
Tolerization with a Novel Dual-Acting Liposomal Tim Agonist Prepares the Immune System for the Success of Gene Therapy


Abstract
1. Introduction
2. Results
2.1. LPX1 Induces Tolerance Without Altering Innate Immunity
2.1.1. Immunogenicity of AAV9null Administered with LPX1
2.1.2. The Effects of LPX1 on Innate Immunity
2.2. Improved Tim Binding of Second-Generation LPX Compounds
2.2.1. Validation of Docking Model
2.2.2. Docking New Drug Candidates
2.3. LPX-TI Induces AAV9null-Dose-Dependent Foxp3+ Upregulation In Vivo
2.4. IM Administration of AAV9null-LPX-TI Induces Highly Tolerogenic Tregs
2.5. AAV9null-LPX-TI Expands Antigen-Specific Tregs in a Recall Response
2.6. IM Administration of AAV9null-LPX-TI Did Not Produce Anti-AAV9 Antibody Titer
3. Discussion
4. Materials and Methods
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| AAV | Adeno-associated viral |
| CTLA4 | Cytotoxic T-Lymphocyte-Associated Protein 4 |
| DMPC | 1,2-Dimyristoyl-sn-glycero-3-phosphocholine |
| ICOS | Inducible T-cell CO-Stimulator |
| Foxp3 | Forkhead box P3 |
| TLR | Toll-Like Receptor |
| Treg | Regulatory T cells |
References
- Nguengang Wakap, S.; Lambert, D.M.; Olry, A.; Rodwell, C.; Gueydan, C.; Lanneau, V.; Murphy, D.; Le Cam, Y.; Rath, A. Estimating cumulative point prevalence of rare diseases: Analysis of the Orphanet database. Eur. J. Hum. Genet. 2020, 28, 165–173. [Google Scholar] [CrossRef] [PubMed]
- Approved Cellular and Gene Therapy Products, USFDA. Available online: https://www.fda.gov/vaccines-blood-biologics/cellular-gene-therapy-products/approved-cellular-and-gene-therapy-products (accessed on 13 April 2025).
- Colella, P.; Ronzitti, G.; Mingozzi, F. Emerging Issues in AAV-Mediated In Vivo Gene Therapy. Mol. Ther. Methods Clin. Dev. 2018, 8, 87–104. [Google Scholar] [CrossRef] [PubMed]
- Zufferey, R.; Dull, T.; Mandel, R.J.; Bukovsky, A.; Quiroz, D.; Naldini, L.; Trono, D. Self-inactivating lentivirus vector for safe and efficient in vivo gene delivery. J. Virol. 1998, 72, 9873–9880. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.S.; Bishop, E.S.; Zhang, R.; Yu, X.; Farina, E.M.; Yan, S.; Zhao, C.; Zeng, Z.; Shu, Y.; Wu, X.; et al. Adenovirus-Mediated Gene Delivery: Potential Applications for Gene and Cell-Based Therapies in the New Era of Personalized Medicine. Genes Dis. 2017, 4, 43–63. [Google Scholar] [CrossRef]
- Costa Verdera, H.; Kuranda, K.; Mingozzi, F. AAV Vector Immunogenicity in Humans: A Long Journey to Successful Gene Transfer. Mol. Ther. 2020, 28, 723–746. [Google Scholar] [CrossRef]
- Vandamme, C.; Adjali, O.; Mingozzi, F. Unraveling the Complex Story of Immune Responses to AAV Vectors Trial After Trial. Hum. Gene Ther. 2017, 28, 1061–1074. [Google Scholar] [CrossRef]
- Martino, A.T.; Suzuki, M.; Markusic, D.M.; Zolotukhin, I.; Ryals, R.C.; Moghimi, B.; Ertl, H.C.; Muruve, D.A.; Lee, B.; Herzog, R.W. The genome of self-complementary adeno-associated viral vectors increases Toll-like receptor 9-dependent innate immune responses in the liver. Blood 2011, 117, 6459–6468. [Google Scholar] [CrossRef]
- Chuecos, M.A.; Lagor, W.R. Liver directed adeno-associated viral vectors to treat metabolic disease. J. Inherit. Metab. Dis. 2024, 47, 22–40. [Google Scholar] [CrossRef]
- Ledford, H. Gene therapy’s comeback: How scientists are trying to make it safer. Nature 2022, 606, 443–444. [Google Scholar] [CrossRef]
- Shirley, J.L.; De Jong, Y.P.; Terhorst, C.; Herzog, R.W. Immune Responses to Viral Gene Therapy Vectors. Mol. Ther. 2020, 28, 709–722. [Google Scholar] [CrossRef]
- ZOLGENSMA (Onasemnogene Abeparvovec-Xioi) Package Inseret; Novartis Gene Therapies, Inc.: Cambridge, MA, USA, 2021.
- Fathallah, A.M.; Oldfield, P.; Fiedler-Kelly, J.; Ramadan, A. Immunogenicity Considerations for Therapeutic Modalities Used in Rare Diseases. J. Clin. Pharmacol. 2022, 62 (Suppl. S2), S110–S118. [Google Scholar] [CrossRef] [PubMed]
- Zubizarreta, I.; Flórez-Grau, G.; Vila, G.; Cabezón, R.; España, C.; Andorra, M.; Saiz, A.; Llufriu, S.; Sepulveda, M.; Sola-Valls, N.; et al. Immune tolerance in multiple sclerosis and neuromyelitis optica with peptide-loaded tolerogenic dendritic cells in a phase 1b trial. Proc. Natl. Acad. Sci. USA 2019, 116, 8463–8470. [Google Scholar] [CrossRef] [PubMed]
- Ilyinskii, P.O.; Michaud, A.M.; Roy, C.J.; Rizzo, G.L.; Elkins, S.L.; Capela, T.; Chowdhury, A.C.; Leung, S.S.; Kishimoto, T.K. Enhancement of liver-directed transgene expression at initial and repeat doses of AAV vectors admixed with ImmTOR nanoparticles. Sci. Adv. 2021, 7, eabd0321. [Google Scholar] [CrossRef]
- Nguyen, N.H.; Glassman, F.Y.; Dingman, R.K.; Shenoy, G.N.; Wohlfert, E.A.; Kay, J.G.; Bankert, R.B.; Balu-Iyer, S.V. Rational design of a nanoparticle platform for oral prophylactic immunotherapy to prevent immunogenicity of therapeutic proteins. Sci. Rep. 2021, 11, 17853. [Google Scholar] [CrossRef]
- Lu, Y.; Wang, J.; Gu, J.; Lu, H.; Li, X.; Qian, X.; Liu, X.; Wang, X.; Zhang, F.; Lu, L. Rapamycin Regulates iTreg Function through CD39 and Runx1 Pathways. J. Immunol. Res. 2014, 2014, 989434. [Google Scholar] [CrossRef]
- Koenecke, C.; Czeloth, N.; Bubke, A.; Schmitz, S.; Kissenpfennig, A.; Malissen, B.; Huehn, J.; Ganser, A.; Förster, R.; Prinz, I. Alloantigen-specific de novo-induced Foxp3+ Treg revert in vivo and do not protect from experimental GVHD. Eur. J. Immunol. 2009, 39, 3091–3096. [Google Scholar] [CrossRef]
- Tran, D.Q.; Ramsey, H.; Shevach, E.M. Induction of FOXP3 expression in naive human CD4+FOXP3 T cells by T-cell receptor stimulation is transforming growth factor-beta dependent but does not confer a regulatory phenotype. Blood 2007, 110, 2983–2990. [Google Scholar] [CrossRef]
- Curotto de Lafaille, M.A.; Lafaille, J.J. Natural and adaptive foxp3+ regulatory T cells: More of the same or a division of labor? Immunity 2009, 30, 626–635. [Google Scholar] [CrossRef]
- DeKruyff, R.H.; Bu, X.; Ballesteros, A.; Santiago, C.; Chim, Y.L.E.; Lee, H.H.; Karisola, P.; Pichavant, M.; Kaplan, G.G.; Umetsu, D.T.; et al. T cell/transmembrane, Ig, and mucin-3 allelic variants differentially recognize phosphatidylserine and mediate phagocytosis of apoptotic cells. J. Immunol. 2010, 184, 1918–1930. [Google Scholar] [CrossRef]
- Sánchez-Fueyo, A.; Tian, J.; Picarella, D.; Domenig, C.; Zheng, X.X.; Sabatos, C.A.; Manlongat, N.; Bender, O.; Kamradt, T.; Kuchroo, V.K.; et al. Tim-3 inhibits T helper type 1-mediated auto- and alloimmune responses and promotes immunological tolerance. Nat. Immunol. 2003, 4, 1093–1101. [Google Scholar] [CrossRef]
- Welsh, R.M.; Kim, S.K.; Cornberg, M.; Clute, S.C.; Selin, L.K.; Naumov, Y.N. The privacy of T cell memory to viruses. Curr. Top. Microbiol. Immunol. 2006, 311, 117–153. [Google Scholar] [PubMed]
- Hösel, M.; Broxtermann, M.; Janicki, H.; Esser, K.; Arzberger, S.; Hartmann, P.; Gillen, S.; Kleeff, J.; Stabenow, D.; Odenthal, M.; et al. Toll-like receptor 2-mediated innate immune response in human nonparenchymal liver cells toward adeno-associated viral vectors. Hepatology 2012, 55, 287–297. [Google Scholar] [CrossRef] [PubMed]
- Kälble, F.; Wu, L.; Lorenz, H.M.; Zeier, M.; Schaier, M.; Steinborn, A. Impaired Differentiation of Highly Proliferative ICOS(+)-Tregs Is Involved in the Transition from Low to High Disease Activity in Systemic Lupus Erythematosus (SLE) Patients. Int. J. Mol. Sci. 2021, 22, 9501. [Google Scholar] [CrossRef]
- Aumeunier, A.; Grela, F.; Ramadan, A.; Pham Van, L.; Bardel, E.; Gomez Alcala, A.; Jeannin, P.; Akira, S.; Bach, J.F.; Thieblemont, N. Systemic Toll-like receptor stimulation suppresses experimental allergic asthma and autoimmune diabetes in NOD mice. PLoS ONE 2010, 5, e11484. [Google Scholar] [CrossRef]
- Frey, A.; Di Canzio, J.; Zurakowski, D. A statistically defined endpoint titer determination method for immunoassays. J. Immunol. Methods 1998, 221, 35–41. [Google Scholar] [CrossRef]
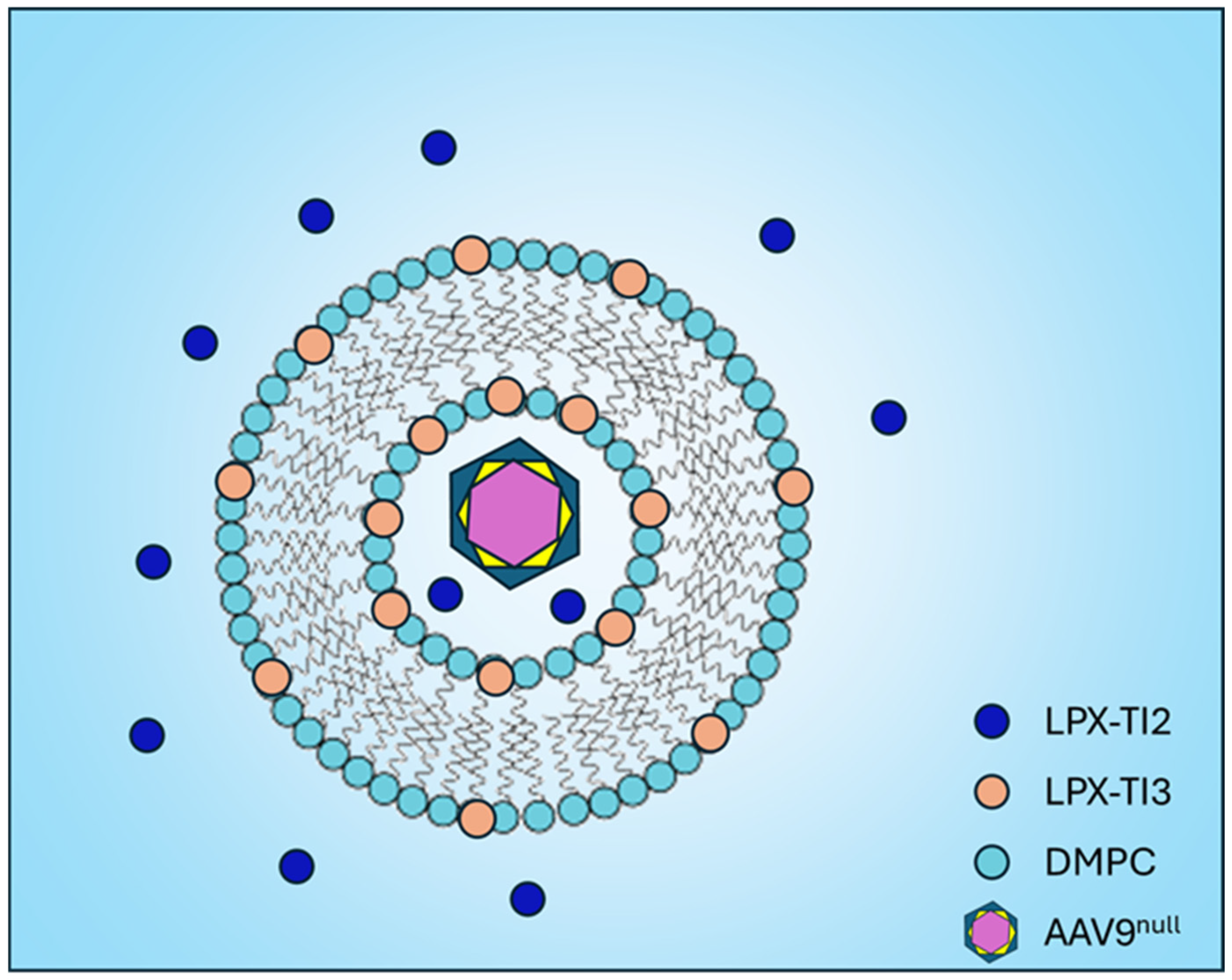


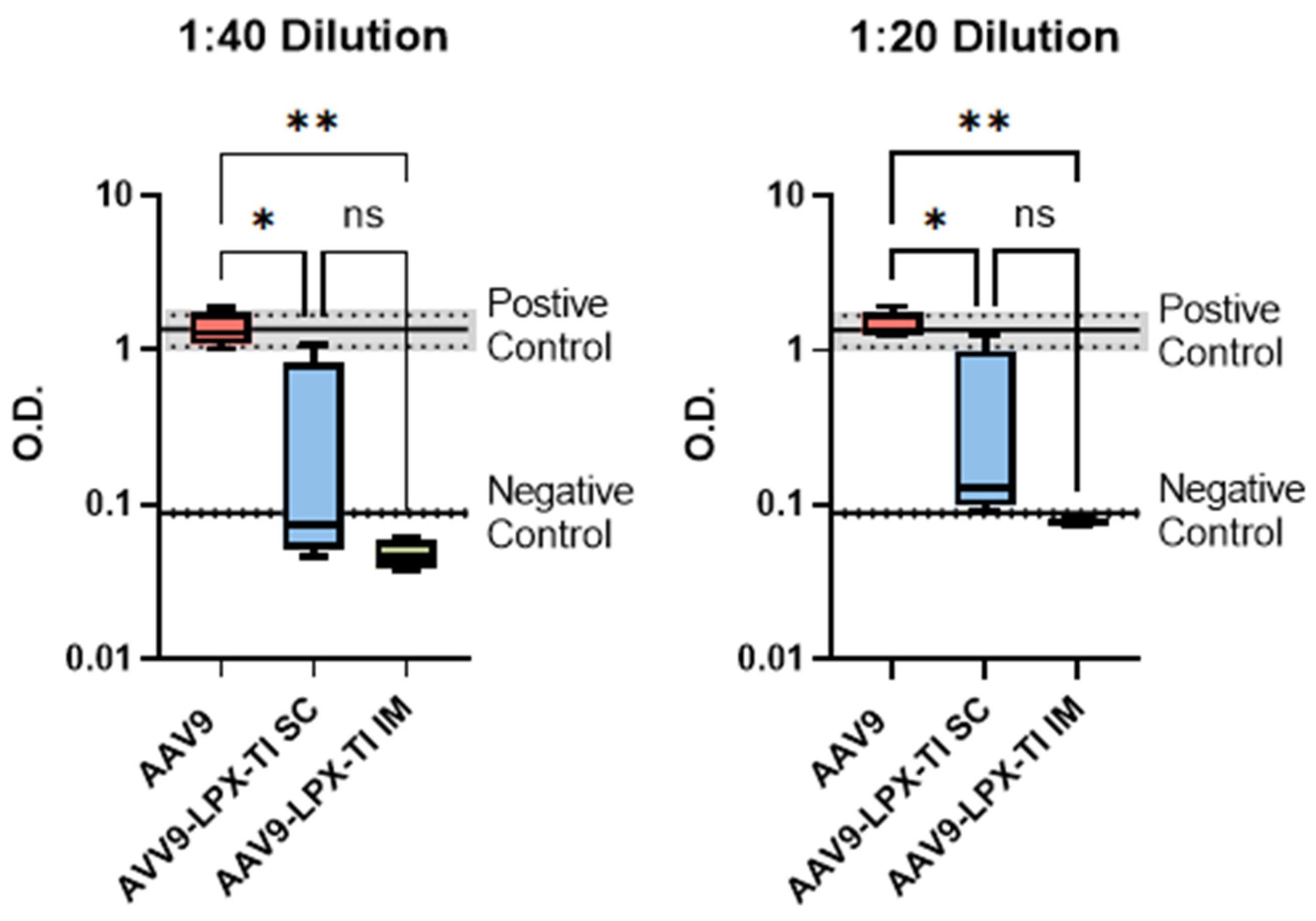


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | OPLS | LPX1 | LPX2 | LPX3 |
|---|---|---|---|---|
| Binding Affinity (kcal/mol) | −5.6 to −3.9 | −6.0 to −4.9 | −7.8 to −6.0 | −5.2 to −4.5 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ramadan, A.; Rao, P.; Allababidi, S.; Khashan, R.; Fathallah, A.M. Tolerization with a Novel Dual-Acting Liposomal Tim Agonist Prepares the Immune System for the Success of Gene Therapy. Int. J. Mol. Sci. 2025, 26, 3830. https://doi.org/10.3390/ijms26083830
Ramadan A, Rao P, Allababidi S, Khashan R, Fathallah AM. Tolerization with a Novel Dual-Acting Liposomal Tim Agonist Prepares the Immune System for the Success of Gene Therapy. International Journal of Molecular Sciences. 2025; 26(8):3830. https://doi.org/10.3390/ijms26083830
Chicago/Turabian StyleRamadan, Abdulraouf, Pushpa Rao, Saleh Allababidi, Raed Khashan, and Anas M. Fathallah. 2025. "Tolerization with a Novel Dual-Acting Liposomal Tim Agonist Prepares the Immune System for the Success of Gene Therapy" International Journal of Molecular Sciences 26, no. 8: 3830. https://doi.org/10.3390/ijms26083830
APA StyleRamadan, A., Rao, P., Allababidi, S., Khashan, R., & Fathallah, A. M. (2025). Tolerization with a Novel Dual-Acting Liposomal Tim Agonist Prepares the Immune System for the Success of Gene Therapy. International Journal of Molecular Sciences, 26(8), 3830. https://doi.org/10.3390/ijms26083830