M.; Rodriguez, P.L.; Vera, P. The Role of ABA in Plant Immunity Is Mediated through the PYR1 Receptor. Int. J. Mol. Sci. 2020, 21, 5852. [Google Scholar] [CrossRef]
- Cao, Z.; Li, Z.; Meng, L.; Cao, D.; Zhao, K.; Hu, S.; Li, Y.; Zhao, K.; Ma, Q.; Li, Y.; et al. Genome-Wide Characterization of Pyrabactin Resistance 1-like (PYL) Family Genes Revealed AhPYL6 Confer the Resistance to Ralstonia Solanacearum in Peanut. Plant Physiol. Biochem. 2024, 217, 109295. [Google Scholar] [CrossRef]
- Lim, C.W.; Kim, J.-H.; Baek, W.; Kim, B.S.; Lee, S.C. Functional Roles of the Protein Phosphatase 2C, AtAIP1, in Abscisic Acid Signaling and Sugar Tolerance in Arabidopsis. Plant Sci. 2012, 187, 83–88. [Google Scholar] [CrossRef]
- Stortenbeker, N.; Bemer, M. The SAUR Gene Family: The Plant’s Toolbox for Adaptation of Growth and Development. J. Exp. Bot. 2019, 70, 17–27. [Google Scholar] [CrossRef]
- Gangappa, S.N.; Berriri, S.; Kumar, S.V. PIF4 Coordinates Thermosensory Growth and Immunity in Arabidopsis. Curr. Biol. 2017, 27, 243–249. [Google Scholar] [CrossRef]
- Zhang, Y.; Cheng, Y.T.; Qu, N.; Zhao, Q.; Bi, D.; Li, X. Negative Regulation of Defense Responses in Arabidopsis by Two NPR1 Paralogs. Plant J. 2006, 48, 647–656. [Google Scholar] [CrossRef]
- Ding, Y.; Sun, T.; Ao, K.; Peng, Y.; Zhang, Y.; Li, X.; Zhang, Y. Opposite Roles of Salicylic Acid Receptors NPR1 and NPR3/NPR4 in Transcriptional Regulation of Plant Immunity. Cell 2018, 173, 1454–1467.e15. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.; Holub, E.B.; Alonso, J.M.; Ecker, J.R.; Fobert, P.R. An Arabidopsis NPR1-like Gene, NPR4, Is Required for Disease Resistance. Plant J. 2005, 41, 304–318. [Google Scholar] [CrossRef] [PubMed]
- Fu, Z.Q.; Yan, S.; Saleh, A.; Wang, W.; Ruble, J.; Oka, N.; Mohan, R.; Spoel, S.H.; Tada, Y.; Zheng, N.; et al. NPR3 and NPR4 Are Receptors for the Immune Signal Salicylic Acid in Plants. Nature 2012, 486, 228–232. [Google Scholar] [CrossRef]
- Canet, J.V.; Dobón, A.; Roig, A.; Tornero, P. Structure-Function Analysis of Npr1 Alleles in Arabidopsis Reveals a Role for Its Paralogs in the Perception of Salicylic Acid. Plant Cell Environ. 2010, 33, 1911–1922. [Google Scholar] [CrossRef] [PubMed]
- Ha, C.M.; Kim, G.-T.; Kim, B.C.; Jun, J.H.; Soh, M.S.; Ueno, Y.; Machida, Y.; Tsukaya, H.; Nam, H.G. The BLADE-ON-PETIOLE 1 Gene Controls Leaf Pattern Formation through the Modulation of Meristematic Activity in Arabidopsis. Development 2003, 130, 161–172. [Google Scholar] [CrossRef]
- Khan, M.; Xu, M.; Murmu, J.; Tabb, P.; Liu, Y.; Storey, K.; McKim, S.M.; Douglas, C.J.; Hepworth, S.R. Antagonistic Interaction of BLADE-ON-PETIOLE1 and 2 with BREVIPEDICELLUS and PENNYWISE Regulates Arabidopsis Inflorescence Architecture. Plant Physiol. 2012, 158, 946–960. [Google Scholar] [CrossRef]
- Canet, J.V.; Dobón, A.; Fajmonová, J.; Tornero, P. The BLADE-ON-PETIOLE Genes of Arabidopsis Are Essential for Resistance Induced by Methyl Jasmonate. BMC Plant Biol. 2012, 12, 199. [Google Scholar] [CrossRef]
- Zhang, Z.; Wang, P.; Luo, X.; Yang, C.; Tang, Y.; Wang, Z.; Hu, G.; Ge, X.; Xia, G.; Wu, J. Cotton Plant Defence against a Fungal Pathogen Is Enhanced by Expanding BLADE-ON-PETIOLE1 Expression beyond Lateral-Organ Boundaries. Commun. Biol. 2019, 2, 238. [Google Scholar] [CrossRef]
- Han, Z.; Xiong, D.; Schneiter, R.; Tian, C. The Function of Plant PR1 and Other Members of the CAP Protein Superfamily in Plant–Pathogen Interactions. Mol. Plant Pathol. 2023, 24, 651–668. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.-L.; Lee, C.-Y.; Cheng, K.-T.; Chang, W.-H.; Huang, R.-N.; Nam, H.G.; Chen, Y.-R. Quantitative Peptidomics Study Reveals That a Wound-Induced Peptide from PR-1 Regulates Immune Signaling in Tomato. Plant Cell 2014, 26, 4135–4148. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Q.-H.; Stephen, S.; Kazan, K.; Jin, G.; Fan, L.; Taylor, J.; Dennis, E.S.; Helliwell, C.A.; Wang, M.-B. Characterization of the Defense Transcriptome Responsive to Fusarium Oxysporum-Infection in Arabidopsis Using RNA-Seq. Gene 2013, 512, 259–266. [Google Scholar] [CrossRef]
- Asai, T.; Tena, G.; Plotnikova, J.; Willmann, M.R.; Chiu, W.-L.; Gomez-Gomez, L.; Boller, T.; Ausubel, F.M.; Sheen, J. MAP Kinase Signalling Cascade in Arabidopsis Innate Immunity. Nature 2002, 415, 977–983. [Google Scholar] [CrossRef]
- Sato, T.; Theologis, A. Cloning the mRNA Encoding 1-Aminocyclopropane-1-Carboxylate Synthase, the Key Enzyme for Ethylene Biosynthesis in Plants. Proc. Natl. Acad. Sci. USA 1989, 86, 6621–6625. [Google Scholar] [CrossRef]
- Lv, B.; Tian, H.; Zhang, F.; Liu, J.; Lu, S.; Bai, M.; Li, C.; Ding, Z. Brassinosteroids Regulate Root Growth by Controlling Reactive Oxygen Species Homeostasis and Dual Effect on Ethylene Synthesis in Arabidopsis. PLoS Genet. 2018, 14, e1007144. [Google Scholar] [CrossRef]
- Li, Z.; Zhang, L.; Yu, Y.; Quan, R.; Zhang, Z.; Zhang, H.; Huang, R. The Ethylene Response Factor AtERF11 That Is Transcriptionally Modulated by the bZIP Transcription Factor HY5 Is a Crucial Repressor for Ethylene Biosynthesis in Arabidopsis. Plant J. 2011, 68, 88–99. [Google Scholar] [CrossRef]
- Penninckx, I.A.; Eggermont, K.; Terras, F.R.; Thomma, B.P.; De Samblanx, G.W.; Buchala, A.; Métraux, J.P.; Manners, J.M.; Broekaert, W.F. Pathogen-Induced Systemic Activation of a Plant Defensin Gene in Arabidopsis Follows a Salicylic Acid-Independent Pathway. Plant Cell 1996, 8, 2309–2323. [Google Scholar] [CrossRef]
- Glazebrook, J. Contrasting Mechanisms of Defense Against Biotrophic and Necrotrophic Pathogens. Annu. Rev. Phytopathol. 2005, 43, 205–227. [Google Scholar] [CrossRef]
- Feys, B.J.F.; Benedetti, C.E.; Penfold, C.N.; Turner, J.G. Arabidopsis Mutants Selected for Resistance to the Phytotoxin Coronatine Are Male Sterile, Insensitive to Methyl Jasmonate, and Resistant to a Bacterial Pathogen. Plant Cell 1994, 6, 751–759. [Google Scholar] [CrossRef]
- Lv, H.; Wang, Q.; Yang, L.; Fang, Z.; Liu, Y.; Zhuang, M.; Zhang, Y.; Yang, Y.; Xie, B.; Wang, X. Breeding of cabbage (Brassica oleracea L.var. capitata) with Fusarium wilt resistance based on microspore culture and biomarker selection. Euphytica 2014, 200, 465–473. [Google Scholar] [CrossRef]
- Liu, X.; Han, F.; Kong, C.; Fang, Z.; Yang, L.; Zhang, Y.; Zhuang, M.; Liu, Y.; Li, Z.; Lv, H. Rapid Introgression of the Fusarium Wilt Resistance Gene into an Elite Cabbage Line through the Combined Application of a Microspore Culture, Genome Background Analysis, and Disease Resistance-Specific Marker Assisted Foreground Selection. Front. Plant Sci. 2017, 8, 354. [Google Scholar] [CrossRef] [PubMed]
- Martin, M. Cutadapt Removes Adapter Sequences from High-Throughput Sequencing Reads. EMBnet. J. 2011, 17, 10–12. [Google Scholar] [CrossRef]
- Babraham, U. Babraham Bioinformatics—FastQC A Quality Control Tool for High Throughput Sequence Data. Soil 2020, 5, 47–81. [Google Scholar]
- Florea, L.; Song, L.; Salzberg, S.L. Thousands of Exon Skipping Events Differentiate among Splicing Patterns in Sixteen Human Tissues. F1000Research 2013, 2, 188. [Google Scholar] [CrossRef]
- Sacco, F.; Seelig, A.; Humphrey, S.J.; Krahmer, N.; Volta, F.; Reggio, A.; Marchetti, P.; Gerdes, J.; Mann, M. Phosphoproteomics Reveals the GSK3-PDX1 Axis as a Key Pathogenic Signaling Node in Diabetic Islets. Cell Metab 2019, 29, 1422–1432.e3. [Google Scholar] [CrossRef]
- Wickham, H. Programming with Ggplot2. In ggplot2: Elegant Graphics for Data Analysis; Wickham, H., Ed.; Springer International Publishing: Cham, Switzerland, 2016; pp. 241–253. ISBN 978-3-319-24277-4. [Google Scholar]
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Butler, H.; Cherry, J.M.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; Eppig, J.T.; et al. Gene Ontology: Tool for the Unification of Biology. Nat. Genet. 2000, 25, 25–29. [Google Scholar] [CrossRef]
- Kanehisa, M.; Goto, S.; Kawashima, S.; Okuno, Y.; Hattori, M. The KEGG Resource for Deciphering the Genome. Nucleic Acids Res. 2004, 32, D277–D280. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of Relative Gene Expression Data Using Real-Time Quantitative PCR and the 2−ΔΔCT Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Rieu, I.; Powers, S.J. Real-Time Quantitative RT-PCR: Design, Calculations, and Statistics. Plant Cell 2009, 21, 1031–1033. [Google Scholar] [CrossRef]
- Han, F.; Yuan, K.; Sun, W.; Zhang, X.; Liu, X.; Zhao, X.; Yang, L.; Wang, Y.; Ji, J.; Liu, Y.; et al. A Natural Mutation in the Promoter of Ms-Cd1 Causes Dominant Male Sterility in Brassica Oleracea. Nat. Commun. 2023, 14, 6212. [Google Scholar] [CrossRef]
- Guo, H.; Ahn, H.-K.; Sklenar, J.; Huang, J.; Ma, Y.; Ding, P.; Menke, F.L.H.; Jones, J.D.G. Phosphorylation-Regulated Activation of the Arabidopsis RRS1-R/RPS4 Immune Receptor Complex Reveals Two Distinct Effector Recognition Mechanisms. Cell Host Microbe 2020, 27, 769–781.e6. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample ID | Reads No. | Bases (bp) | Clean Reads No. | Clean Data (bp) | Clean Reads (%) | Q20 (%) | Q30 (%) |
|---|---|---|---|---|---|---|---|
| S-Foc-1 | 44,603,366 | 6,735,108,266 | 41,427,642 | 6,255,573,942 | 92.88 | 97.54 | 94.22 |
| S-Foc-2 | 43,900,734 | 6,629,010,834 | 40,789,702 | 6,159,245,002 | 92.91 | 97.52 | 93.95 |
| S-Foc-3 | 42,642,538 | 6,439,023,238 | 39,572,722 | 5,975,481,022 | 92.8 | 97.69 | 93.79 |
| S-MOCK1 | 39,137,790 | 5,909,806,290 | 36,291,182 | 5,479,968,482 | 92.72 | 97.65 | 94.06 |
| S-MOCK2 | 39,511,534 | 5,966,241,634 | 36,646,568 | 5,533,631,768 | 92.74 | 97.48 | 93.93 |
| S-MOCK3 | 48,918,954 | 7,386,762,054 | 45,412,050 | 6,857,219,550 | 92.83 | 97.41 | 94.3 |
| YR-Foc-1 | 39,549,064 | 5,971,908,664 | 36,671,228 | 5,537,355,428 | 92.72 | 97.54 | 94.08 |
| YR-Foc-2 | 41,756,658 | 6,305,255,358 | 38,804,600 | 5,859,494,600 | 92.93 | 97.01 | 93.94 |
| YR-Foc-3 | 49,111,204 | 7,415,791,804 | 45,588,484 | 6,883,861,084 | 92.82 | 97.1 | 93.97 |
| YR-MOCK1 | 39,428,946 | 5,953,770,846 | 36,585,610 | 5,524,427,110 | 92.78 | 97.58 | 93.98 |
| YR-MOCK2 | 39,042,744 | 5,895,454,344 | 35,950,636 | 5,428,546,036 | 92.08 | 97.51 | 93.37 |
| YR-MOCK3 | 41,046,088 | 6,197,959,288 | 37,813,296 | 5,709,807,696 | 92.12 | 97.55 | 93.56 |
| Total | 508,649,620 | 76,806,092,620 | 471,553,720 | 71,204,611,720 | 92.71 | 97.47 | 93.94 |
| Pathway | Gene | Expression | Annotation |
|---|---|---|---|
| Plant hormone signal transduction | BolC03g044440.2J | Up | Encodes histidine phosphate transferase, cytokinin |
| BolC07g046020.2J | Up | Encodes an Arabidopsis response regulator (ARR) protein, cytokinin | |
| BolC08g057620.2J | Up | Encodes a member of the PP2C family, abscisic acid | |
| BolC07g057090.2J | Down | Encodes a CAP (cysteine-rich secretory proteins, antigen 5, and pathogenesis-related 1 protein) superfamily protein, salicylic acid | |
| BolC01g011300.2J | Down | Encodes a PYR/PYL/RCAR family protein, abscisic acid | |
| BolC02g031960.2J | Down | SAUR-like auxin-responsive protein family, auxin | |
| BolC03g061170.2J | Down | Encodes a transcription repressor that mediates a negative feedback loop in cytokinin signalling, cytokinin | |
| BolC03g026110.2J | Down | Encodes a nuclear localized bHLH protein, auxin | |
| BolC03g024500.2J | Down | Encodes a PYR/PYL/RCAR family protein, abscisic acid | |
| BolC04g064360.2J | Down | Encodes a PYR/PYL/RCAR family protein, abscisic acid | |
| BolC08g041070.2J | Down | Encodes a protein containing ankyrin repeat domains and BTB/POZ domains, jasmonic acid | |
| MAPK signaling | BolC06g030650.2J | Up | Encodes an ethylene- and jasmonate-responsive plant defensin, belongs to the plant defensin (PDF) family |
| BolC08g044590.2J | Up | Encodes a member of the 1-aminocyclopropane-1-carboxylate (ACC) synthase gene family | |
| BolC03g034380.2J | Down | Encodes a member of the A1 subgroup of the MEKK (MAPK/ERK kinase kinase) family |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhao, C.; Liu, X.; Zhou, A.; Ji, J.; Wang, Y.; Zhuang, M.; Zhang, Y.; Yang, L.; Ma, L.; Chellappan, B.V.; et al. Transcriptome Analysis of Cabbage Near-Isogenic Lines Reveals the Involvement of the Plant Defensin Gene PDF1.2 in Fusarium Wilt Resistance. Int. J. Mol. Sci. 2025, 26, 3770. https://doi.org/10.3390/ijms26083770
Zhao C, Liu X, Zhou A, Ji J, Wang Y, Zhuang M, Zhang Y, Yang L, Ma L, Chellappan BV, et al. Transcriptome Analysis of Cabbage Near-Isogenic Lines Reveals the Involvement of the Plant Defensin Gene PDF1.2 in Fusarium Wilt Resistance. International Journal of Molecular Sciences. 2025; 26(8):3770. https://doi.org/10.3390/ijms26083770
Chicago/Turabian StyleZhao, Cunbao, Xing Liu, Ailing Zhou, Jialei Ji, Yong Wang, Mu Zhuang, Yangyong Zhang, Limei Yang, Lisong Ma, Biju V. Chellappan, and et al. 2025. "Transcriptome Analysis of Cabbage Near-Isogenic Lines Reveals the Involvement of the Plant Defensin Gene PDF1.2 in Fusarium Wilt Resistance" International Journal of Molecular Sciences 26, no. 8: 3770. https://doi.org/10.3390/ijms26083770
APA StyleZhao, C., Liu, X., Zhou, A., Ji, J., Wang, Y., Zhuang, M., Zhang, Y., Yang, L., Ma, L., Chellappan, B. V., Artemyeva, A. M., & Lv, H. (2025). Transcriptome Analysis of Cabbage Near-Isogenic Lines Reveals the Involvement of the Plant Defensin Gene PDF1.2 in Fusarium Wilt Resistance. International Journal of Molecular Sciences, 26(8), 3770. https://doi.org/10.3390/ijms26083770