
Inhibition of ABCG2 by SCO-101 Enhances Chemotherapy Efficacy in Cancer

 , , , , , ,
, , , , , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
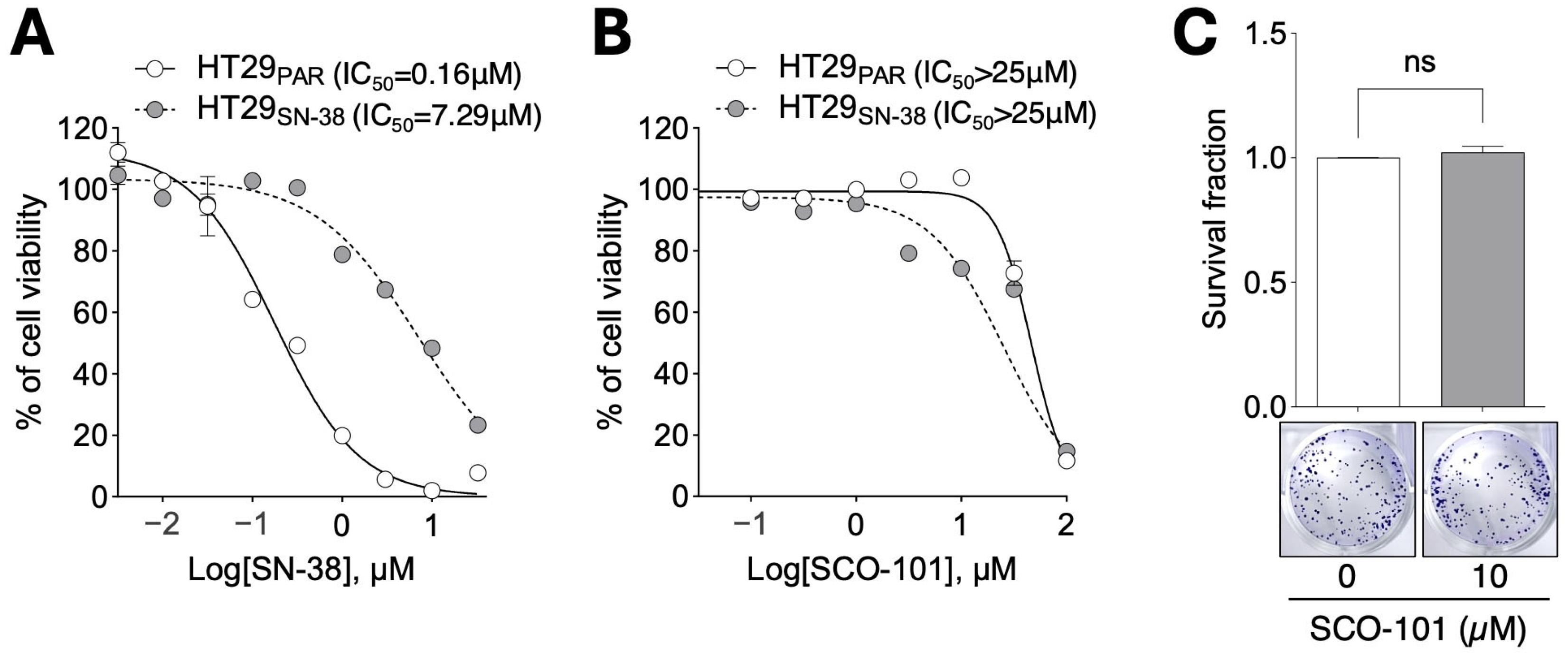
2.1. SCO-101 Treatment Displays Low Toxicity in Cellular Models of Drug Resistance
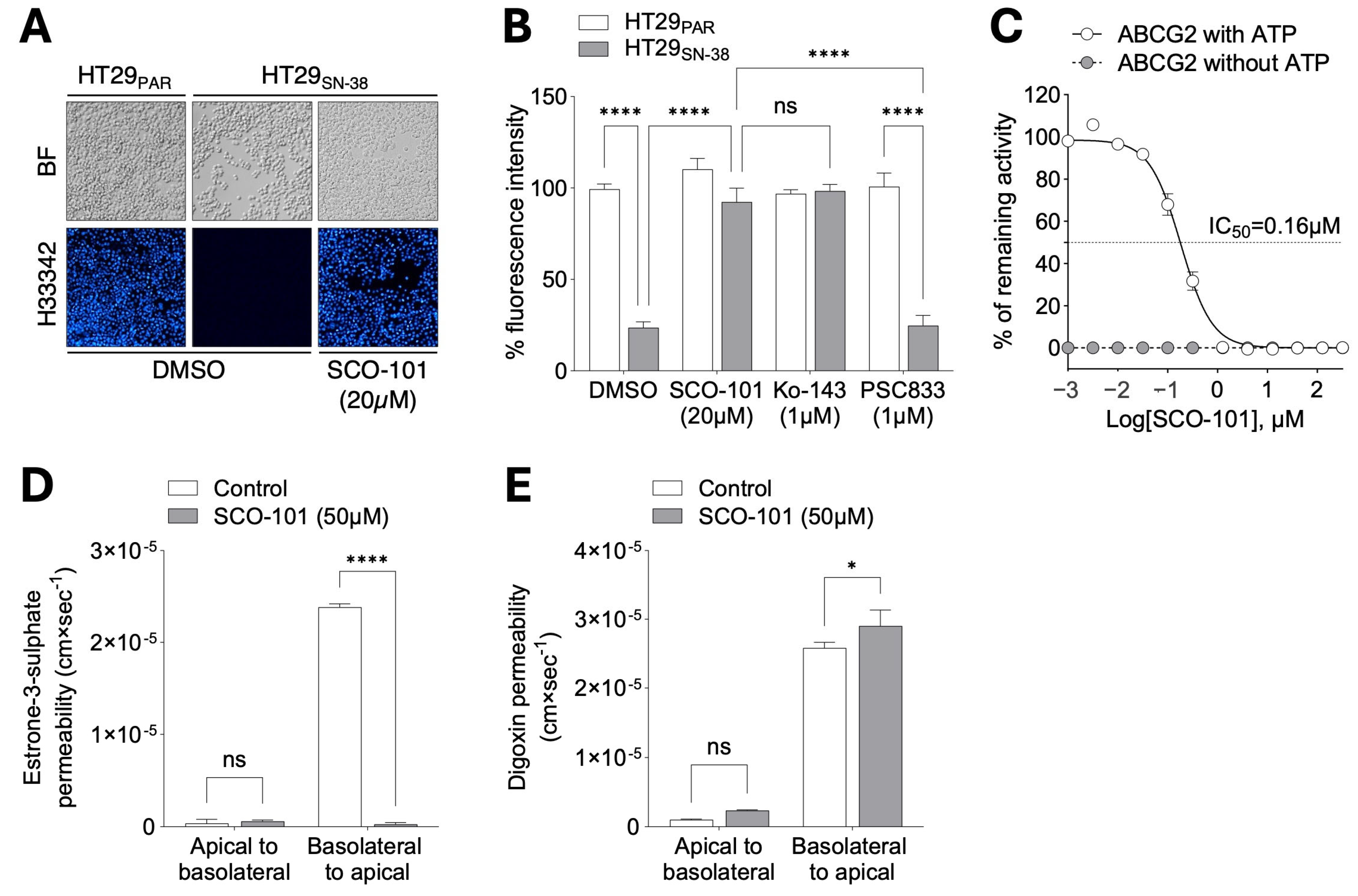
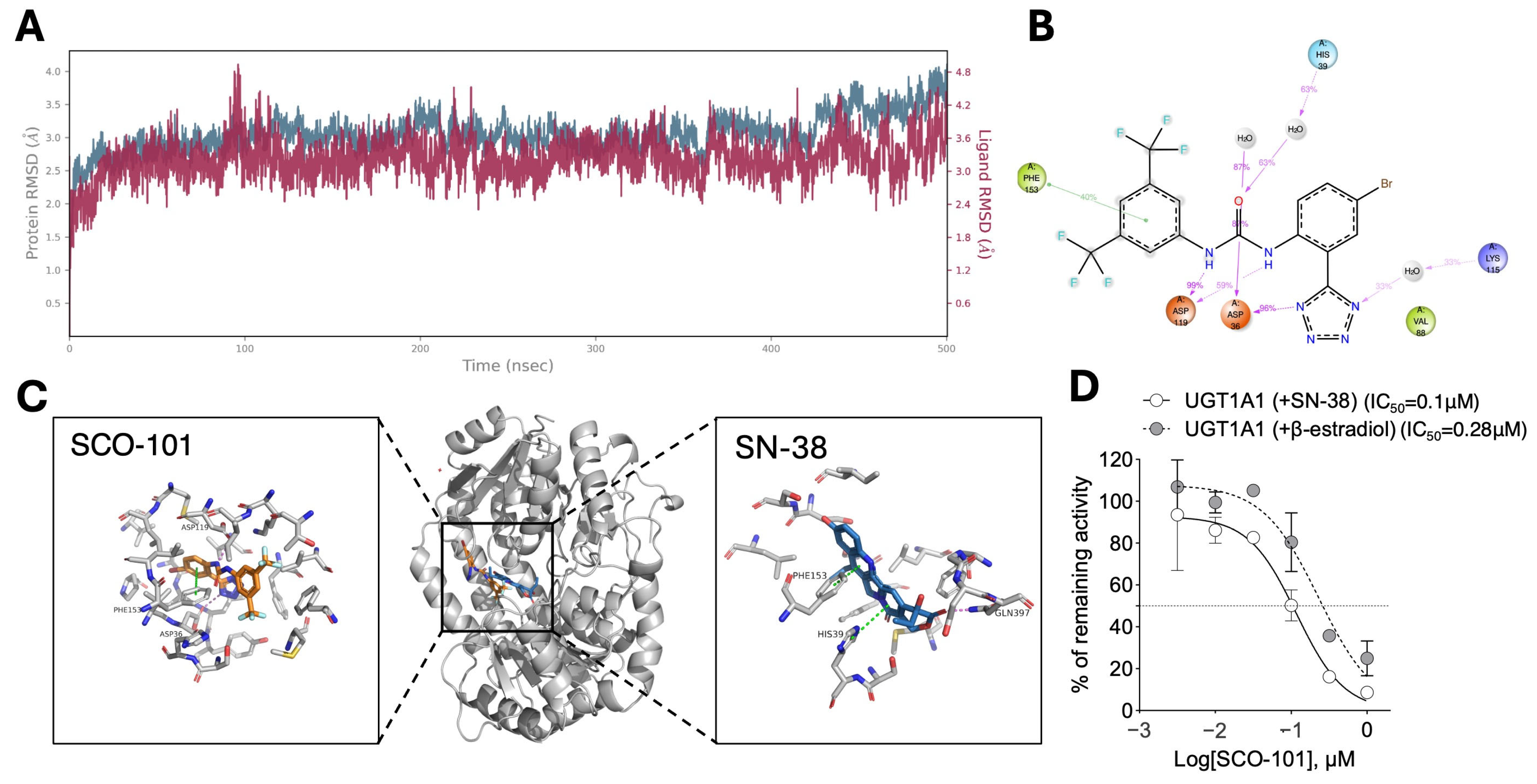
2.2. SCO-101 Is an Inhibitor of Both ABCG2 and UGT1A1
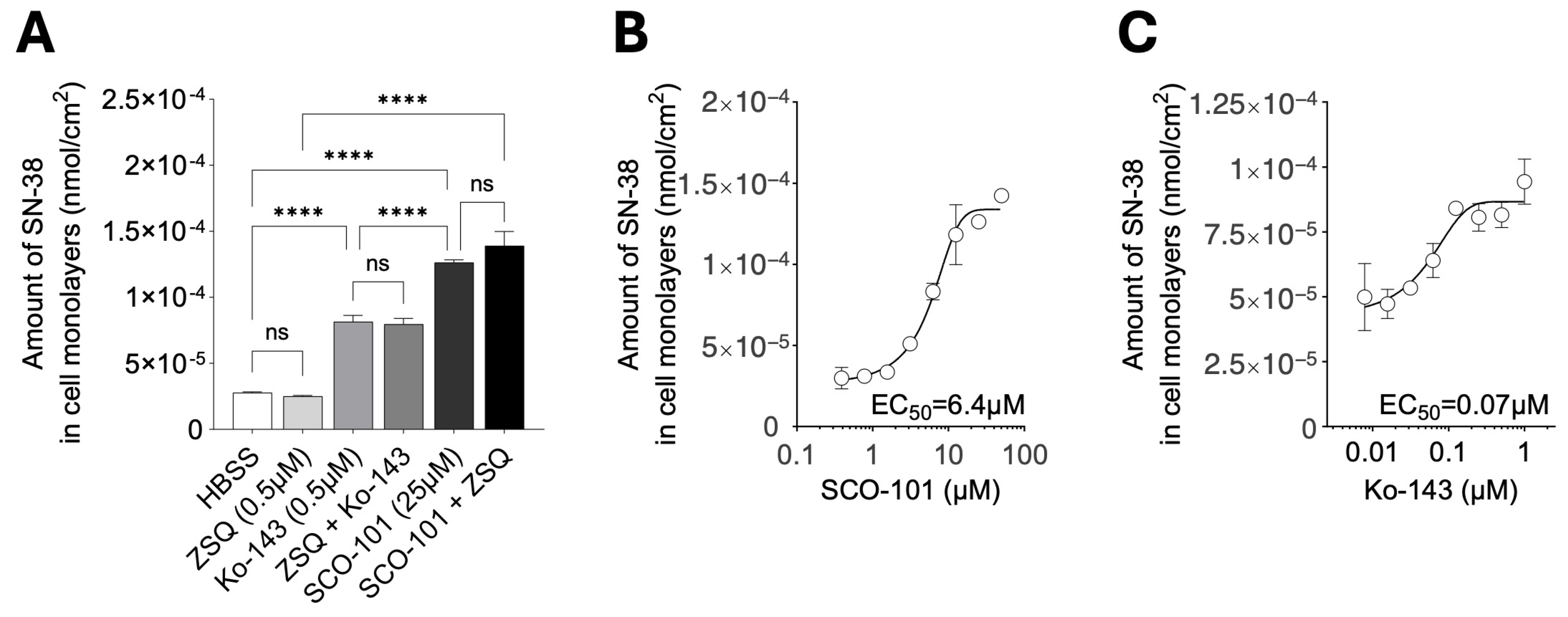
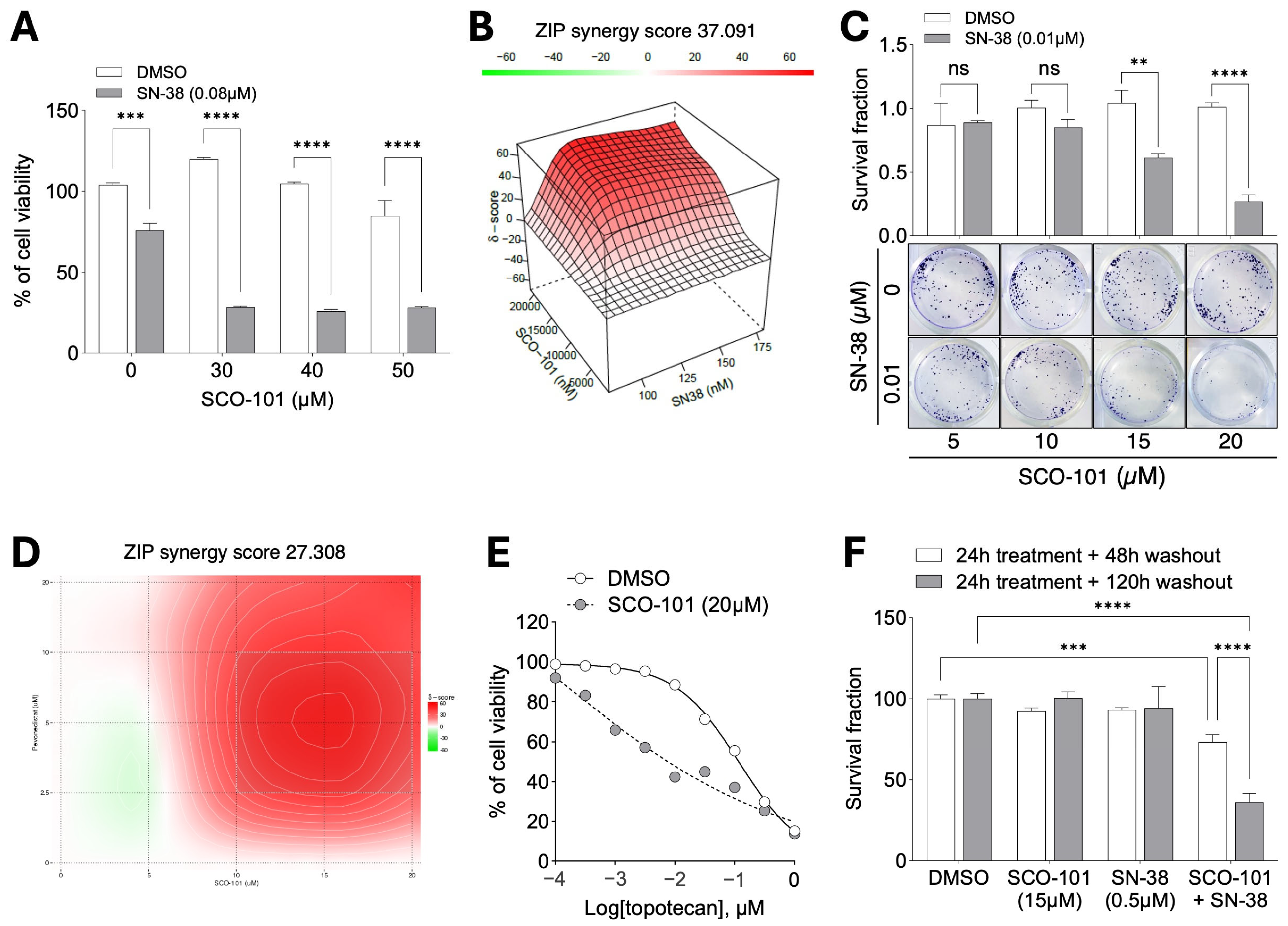
2.3. SCO-101 Re-Sensitizes Resistant Cancer Cells to Chemotherapy Agents
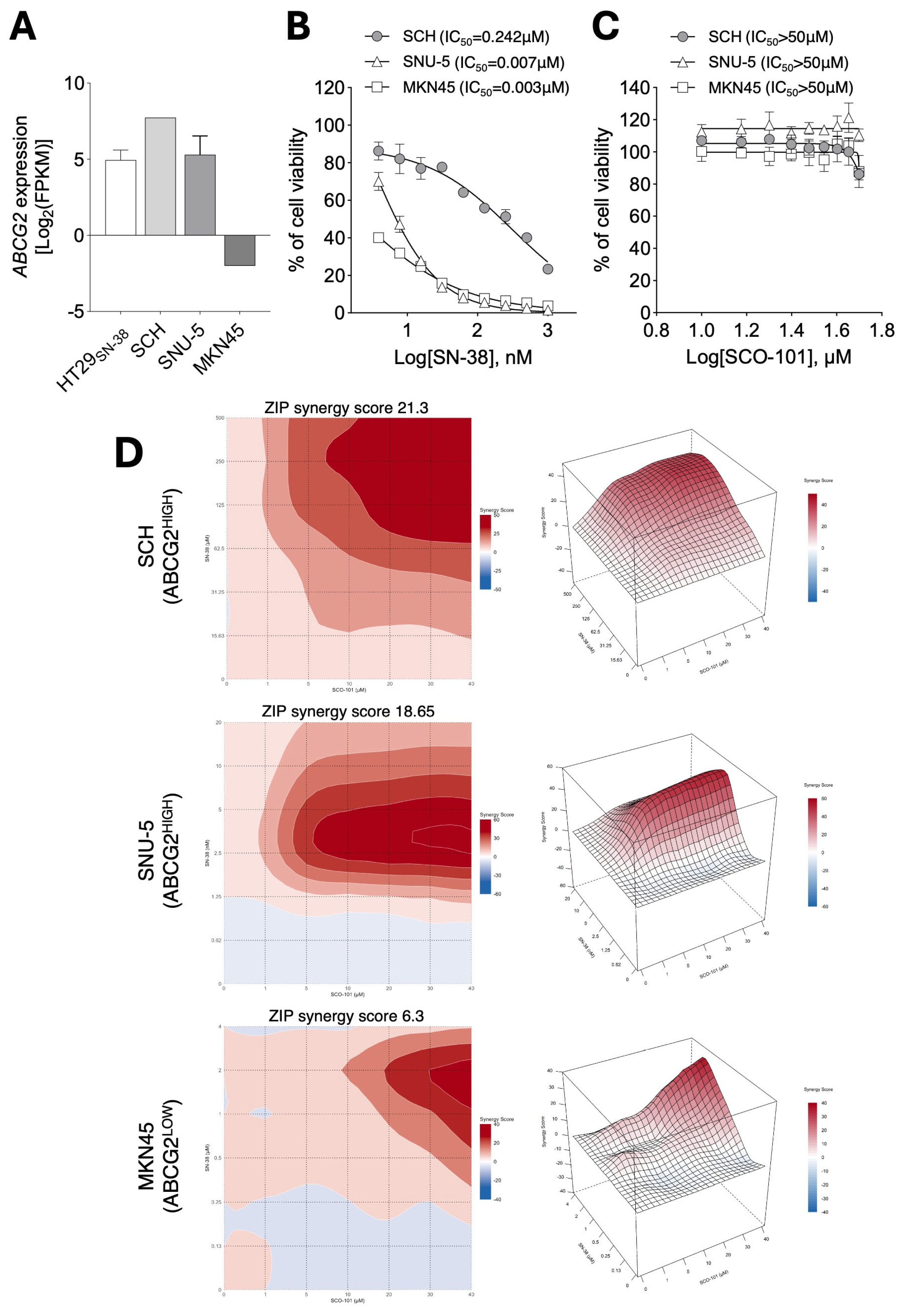
2.4. ABCG2 Expression Levels Correlate with Response to SCO-101 in Combination with Anti-Cancer Agents
3. Discussion
4. Materials and Methods
4.1. Reagents
4.2. Cell Lines
4.3. Cell Viability Assays
4.4. Colony Formation Assay
4.5. Off-Target Analysis
4.6. Docking
4.7. mRNA Expression Analysis
4.8. Western Blot Analysis
4.9. Flow Cytometry Analysis
4.10. Cellular Dye Efflux Assay
4.11. Bidirectional Transport Assay
4.12. Efflux Transporter Inhibition Assays
4.13. Inhibition of UGT1A1 Activity
4.14. 3H-SN-38 Uptake in Cell Monolayers
4.15. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| MDR | Multidrug resistance |
| ABC | ATP-binding cassette |
| BCRP/ABCG2 | Breast cancer resistant protein/ABC transporter G family member 2 |
| MDR1/ABCB1 | Multidrug resistance protein 1/ATP-binding cassette sub-family B member 1 |
| MRP1/ABCC1 | Multidrug resistance-associated protein 1/ABC transporter C family member 1 |
| MRP2/ABCC2 | Multidrug resistance-associated protein 2/ATP-binding cassette sub-family C member 2 |
| UGT1A1 | UDP Glucuronosyltransferase Family 1 Member A1 |
| FOLFIRI | Folinic acid, 5FU, irinotecan |
| AML | Acute myeloid leukemia |
| CDX | Cell line-derived xenograft |
| PDX | Patient-derived xenograft |
| ZSQ | Zosuquidar |
References
- Bray, F.; Laversanne, M.; Sung, H.; Ferlay, J.; Siegel, R.L.; Soerjomataram, I.; Jemal, A. Global cancer statistics 2022: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2024, 74, 229–263. [Google Scholar] [CrossRef] [PubMed]
- International Agency for Research on Cancer. Estimated Number of Deaths from 2022 to 2050, Both Sexes, Age [0–85+]. Available online: https://gco.iarc.fr/tomorrow/en/dataviz/isotype?types=1&single_unit=500000&years=2050 (accessed on 20 January 2025).
- Meads, M.B.; Gatenby, R.A.; Dalton, W.S. Environment-mediated drug resistance: A major contributor to minimal residual disease. Nat. Rev. Cancer 2009, 9, 665–674. [Google Scholar] [CrossRef] [PubMed]
- Vasan, N.; Baselga, J.; Hyman, D.M. A view on drug resistance in cancer. Nature 2019, 575, 299–309. [Google Scholar] [CrossRef] [PubMed]
- Ramos, A.; Sadeghi, S.; Tabatabaeian, H. Battling Chemoresistance in Cancer: Root Causes and Strategies to Uproot Them. Int. J. Mol. Sci. 2021, 22, 9451. [Google Scholar] [CrossRef]
- Hangauer, M.J.; Viswanathan, V.S.; Ryan, M.J.; Bole, D.; Eaton, J.K.; Matov, A.; Galeas, J.; Dhruv, H.D.; Berens, M.E.; Schreiber, S.L.; et al. Drug-tolerant persister cancer cells are vulnerable to GPX4 inhibition. Nature 2017, 551, 247–250. [Google Scholar] [CrossRef]
- Lim, S.Y.; Menzies, A.M.; Rizos, H. Mechanisms and strategies to overcome resistance to molecularly targeted therapy for melanoma. Cancer 2017, 123, 2118–2129. [Google Scholar] [CrossRef]
- Sharma, P.; Hu-Lieskovan, S.; Wargo, J.A.; Ribas, A. Primary, Adaptive, and Acquired Resistance to Cancer Immunotherapy. Cell 2017, 168, 707–723. [Google Scholar] [CrossRef]
- Rambow, F.; Rogiers, A.; Marin-Bejar, O.; Aibar, S.; Femel, J.; Dewaele, M.; Karras, P.; Brown, D.; Chang, Y.H.; Debiec-Rychter, M.; et al. Toward Minimal Residual Disease-Directed Therapy in Melanoma. Cell 2018, 174, 843–855.e819. [Google Scholar] [CrossRef]
- Cree, I.A.; Charlton, P. Molecular chess? Hallmarks of anti-cancer drug resistance. BMC Cancer 2017, 17, 10. [Google Scholar] [CrossRef]
- Candeil, L.; Gourdier, I.; Peyron, D.; Vezzio, N.; Copois, V.; Bibeau, F.; Orsetti, B.; Scheffer, G.L.; Ychou, M.; Khan, Q.A.; et al. ABCG2 overexpression in colon cancer cells resistant to SN38 and in irinotecan-treated metastases. Int. J. Cancer 2004, 109, 848–854. [Google Scholar] [CrossRef]
- Tada, Y.; Wada, M.; Kuroiwa, K.; Kinugawa, N.; Harada, T.; Nagayama, J.; Nakagawa, M.; Naito, S.; Kuwano, M. MDR1 gene overexpression and altered degree of methylation at the promoter region in bladder cancer during chemotherapeutic treatment. Clin. Cancer Res. 2000, 6, 4618–4627. [Google Scholar] [PubMed]
- Ambudkar, S.V.; Dey, S.; Hrycyna, C.A.; Ramachandra, M.; Pastan, I.; Gottesman, M.M. Biochemical, cellular, and pharmacological aspects of the multidrug transporter. Annu. Rev. Pharmacol. Toxicol. 1999, 39, 361–398. [Google Scholar] [CrossRef] [PubMed]
- Albermann, N.; Schmitz-Winnenthal, F.H.; Z’Graggen, K.; Volk, C.; Hoffmann, M.M.; Haefeli, W.E.; Weiss, J. Expression of the drug transporters MDR1/ABCB1, MRP1/ABCC1, MRP2/ABCC2, BCRP/ABCG2, and PXR in peripheral blood mononuclear cells and their relationship with the expression in intestine and liver. Biochem. Pharmacol. 2005, 70, 949–958. [Google Scholar] [CrossRef]
- Alam, A.; Locher, K.P. Structure and Mechanism of Human ABC Transporters. Annu. Rev. Biophys. 2023, 52, 275–300. [Google Scholar] [CrossRef]
- Gottesman, M.M.; Fojo, T.; Bates, S.E. Multidrug resistance in cancer: Role of ATP-dependent transporters. Nat. Rev. Cancer 2002, 2, 48–58. [Google Scholar] [CrossRef]
- Fletcher, J.I.; Haber, M.; Henderson, M.J.; Norris, M.D. ABC transporters in cancer: More than just drug efflux pumps. Nat. Rev. Cancer 2010, 10, 147–156. [Google Scholar] [CrossRef]
- Robey, R.W.; Pluchino, K.M.; Hall, M.D.; Fojo, A.T.; Bates, S.E.; Gottesman, M.M. Revisiting the role of ABC transporters in multidrug-resistant cancer. Nat. Rev. Cancer 2018, 18, 452–464. [Google Scholar] [CrossRef]
- Pote, M.S.; Gacche, R.N. ATP-binding cassette efflux transporters and MDR in cancer. Drug Discov. Today 2023, 28, 103537. [Google Scholar] [CrossRef]
- Duvivier, L.; Gerard, L.; Diaz, A.; Gillet, J.P. Linking ABC transporters to the hallmarks of cancer. Trends Cancer 2024, 10, 124–134. [Google Scholar] [CrossRef]
- Engle, K.; Kumar, G. Cancer multidrug-resistance reversal by ABCB1 inhibition: A recent update. Eur. J. Med. Chem. 2022, 239, 114542. [Google Scholar] [CrossRef]
- Dong, J.; Yuan, L.; Hu, C.; Cheng, X.; Qin, J.J. Strategies to overcome cancer multidrug resistance (MDR) through targeting P-glycoprotein (ABCB1): An updated review. Pharmacol. Ther. 2023, 249, 108488. [Google Scholar] [CrossRef] [PubMed]
- Cole, S.P. Targeting multidrug resistance protein 1 (MRP1, ABCC1): Past, present, and future. Annu. Rev. Pharmacol. Toxicol. 2014, 54, 95–117. [Google Scholar] [CrossRef] [PubMed]
- Hanssen, K.M.; Haber, M.; Fletcher, J.I. Targeting multidrug resistance-associated protein 1 (MRP1)-expressing cancers: Beyond pharmacological inhibition. Drug Resist. Updates 2021, 59, 100795. [Google Scholar] [CrossRef] [PubMed]
- Robey, R.W.; Polgar, O.; Deeken, J.; To, K.W.; Bates, S.E. ABCG2: Determining its relevance in clinical drug resistance. Cancer Metastasis Rev. 2007, 26, 39–57. [Google Scholar] [CrossRef]
- Stacy, A.E.; Jansson, P.J.; Richardson, D.R. Molecular pharmacology of ABCG2 and its role in chemoresistance. Mol. Pharmacol. 2013, 84, 655–669. [Google Scholar] [CrossRef]
- Sarkadi, B.; Ozvegy-Laczka, C.; Nemet, K.; Varadi, A. ABCG2—A transporter for all seasons. FEBS Lett. 2004, 567, 116–120. [Google Scholar] [CrossRef]
- Kathawala, R.J.; Espitia, C.M.; Jones, T.M.; Islam, S.; Gupta, P.; Zhang, Y.K.; Chen, Z.S.; Carew, J.S.; Nawrocki, S.T. ABCG2 Overexpression Contributes to Pevonedistat Resistance. Cancers 2020, 12, 429. [Google Scholar] [CrossRef]
- Allain, E.P.; Rouleau, M.; Levesque, E.; Guillemette, C. Emerging roles for UDP-glucuronosyltransferases in drug resistance and cancer progression. Br. J. Cancer 2020, 122, 1277–1287. [Google Scholar] [CrossRef]
- Takano, M.; Sugiyama, T. UGT1A1 polymorphisms in cancer: Impact on irinotecan treatment. Pharmgenomics Pers. Med. 2017, 10, 61–68. [Google Scholar] [CrossRef]
- Nelson, R.S.; Seligson, N.D.; Bottiglieri, S.; Carballido, E.; Cueto, A.D.; Imanirad, I.; Levine, R.; Parker, A.S.; Swain, S.M.; Tillman, E.M.; et al. UGT1A1 Guided Cancer Therapy: Review of the Evidence and Considerations for Clinical Implementation. Cancers 2021, 13, 1566. [Google Scholar] [CrossRef]
- Lai, J.I.; Tseng, Y.J.; Chen, M.H.; Huang, C.F.; Chang, P.M. Clinical Perspective of FDA Approved Drugs With P-Glycoprotein Inhibition Activities for Potential Cancer Therapeutics. Front. Oncol. 2020, 10, 561936. [Google Scholar] [CrossRef] [PubMed]
- Toyoda, Y.; Takada, T.; Suzuki, H. Inhibitors of Human ABCG2: From Technical Background to Recent Updates With Clinical Implications. Front. Pharmacol. 2019, 10, 208. [Google Scholar] [CrossRef] [PubMed]
- A/S, S.O. A Phase 2 Trial of SCO-101 in Combination with FOLFIRI for Patients with Metastatic Colorectal Cancer (mCRC) With Acquired Resistance to FOLFIRI. Available online: https://clinicaltrials.gov/study/NCT04247256?cond=Cancer&term=sco-101&rank=1 (accessed on 10 February 2025).
- Helix, N.; Strobaek, D.; Dahl, B.H.; Christophersen, P. Inhibition of the endogenous volume-regulated anion channel (VRAC) in HEK293 cells by acidic di-aryl-ureas. J. Membr. Biol. 2003, 196, 83–94. [Google Scholar] [CrossRef]
- Nohr-Nielsen, A.; Bagger, S.O.; Brunner, N.; Stenvang, J.; Lund, T.M. Pharmacodynamic modelling reveals synergistic interaction between docetaxel and SCO-101 in a docetaxel-resistant triple negative breast cancer cell line. Eur. J. Pharm. Sci. 2020, 148, 105315. [Google Scholar] [CrossRef]
- Bergmann, T.K.; Stage, T.B.; Stenvang, J.; Christophersen, P.; Jacobsen, T.A.; Roest, N.L.; Vestlev, P.M.; Brunner, N. Four phase 1 trials to evaluate the safety and pharmacokinetic profile of single and repeated dosing of SCO-101 in adult male and female volunteers. Basic. Clin. Pharmacol. Toxicol. 2020, 127, 329–337. [Google Scholar] [CrossRef]
- Jensen, N.F.; Stenvang, J.; Beck, M.K.; Hanakova, B.; Belling, K.C.; Do, K.N.; Viuff, B.; Nygard, S.B.; Gupta, R.; Rasmussen, M.H.; et al. Establishment and characterization of models of chemotherapy resistance in colorectal cancer: Towards a predictive signature of chemoresistance. Mol. Oncol. 2015, 9, 1169–1185. [Google Scholar] [CrossRef]
- Wei, L.Y.; Wu, Z.X.; Yang, Y.; Zhao, M.; Ma, X.Y.; Li, J.S.; Yang, D.H.; Chen, Z.S.; Fan, Y.F. Overexpression of ABCG2 confers resistance to pevonedistat, an NAE inhibitor. Exp. Cell Res. 2020, 388, 111858. [Google Scholar] [CrossRef]
- Zander, S.A.; Kersbergen, A.; van der Burg, E.; de Water, N.; van Tellingen, O.; Gunnarsdottir, S.; Jaspers, J.E.; Pajic, M.; Nygren, A.O.; Jonkers, J.; et al. Sensitivity and acquired resistance of BRCA1;p53-deficient mouse mammary tumors to the topoisomerase I inhibitor topotecan. Cancer Res. 2010, 70, 1700–1710. [Google Scholar] [CrossRef]
- Sorf, A.; Sucha, S.; Morell, A.; Novotna, E.; Staud, F.; Zavrelova, A.; Visek, B.; Wsol, V.; Ceckova, M. Targeting Pharmacokinetic Drug Resistance in Acute Myeloid Leukemia Cells with CDK4/6 Inhibitors. Cancers 2020, 12, 1596. [Google Scholar] [CrossRef]
- Sucha, S.; Sorf, A.; Svoren, M.; Vagiannis, D.; Ahmed, F.; Visek, B.; Ceckova, M. ABCB1 as a potential beneficial target of midostaurin in acute myeloid leukemia. Biomed. Pharmacother. 2022, 150, 112962. [Google Scholar] [CrossRef]
- Hanahan, D. Hallmarks of Cancer: New Dimensions. Cancer Discov. 2022, 12, 31–46. [Google Scholar] [CrossRef] [PubMed]
- Pilotto Heming, C.; Muriithi, W.; Wanjiku Macharia, L.; Niemeyer Filho, P.; Moura-Neto, V.; Aran, V. P-glycoprotein and cancer: What do we currently know? Heliyon 2022, 8, e11171. [Google Scholar] [CrossRef] [PubMed]
- Mo, W.; Zhang, J.T. Human ABCG2: Structure, function, and its role in multidrug resistance. Int. J. Biochem. Mol. Biol. 2012, 3, 1–27. [Google Scholar] [PubMed]
- Henrich, C.J.; Bokesch, H.R.; Dean, M.; Bates, S.E.; Robey, R.W.; Goncharova, E.I.; Wilson, J.A.; McMahon, J.B. A high-throughput cell-based assay for inhibitors of ABCG2 activity. J. Biomol. Screen. 2006, 11, 176–183. [Google Scholar] [CrossRef]
- Henrich, C.J.; Robey, R.W.; Bokesch, H.R.; Bates, S.E.; Shukla, S.; Ambudkar, S.V.; Dean, M.; McMahon, J.B. New inhibitors of ABCG2 identified by high-throughput screening. Mol. Cancer Ther. 2007, 6, 3271–3278. [Google Scholar] [CrossRef]
- Le, M.T.; Hoang, V.N.; Nguyen, D.N.; Bui, T.H.; Phan, T.V.; Huynh, P.N.; Tran, T.D.; Thai, K.M. Structure-Based Discovery of ABCG2 Inhibitors: A Homology Protein-Based Pharmacophore Modeling and Molecular Docking Approach. Molecules 2021, 26, 3115. [Google Scholar] [CrossRef]
- Ahmed-Belkacem, A.; Pozza, A.; Munoz-Martinez, F.; Bates, S.E.; Castanys, S.; Gamarro, F.; Di Pietro, A.; Perez-Victoria, J.M. Flavonoid structure-activity studies identify 6-prenylchrysin and tectochrysin as potent and specific inhibitors of breast cancer resistance protein ABCG2. Cancer Res. 2005, 65, 4852–4860. [Google Scholar] [CrossRef]
- Zhang, S.; Yang, X.; Morris, M.E. Flavonoids are inhibitors of breast cancer resistance protein (ABCG2)-mediated transport. Mol. Pharmacol. 2004, 65, 1208–1216. [Google Scholar] [CrossRef]
- Allen, J.D.; van Loevezijn, A.; Lakhai, J.M.; van der Valk, M.; van Tellingen, O.; Reid, G.; Schellens, J.H.; Koomen, G.J.; Schinkel, A.H. Potent and specific inhibition of the breast cancer resistance protein multidrug transporter in vitro and in mouse intestine by a novel analogue of fumitremorgin C. Mol. Cancer Ther. 2002, 1, 417–425. [Google Scholar]
- Brackman, D.J.; Giacomini, K.M. Reverse Translational Research of ABCG2 (BCRP) in Human Disease and Drug Response. Clin. Pharmacol. Ther. 2018, 103, 233–242. [Google Scholar] [CrossRef]
- Adamska, A.; Falasca, M. ATP-binding cassette transporters in progression and clinical outcome of pancreatic cancer: What is the way forward? World J. Gastroenterol. 2018, 24, 3222–3238. [Google Scholar] [CrossRef] [PubMed]
- Scandion Oncology A/S Scandion Oncology Reports Promising Updated Phase II CORIST Data, Including Topline Overall Survival. Available online: https://storage.mfn.se/0bae93eb-3725-4176-9faa-2d2197a23dbe/scandion-oncology-reports-promising-updated-corist-phase-ii-data.pdf (accessed on 10 February 2025).
- Osborne, M.J.; Coutinho de Oliveira, L.; Volpon, L.; Zahreddine, H.A.; Borden, K.L.B. Overcoming Drug Resistance through the Development of Selective Inhibitors of UDP-Glucuronosyltransferase Enzymes. J. Mol. Biol. 2019, 431, 258–272. [Google Scholar] [CrossRef] [PubMed]
- Marques, S.C.; Ikediobi, O.N. The clinical application of UGT1A1 pharmacogenetic testing: Gene-environment interactions. Hum. Genom. 2010, 4, 238–249. [Google Scholar] [CrossRef]
- Vestlev, P.M. Clinical Trial to Investigate Safety, Tolerability and MTD for SCO-101 in Combination with Gemcitabine and Nab-paclitaxel in Inoperable Pancreatic Cancer Patients. (PANTAX-Ib). Available online: https://clinicaltrials.gov/study/NCT04652206?term=pantax&rank=1 (accessed on 10 February 2025).
- Scandion Oncology A/S Scandion Oncology Announces Final Data from the CORIST Trial and Is Ready to Move into a Randomized Phase II Proof of Concept Trial in Colorectal Cancer. Available online: https://scandiononcology.com/mfn_news/scandion-oncology-announces-final-data-from-the-corist-trial-and-is-ready-to-move-into-a-randomized-phase-ii-proof-of-concept-trial-in-colorectal-cancer/ (accessed on 13 January 2025).
- Friesner, R.A.; Murphy, R.B.; Repasky, M.P.; Frye, L.L.; Greenwood, J.R.; Halgren, T.A.; Sanschagrin, P.C.; Mainz, D.T. Extra precision glide: Docking and scoring incorporating a model of hydrophobic enclosure for protein-ligand complexes. J. Med. Chem. 2006, 49, 6177–6196. [Google Scholar] [CrossRef]
- Halgren, T.A.; Murphy, R.B.; Friesner, R.A.; Beard, H.S.; Frye, L.L.; Pollard, W.T.; Banks, J.L. Glide: A new approach for rapid, accurate docking and scoring. 2. Enrichment factors in database screening. J. Med. Chem. 2004, 47, 1750–1759. [Google Scholar] [CrossRef]
- Sastry, G.M.; Adzhigirey, M.; Day, T.; Annabhimoju, R.; Sherman, W. Protein and ligand preparation: Parameters, protocols, and influence on virtual screening enrichments. J. Comput. Aided Mol. Des. 2013, 27, 221–234. [Google Scholar] [CrossRef]
- Jackson, S.M.; Manolaridis, I.; Kowal, J.; Zechner, M.; Taylor, N.M.I.; Bause, M.; Bauer, S.; Bartholomaeus, R.; Bernhardt, G.; Koenig, B.; et al. Structural basis of small-molecule inhibition of human multidrug transporter ABCG2. Nat. Struct. Mol. Biol. 2018, 25, 333–340. [Google Scholar] [CrossRef]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Zidek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef]
- Ambjorner, S.E.B.; Wiese, M.; Kohler, S.C.; Svindt, J.; Lund, X.L.; Gajhede, M.; Saaby, L.; Brodin, B.; Rump, S.; Weigt, H.; et al. The Pyrazolo [3,4-d]pyrimidine Derivative, SCO-201, Reverses Multidrug Resistance Mediated by ABCG2/BCRP. Cells 2020, 9, 613. [Google Scholar] [CrossRef]
- Petersen, M.J.; Lund, X.L.; Semple, S.J.; Buirchell, B.; Franzyk, H.; Gajhede, M.; Kongstad, K.T.; Stenvang, J.; Staerk, D. Reversal of ABCG2/BCRP-Mediated Multidrug Resistance by 5,3′,5′-Trihydroxy-3,6,7,4′-Tetramethoxyflavone Isolated from the Australian Desert Plant Eremophila galeata Chinnock. Biomolecules 2021, 11, 1534. [Google Scholar] [CrossRef]
- Bowers, K.J.; Chow, E.; Xu, H.; Dror, R.O.; Eastwood, M.P.; Gregersen, B.A.; Klepeis, J.L.; Kolossvary, I.; Moraes, M.A.; Sacerdoti, F.D.; et al. Scalable Algorithms for Molecular Dynamics Simulations on Commodity Clusters. In Proceedings of the 2006 ACM/IEEE Conference on Supercomputing (SC06), Tampa, FL, USA, 11–17 November 2006; IEEE: New York, NY, USA, 2007. [Google Scholar]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pfeiffer, A.; Di Leo, L.; Bechmann, M.B.; Nawabi, M.; Ambjørner, S.; Ardeshir-Larijani, D.; Colstrup, L.T.; Borchert, S.V.; Saaby, L.; Brodin, B.; et al. Inhibition of ABCG2 by SCO-101 Enhances Chemotherapy Efficacy in Cancer. Int. J. Mol. Sci. 2025, 26, 3790. https://doi.org/10.3390/ijms26083790
Pfeiffer A, Di Leo L, Bechmann MB, Nawabi M, Ambjørner S, Ardeshir-Larijani D, Colstrup LT, Borchert SV, Saaby L, Brodin B, et al. Inhibition of ABCG2 by SCO-101 Enhances Chemotherapy Efficacy in Cancer. International Journal of Molecular Sciences. 2025; 26(8):3790. https://doi.org/10.3390/ijms26083790
Chicago/Turabian StylePfeiffer, Anamarija, Luca Di Leo, Marc Baker Bechmann, Mubeen Nawabi, Sophie Ambjørner, Diba Ardeshir-Larijani, Louise Thybo Colstrup, Signe Vedel Borchert, Lasse Saaby, Birger Brodin, and et al. 2025. "Inhibition of ABCG2 by SCO-101 Enhances Chemotherapy Efficacy in Cancer" International Journal of Molecular Sciences 26, no. 8: 3790. https://doi.org/10.3390/ijms26083790
APA StylePfeiffer, A., Di Leo, L., Bechmann, M. B., Nawabi, M., Ambjørner, S., Ardeshir-Larijani, D., Colstrup, L. T., Borchert, S. V., Saaby, L., Brodin, B., Gajhede, M., Lund, X. L., Čečková, M., Brünner, N., & Stenvang, J. (2025). Inhibition of ABCG2 by SCO-101 Enhances Chemotherapy Efficacy in Cancer. International Journal of Molecular Sciences, 26(8), 3790. https://doi.org/10.3390/ijms26083790