
Modularized Genes in an Adrenal Pathway Reveal a Novel Mechanism in Hypertension Pathogenesis
Abstract
1. Introduction
1.1. Background and Questions Raised
1.2. Revealing Mechanisms of Blood Pressure Regulation in Physiology
1.3. Objectives
2. Results
2.1. Congenic Knock-Ins Mediate a Transition of Associative Results to Molecular Mechanisms Determining BP Physiology
2.2. Physiologically, a QTL Has a Major Effect on Blood Pressure
2.3. Three QTLs Act by Modularity/Common Pathways in Physiologically Controlling Blood Pressure
2.4. Revelation of a Pathway via Molecularly Identifying a QTL
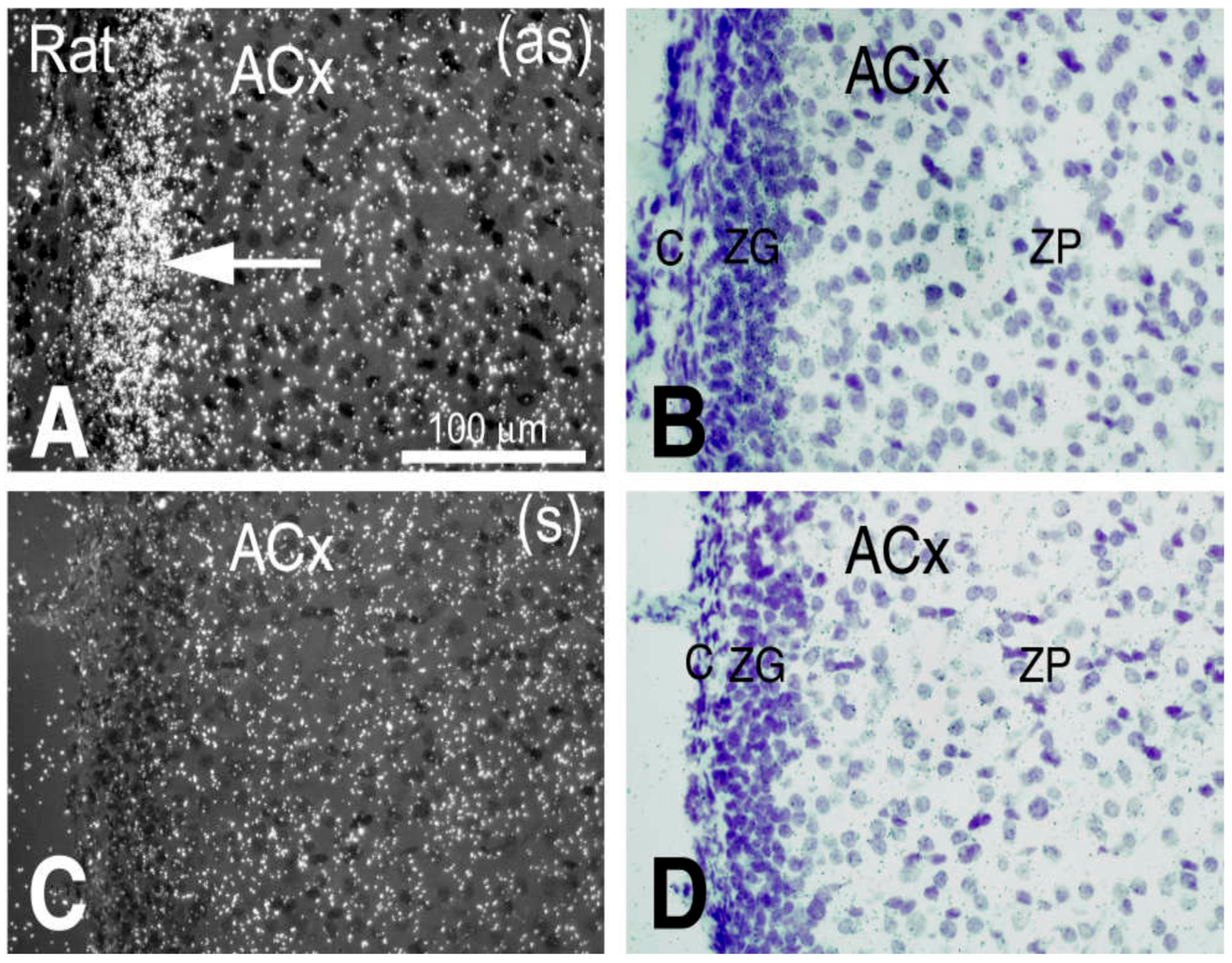
2.5. The Gene (Cuedc1) Encoding CUE Domain Containing 1 Protein Is the Strongest Candidate for C10QTL6 in Hypertension Pathogenesis
2.6. Direct Translation of the Hypertension Pathogenic Pathway of Cuedc1 from Rodents to Humans
3. Discussion
3.1. Major Findings from Our Current Work
3.2. Physiological Impact from a QTL on Blood Pressure Is Major
3.3. Combined Effects from Multiple QTLs Are Physiologically Epistatic and Suggest a Shared Pathway Merging Them in Regulating Blood Pressure Physiologically
3.4. A Novel Pathway of Hypertension Pathogenesis as Part of Polygenic Architecture Determining Blood Pressure Physiology
3.5. Discovering a New Role Played by Cuedc1 in Physiologically Controlling Blood Pressure via Regulating Aldosterone Synthesis and Kidney and Cardiac Functions
3.6. Direct Translation of CUEDC1 into a Human Mechanism in Polygenic Hypertension
3.7. Human Medical Applications Can Be Generated in Personalized Diagnosis and Treatment of Hypertension by Targeting CUEDC1
3.8. Multiple Steps of a Pathway Acting Together as a Module in Hypertension Pathogenesis
3.9. Caveats
4. Materials and Methods
4.1. Animals
4.2. Mutation Screening and Verifications
4.3. BP Experimental Protocols and Analyses
4.4. Assessing Cardiac and Renal Functions and In Situ Hybridization
4.5. Aldosterone Measurements
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Evangelou, E.; Warren, H.R.; Mosen-Ansorena, D.; Mifsud, B.; Pazoki, R.; Gao, H.; Ntritsos, G.; Dimou, N.; Cabrera, C.P.; Karaman, I.; et al. Genetic analysis of over 1 million people identifies 535 new loci associated with blood pressure traits. Nat. Genet. 2018, 50, 1412–1425. [Google Scholar] [CrossRef]
- Oliveros, W.; Delfosse, K.; Lato, D.F.; Kiriakopulos, K.; Mokhtaridoost, M.; Said, A.; McMurray, B.J.; Browning, J.W.L.; Mattioli, K.; Meng, G.; et al. Systematic characterization of regulatory variants of blood pressure genes. Cell Genom. 2023, 3, 100330. [Google Scholar] [CrossRef]
- Olczak, K.J.; Taylor-Bateman, V.; Nicholls, H.L.; Traylor, M.; Cabrera, C.P.; Munroe, P.B. Hypertension genetics past, present and future applications. J. Intern. Med. 2021, 290, 1130–1152. [Google Scholar] [CrossRef]
- Yang, M.-L.; Xu, C.; Gupte, T.; Hoffmann, T.J.; Iribarren, C.; Zhou, X.; Ganesh, S.K. Sex-specific genetic architecture of blood pressure. Nat. Med. 2024, 30, 818–828. [Google Scholar] [CrossRef]
- Deng, A.; Ménard, A. Animal Model Studies Reveal that Common Human-Centric Non-Coding Variants from Epidemiology are By-products of Primate Evolution Unrelated to Physiological Control of Blood Pressure. Cardiol. Cardiovasc. Med. 2021, 5, 471–501. [Google Scholar] [CrossRef]
- Cowley, A.W. Chrm3 Gene and M3 Muscarinic Receptors Contribute to Salt-Sensitive Hypertension, but Now a Physiological Puzzle. Hypertension 2018, 72, 588–591. [Google Scholar] [CrossRef]
- Deng, A.Y.; deBlois, D.; Laporte, S.A.; Gelinas, D.; Tardif, J.C.; Thorin, E.; Shi, Y.; Raignault, A.; Ménard, A. Novel Pathogenesis of Hypertension and Diastolic Dysfunction Caused by M3R (Muscarinic Cholinergic 3 Receptor) Signaling. Hypertension 2018, 72, 755–764. [Google Scholar] [CrossRef]
- Deng, A.Y. Modularity/non-cumulativity of quantitative trait loci on blood pressure. J. Hum. Hypertens. 2020, 34, 432–439. [Google Scholar] [CrossRef]
- Chauvet, C.; Crespo, K.; Menard, A.; Roy, J.; Deng, A.Y. Modularization and epistatic hierarchy determine homeostatic actions of multiple blood pressure quantitative trait loci. Hum. Mol. Genet. 2013, 22, 4451–4459. [Google Scholar] [CrossRef]
- Chauvet, C.; Menard, A.; Deng, A.Y. Two candidate genes for two quantitative trait loci epistatically attenuate hypertension in a novel pathway. J. Hypertens. 2015, 33, 1791–1801. [Google Scholar] [CrossRef]
- Deng, A.Y. Genetic mechanisms of polygenic hypertension: Fundamental insights from experimental models. J. Hypertens. 2015, 33, 669–680. [Google Scholar] [CrossRef]
- Deng, A.Y.; Huot-Marchard, J.-É.; deBlois, D.; Thorin, E.; Chauvet, C.; Menard, A. Functional Dosage of Muscarinic Cholinergic Receptor 3 Signalling, Not the Gene Dose, Determines Its Hypertension Pathogenesis. Can. J. Cardiol. 2019, 35, 661–670. [Google Scholar] [CrossRef]
- Alves-Lopes, R.; Neves, K.B.; Touyz, R.M. Muscarinic Receptor Type-3 in Hypertension and Cholinergic-Adrenergic Crosstalk: Genetic Insights and Potential for New Antihypertensive Targets. Can. J. Cardiol. 2019, 35, 555–557. [Google Scholar] [CrossRef]
- Te Riet, L.; van Esch, J.H.; Roks, A.J.; van den Meiracker, A.H.; Danser, A.H. Hypertension: Renin–Angiotensin–Aldosterone System Alterations. Circ. Res. 2015, 116, 960–975. [Google Scholar] [CrossRef]
- Boyle, E.A.; Li, Y.I.; Pritchard, J.K. An Expanded View of Complex Traits: From Polygenic to Omnigenic. Cell 2017, 169, 1177–1186. [Google Scholar] [CrossRef]
- Deng, A.Y.; Menard, A.; Deng, D.W. Shifting Paradigm from Gene Expressions to Pathways Reveals Physiological Mechanisms in Blood Pressure Control in Causation. Int. J. Mol. Sci. 2023, 24, 1262. [Google Scholar] [CrossRef]
- O’Leary, M.A.; Bloch, J.I.; Flynn, J.J.; Gaudin, T.J.; Giallombardo, A.; Giannini, N.P.; Goldberg, S.L.; Kraatz, B.P.; Luo, Z.X.; Meng, J.; et al. The Placental Mammal Ancestor and the Post–K-Pg Radiation of Placentals. Science 2013, 339, 662–667. [Google Scholar] [CrossRef]
- White, C.R.; Seymour, R.S. The role of gravity in the evolution of mammalian blood pressure. Evolution 2014, 68, 901–908. [Google Scholar] [CrossRef]
- Agarwal, M.K.; Mirshahi, M. General overview of mineralocorticoid hormone action. Pharmacol. Ther. 1999, 84, 273–326. [Google Scholar] [CrossRef]
- Lopes, R.; Korkmaz, G.; Revilla, S.A.; van Vliet, R.; Nagel, R.; Custers, L.; Kim, Y.; van Breugel, P.C.; Zwart, W.; Moumbeini, B.; et al. CUEDC1 is a primary target of ERα essential for the growth of breast cancer cells. Cancer Lett. 2018, 436, 87–95. [Google Scholar] [CrossRef]
- Delcayre, C.; Silvestre, J.S.; Garnier, A.; Oubenaissa, A.; Cailmail, S.; Tatara, E.; Swynghedauw, B.; Robert, V. Cardiac aldosterone production and ventricular remodeling. Kidney Int. 2000, 57, 1346–1351. [Google Scholar] [CrossRef]
- Huang, B.S.; White, R.A.; Ahmad, M.; Jeng, A.Y.; Leenen, F.H.H. Central infusion of aldosterone synthase inhibitor prevents sympathetic hyperactivity and hypertension by central Na+ in Wistar rats. AJP Regul. Integr. Comp. Physiol. 2008, 295, R166–R172. [Google Scholar] [CrossRef]
- Xu, G.; Shen, H.; Nibona, E.; Wu, K.; Ke, X.; Al Hafiz, A.; Liang, X.; Zhong, X.; Zhou, Q.; Qi, C.; et al. Fundc1 is necessary for proper body axis formation during embryogenesis in zebrafish. Sci. Rep. 2019, 9, 18910. [Google Scholar] [CrossRef]
- Cicila, G.T.; Rapp, J.P.; Wang, J.M.; St Lezin, E.; Ng, S.C.; Kurtz, T.W. Linkage of 11 beta-hydroxylase mutations with altered steroid biosynthesis and blood pressure in the Dahl rat. Nat. Genet. 1993, 3, 346–353. [Google Scholar] [CrossRef]
- Crespo, K.; Chauvet, C.; Blain, M.; Menard, A.; Roy, J.; Deng, A.Y. Normotension in Lewis and Dahl salt-resistant rats is governed by different genes. J. Hypertens. 2011, 29, 460–465. [Google Scholar] [CrossRef]
- Atanur, S.S.; Diaz, A.G.; Maratou, K.; Sarkis, A.; Rotival, M.; Game, L.; Tschannen, M.R.; Kaisaki, P.J.; Otto, G.W.; Ma, M.C.; et al. Genome Sequencing Reveals Loci under Artificial Selection that Underlie Disease Phenotypes in the Laboratory Rat. Cell 2013, 154, 691–703. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
| C10QTLs | Blood Pressure Effect in Physiology | Gene | Mutation Detected Lew/DSS | Change in Amino Acid (AA) Lew/DSS | % Conservation in Amino Acid Comparing to Rat | |
|---|---|---|---|---|---|---|
| Human | Tasmania Devil | |||||
| C10QTL6 | 37.5% | Cuedc1 | G510T | A170S | 88.9% | 87.9% |
| MrpS23 | none | None | ||||
| C10QTL1 | 62.5% | Ppm1e | 1 missensed mutation | |||
| C10QTL5 | 58.8% | Prr11 | 2 missensed mutations | |||
| C10QTL4 | 33.8% | Schlafen 2 Schlafen 1 Schlafen 3 Schlafen 4 Schlafen 14 | None Multiple Multiple G330C T1857C T3421C | None 6 amino acids 24 amino acids None None Tryp1141Arg | ||
| Cardiac Phenotypes Measured by Echocardiography | DSS (n = 17) | Lewis (n = 13) | C10S.L18 (n = 10) | |
|---|---|---|---|---|
| Heart rate (beats per min) | 419.1 ± 28.0 | 388.6 ± 34.4 * | 395.2.9 ± 37.8 | |
| Aortic peak velocity (cm/s) | 107.2 ± 14.2 | 94.6 ± 11.6 * | 89.1 ± 9.9 * | |
| Left ventricular (LV) wall thickness and mass | LVAW (mm) | 0.19 ± 0.02 | 0.15 ± 0.01 * | 0.18 ± 0.02 |
| LVPW (mm) | 0.19 ± 0.02 | 0.15 ± 0.01 * | 0.17 ± 0.02 | |
| Mass (g) | 1.38 ± 0.09 | 1.18 ± 0.13 * | 1.23 ± 0.08 * | |
| LV mass (g) | 0.91 ± 0.09 | 0.72 ± 0.13 * | 0.88 ± 0.09 * | |
| LV hypertrophy and hyperdynamic state | Yes | No | Normalized | |
| Left atrial (LA) dimension | Systolic (mm) | 5.16 ± 0.63 | 4.39 ± 0.48 * | 4.6 ± 0.23 * |
| Diastolic (mm) | 3.54 ± 0.62 | 2.89 ± 0.38 * | 2.92 ± 0.26 * | |
| Fractional shortening (%) | 31.4 ± 7.24 | 34.1 ± 6.08 | 36.7 ± 5.49 | |
| LA structural remodeling | Yes | No | Normalized | |
| Pulse Doppler mitral filling pattern | E velocity (cm/s) | 113.0 ± 14.9 | 92.8 ± 12.7 * | 102.3 ± 7.73 * |
| DT (ms) | 45.9 ± 6.54 | 59.3 ± 11.5 * | 53.2 ± 6.02 | |
| DR (cm/s2) | 2446 ± 460.6 | 1548 ± 244.8 * | 1914 ± 246.1 * | |
| Left ventricular isovolumetric relaxation time | IVRT (ms) | 20.2 ± 3.12 | 16.3 ± 2.66 * | 17.7 ± 4.58 |
| RR (ms) | 144.3 ± 10.6 | 153.8 ± 12.9 | 153.0 ± 15.8 | |
| IVRT/(RR)1/2 | 1.68 ± 0.25 | 1.33 ± 0.23 * | 1.42 ± 0.30 | |
| Left ventricular global myocardial performance index | MD (ms) | 53.8 ± 7.84 | 62.0 ± 8.07 * | 61.2 ± 5.60 |
| DD (ms) | 77.9 ± 9.63 | 79.3 ± 8.72 | 83.4 ± 10.6 | |
| ET (ms) | 70.5 ± 6.32 | 79.4 ± 4.88 * | 80.4 ± 6.15 * | |
| MPI | 0.35 ± 0.12 | 0.22 ± 0.05 * | 0.27 ± 0.03 * | |
| LV diastolic dysfunction | Yes | No | Normalized | |
| DSS_Rat | MTSLFRRSSSGSGGGGATGARGAGTGTGDGSAAPQELNNSRPARQVRRLEFNQAMDDFKT | 60 |
| Human | MTSLFRRSSSGSGGGGTAGARGGG----GGTAAPQELNNSRPARQVRRLEFNQAMDDFKT | 56 |
| Tasmanian | MTSLFRRSSSNGGS------------RGGGNASAQELNNSRPARQVRRLEFNQAMEDFKT | 48 |
| **********..*. .*.*: *********************:**** | ||
| DSS_Rat | MFPNMDYDIIECVLRANSGAVDATIDQLLQMNLEAGGVS--AYEDSSDSEDSIPPEILER | 118 |
| Human | MFPNMDYDIIECVLRANSGAVDATIDQLLQMNLEGGGSSGGVYEDSSDSEDSIPPEILER | 116 |
| Tasmanian | MFPNMDYDIIECVLRANNGAVDATIDQLLQMNLDG-----SSYDDSSDSDDSIPPEILER | 103 |
| *****************.***************:. *:*****:********** | ||
| DSS_Rat | TLEPDSSEEEPPPVYSPPAYHMHVFDRPYLTAPPTPPPRIDVLGSGQPASQSRYRNWNPP | 178 |
| Human | TLEPDSSDEEPPPVYSPPAYHMHVFDRPYPLAPPTPPPRIDALGSGAPTSQRRYRNWNPP | 176 |
| Tasmanian | TLEPDSSDEEPPPVYSPPAYHMHMFDRPYPLAPPTPPPRIDVPSAGVPLTQRRYRNWNPP | 163 |
| *******:***************:***** **********. .:* * :* ******** | ||
| DSS_Rat | LLGNLPDDFLRILPQQMDSIQGHPGGSKPM-SGEGVPPVAPGPMACDQDSRWKQYLEDER | 237 |
| Human | LLGNLPDDFLRILPQQLDSIQGNAGGPKPG-SGEGCPPAMAGPGPGDQESRWKQYLEDER | 235 |
| Tasmanian | LLGNLPDDFLRILPQQLDSLQNTQSGPPKLGLGEVSQP---MVGNLEEECRWKQYLEDER | 220 |
| ****************:**:*. .* ** * :::.********** | ||
| DSS_Rat | IALFLQNEEFMKELQRNRDFLLALERDRLKYESQKSKSSNVAVGSDVGFPSSVPG----- | 292 |
| Human | IALFLQNEEFMKELQRNRDFLLALERDRLKYESQKSKSSSVAVGNDFGFSSPVPG----- | 290 |
| Tasmanian | IALFLQNEEFMKELQRNRDFLLALERDRLKYESQKSKSSNMAVSNDFGFPSTVTGDAALG | 280 |
| ***************************************.:**..*.** * * * | ||
| DSS_Rat | INDTNPTVSEDALFRDKLKHMGKSTRRKLFELARAFSEKTKMRKSKKKHLPKLQSLGAAA | 352 |
| Human | TGDANPAVSEDALFRDKLKHMGKSTRRKLFELARAFSEKTKMRKSKRKHLLKHQSLGAAA | 350 |
| Tasmanian | ASEANPAVSEDALFRDKLKHMGKSTRRKLFELARAFSEKTKMRKTKRKQLLKHQSAGWGL | 340 |
| .::**:*************************************:*:*:* * ** * . | ||
| DSS_Rat | STANLLDDVEGHAYEED--------FRGRRQEEPKVEE---------TLREGQ------- | 388 |
| Human | STANLLDDVEGHACDED--------FRGRRQEAPKVEE---------GLREGQ------- | 386 |
| Tasmanian | QHRQLISWMTWKAMRVKKTSGQGSRRHSRRRKHPEKDSKRCWSSPEMKCPNGQPNSEGLA | 400 |
| . :*:. : :* . :.**:: *: :. :** | ||
| DSS_Rat | ------------- | 388 |
| Human | ------------- | 386 |
| Tasmanian | LAAGTCCSKTEGV | 413 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Deng, D.W.; Ménard, A.; Deng, A.Y. Modularized Genes in an Adrenal Pathway Reveal a Novel Mechanism in Hypertension Pathogenesis. Int. J. Mol. Sci. 2025, 26, 3782. https://doi.org/10.3390/ijms26083782
Deng DW, Ménard A, Deng AY. Modularized Genes in an Adrenal Pathway Reveal a Novel Mechanism in Hypertension Pathogenesis. International Journal of Molecular Sciences. 2025; 26(8):3782. https://doi.org/10.3390/ijms26083782
Chicago/Turabian StyleDeng, David W., Annie Ménard, and Alan Y. Deng. 2025. "Modularized Genes in an Adrenal Pathway Reveal a Novel Mechanism in Hypertension Pathogenesis" International Journal of Molecular Sciences 26, no. 8: 3782. https://doi.org/10.3390/ijms26083782
APA StyleDeng, D. W., Ménard, A., & Deng, A. Y. (2025). Modularized Genes in an Adrenal Pathway Reveal a Novel Mechanism in Hypertension Pathogenesis. International Journal of Molecular Sciences, 26(8), 3782. https://doi.org/10.3390/ijms26083782