
Establishment of a Human iPSC Line from Mucolipidosis Type II That Expresses the Key Markers of the Disease
 , ,
, ,  , ,
, ,  ,
,  ,
,  ,
,  and
and
Abstract
1. Introduction
2. Results
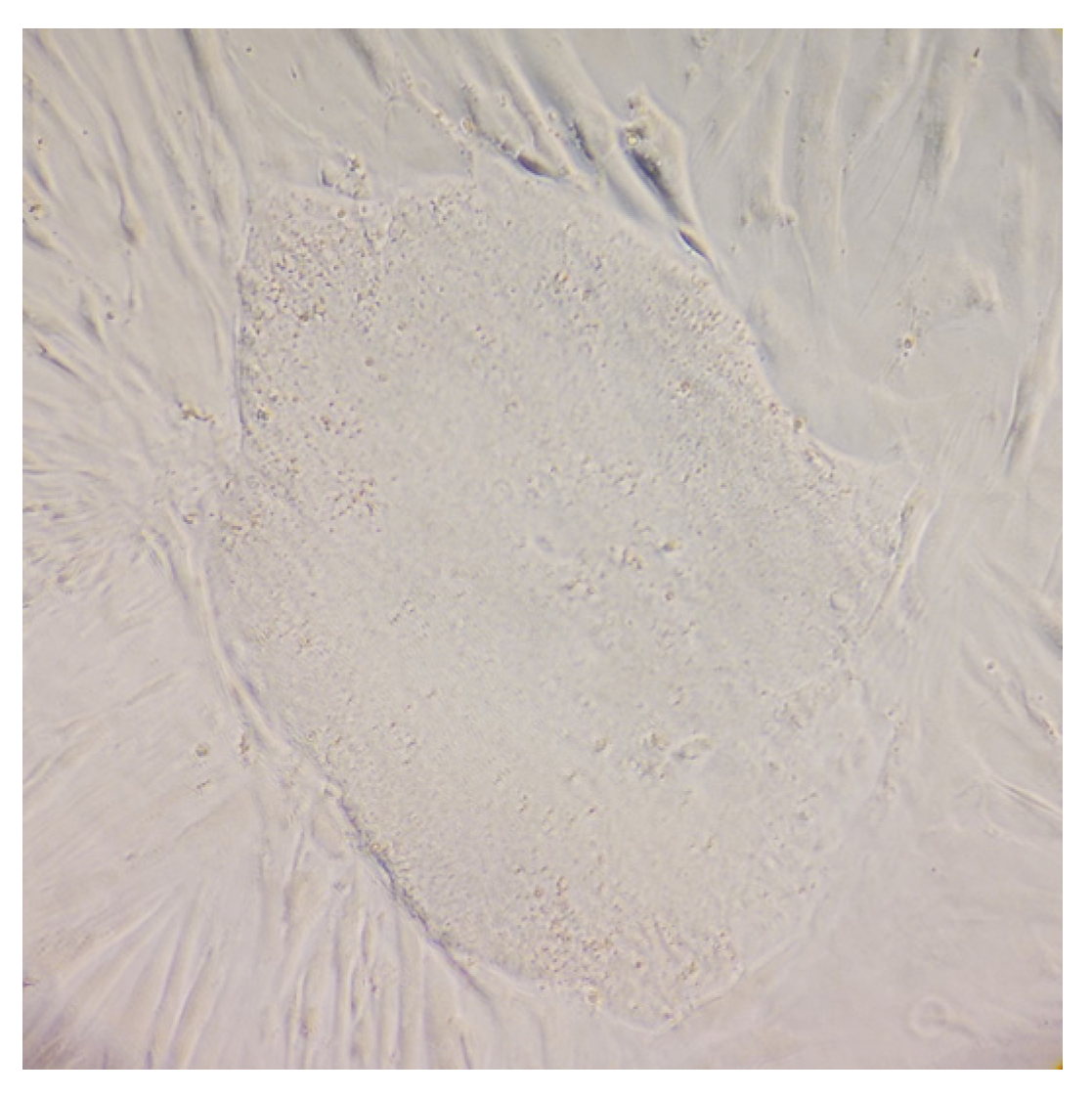
2.1. Human ML II Fibroblasts Were Reprogrammed into iPSC-like Colonies
2.2. iPSC Pluripotency Characterization
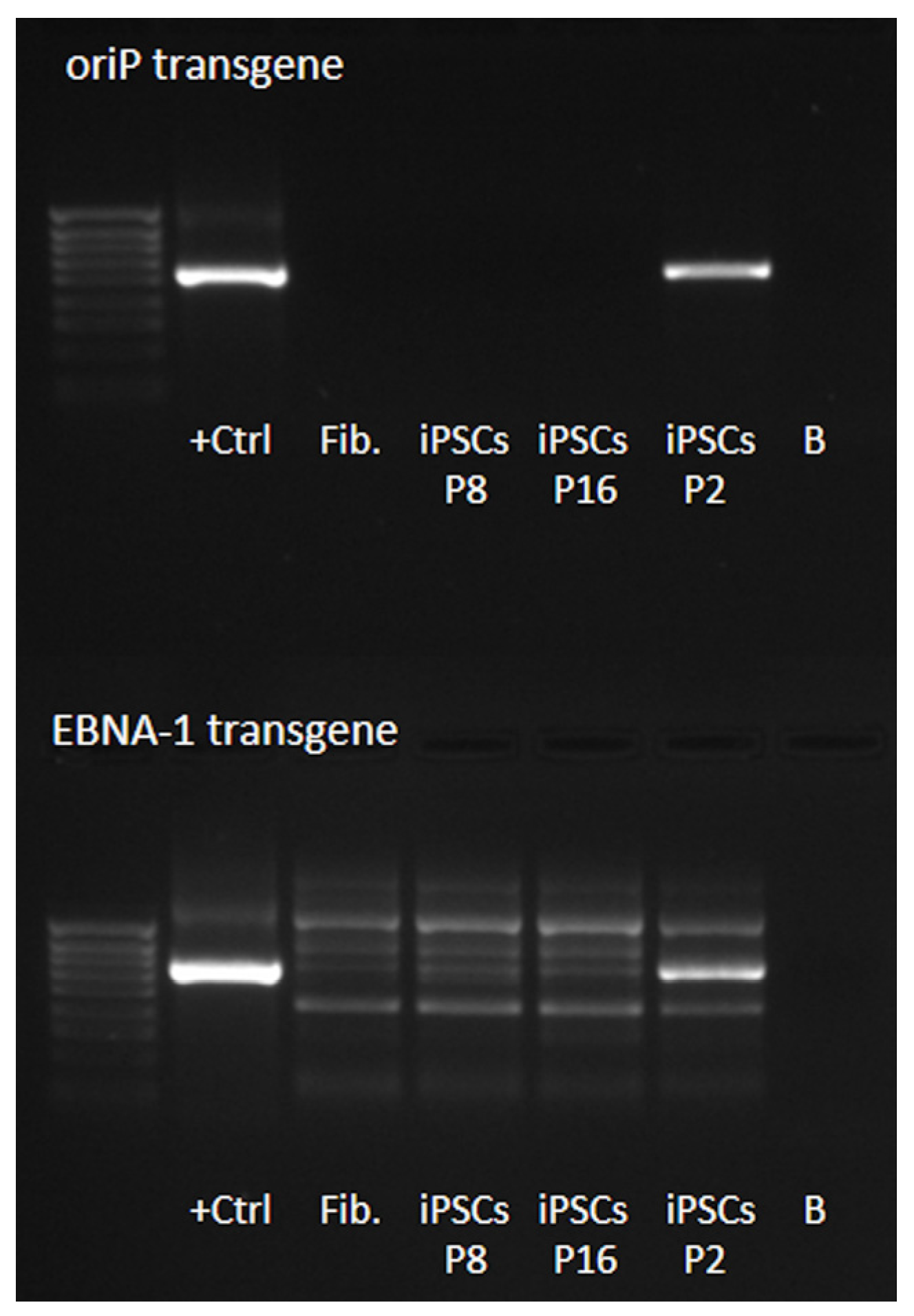
2.2.1. iPSCs Were Free from Reprogramming Vectors and Expressed the Key Pluripotency Markers Endogenously
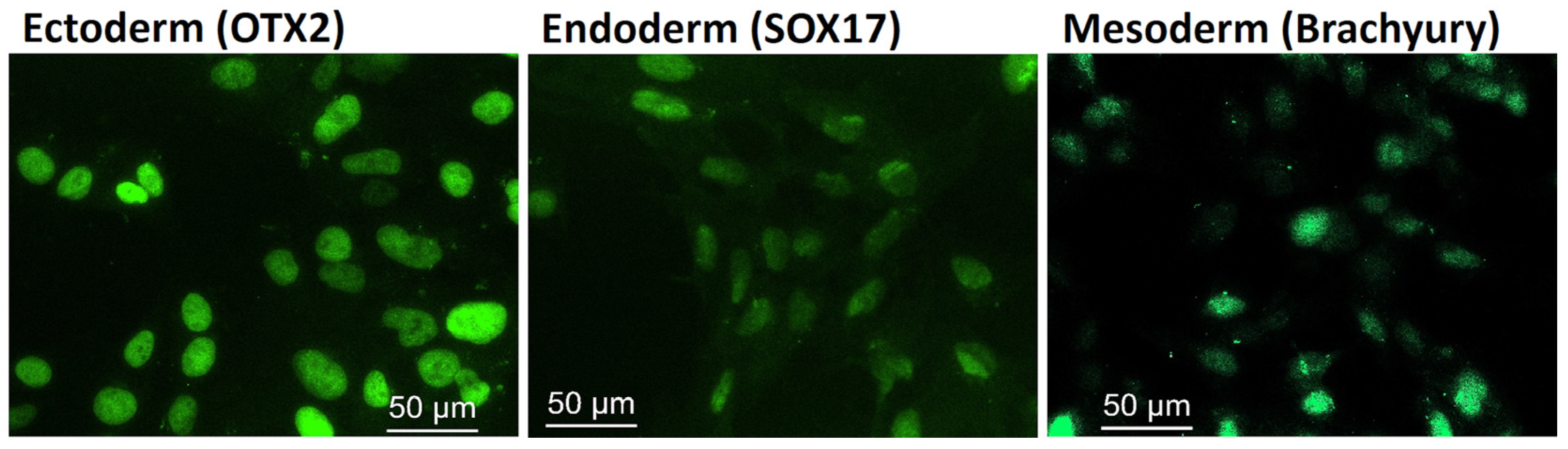
2.2.2. ML II iPSCs Are Capable of Differentiating into the Three Embryonic Germ Layers
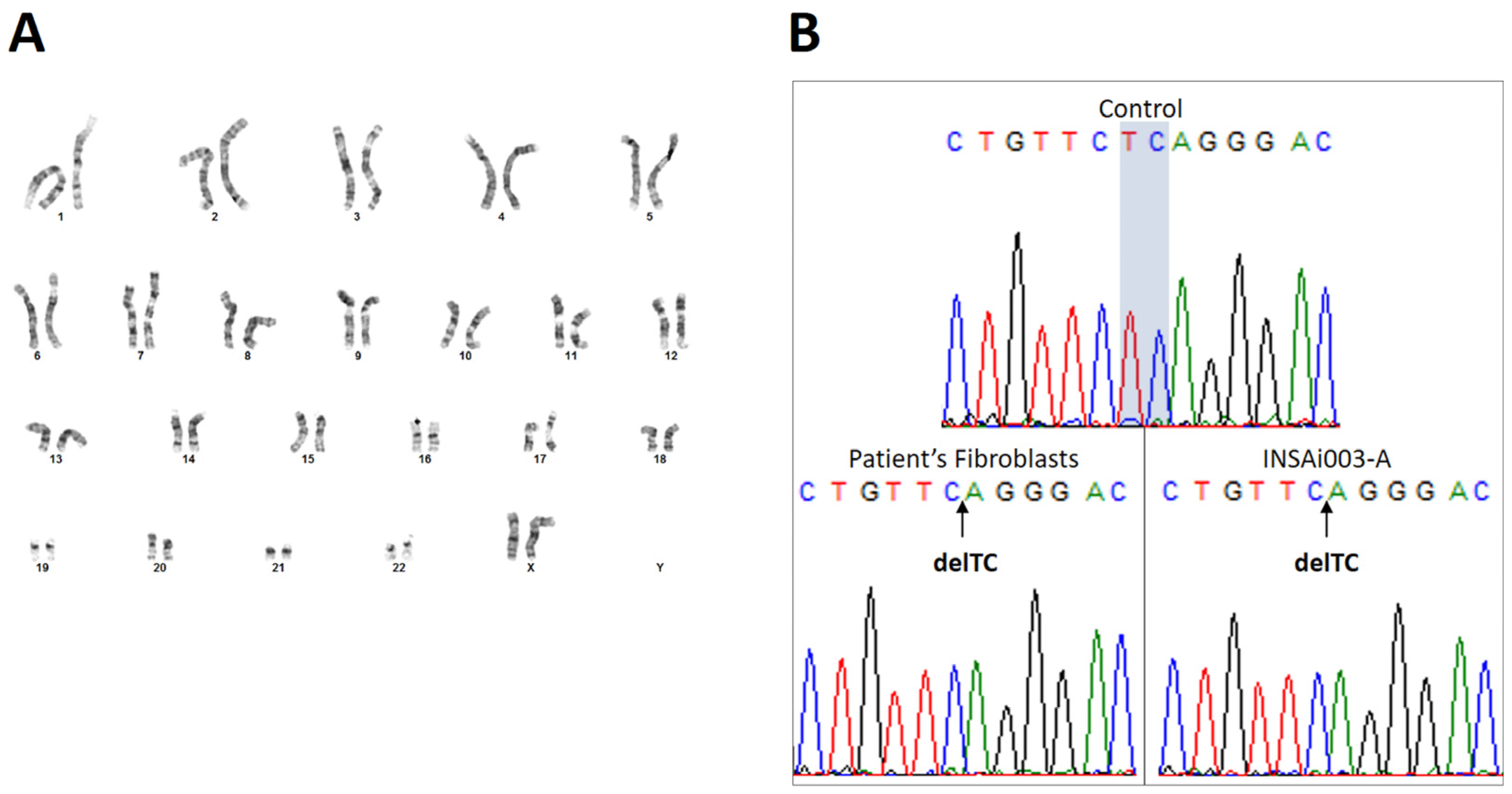
2.3. Genetic Characterization Confirmed That ML II iPSCs Have a Normal Karyotype, Were Derived from ML II Patient’s Fibroblasts, and Harbor the Most Frequent Pathogenic Variant
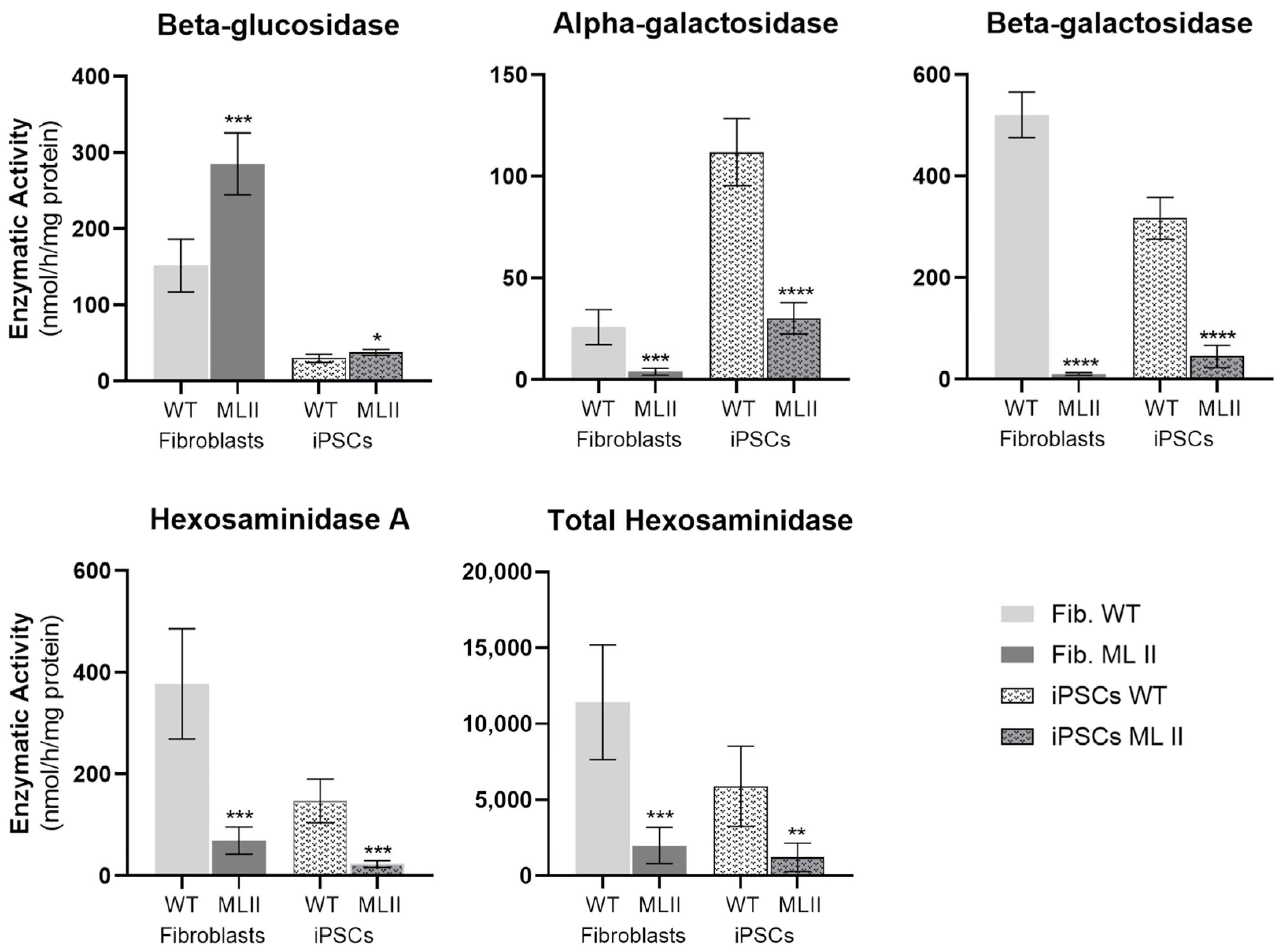
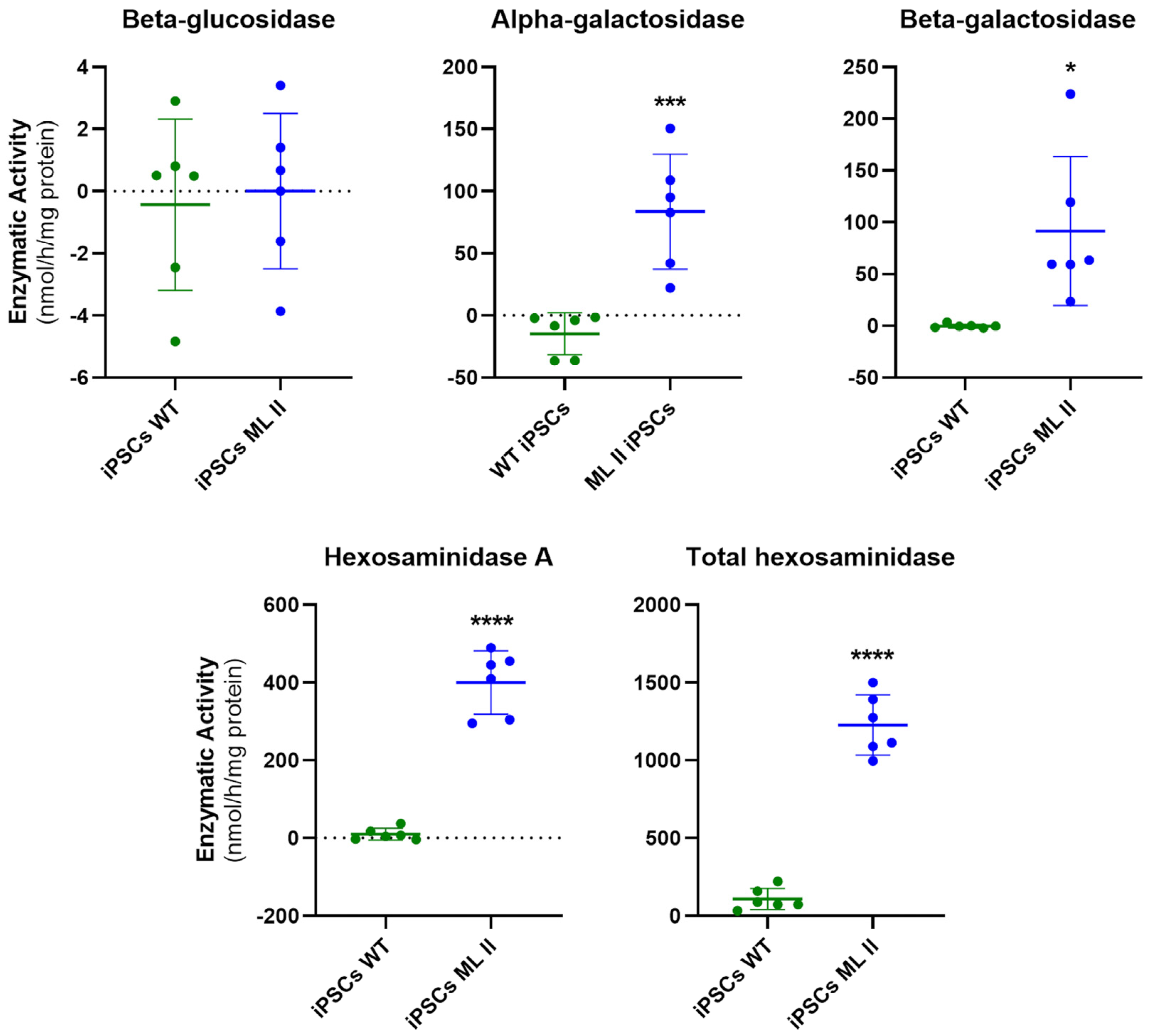
2.4. Enzymatic Activity of M6P-Dependent Hydrolases Is Impaired in ML II iPSCs as in ML II Fibroblasts
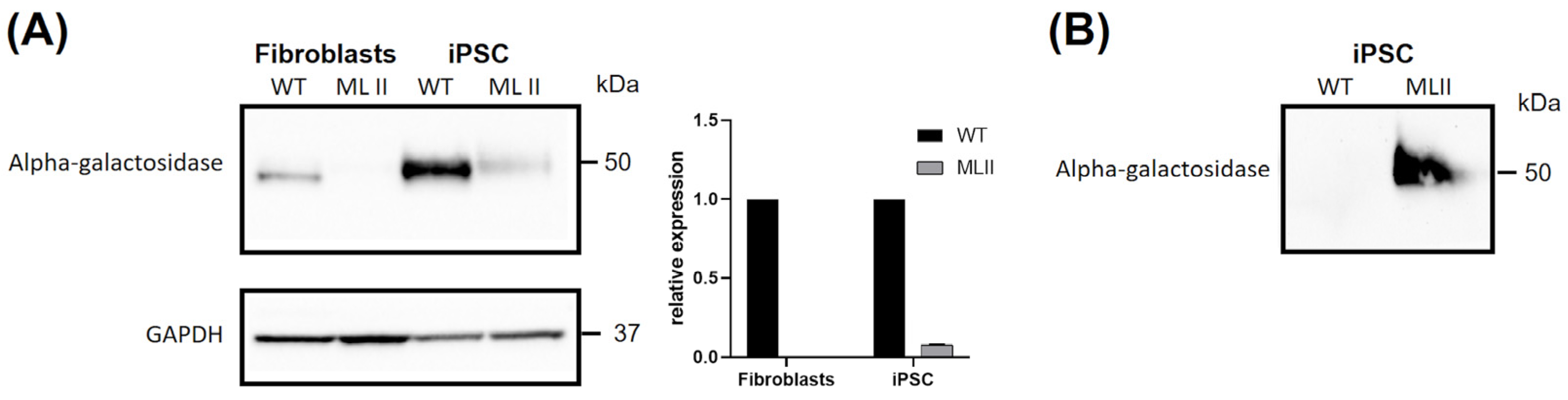
2.5. Western Blot Results Further Support Abnormal Trafficking of a M6P-Dependent Enzyme, Alpha-Galactosidase
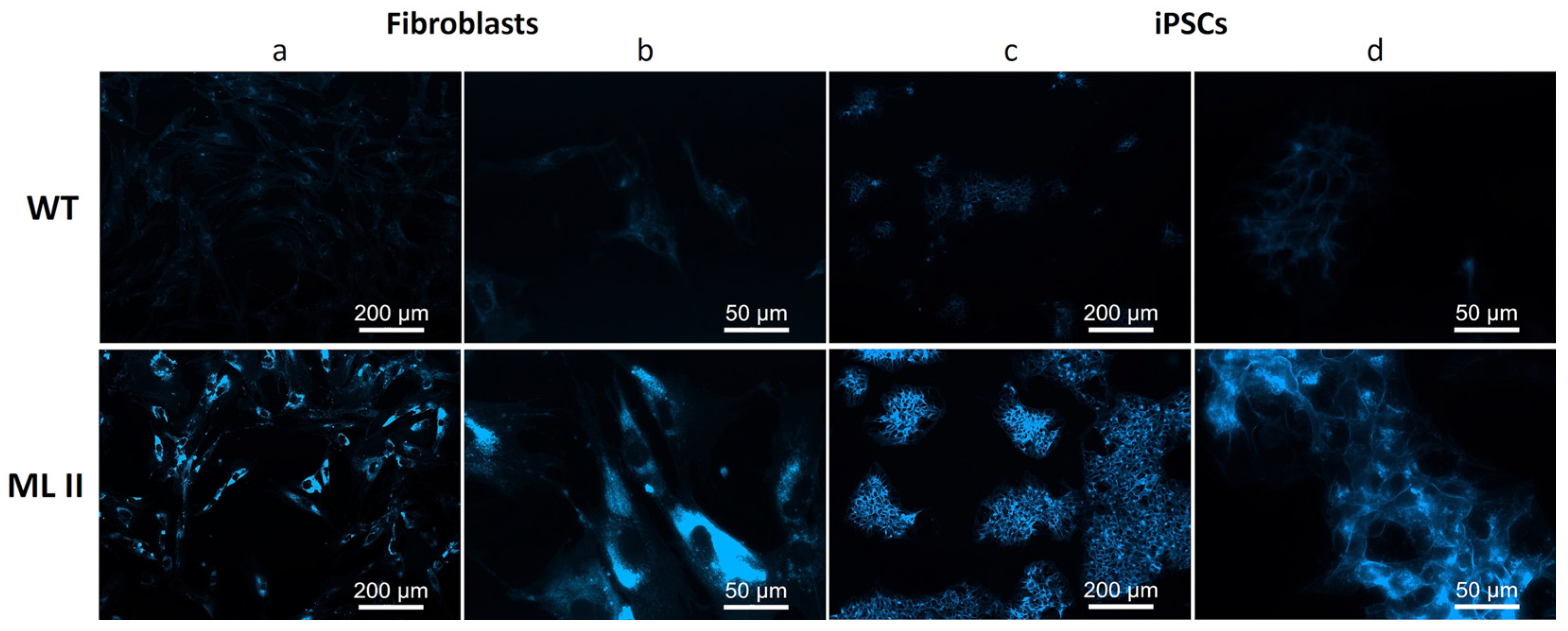
2.6. Filipin Staining Suggests Accumulation of Free Cholesterol in ML II iPSCs
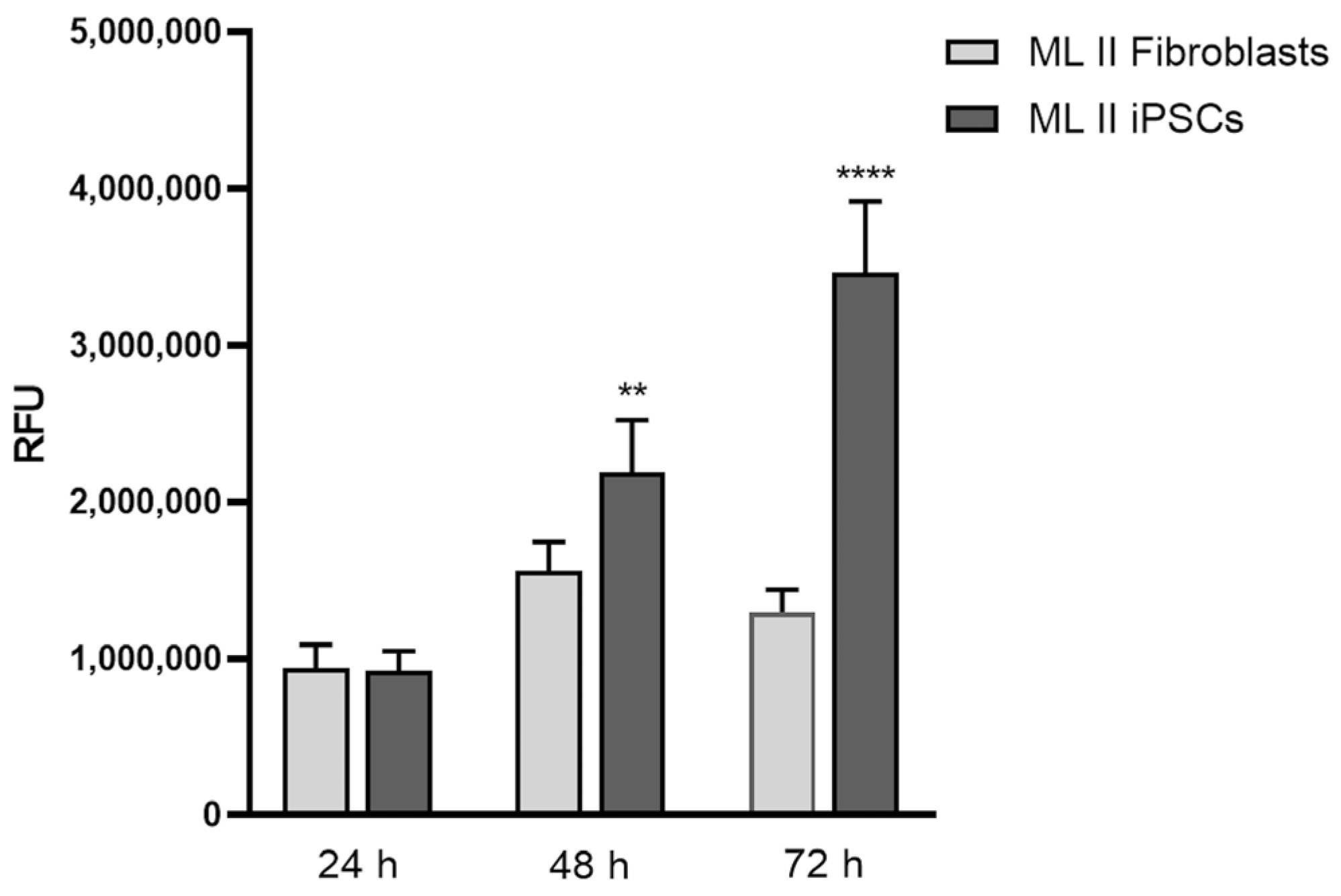
2.7. ML II iPSCs Have a Higher Proliferation Rate than ML II Fibroblasts
3. Discussion
4. Materials and Methods
4.1. Cell Cultures
4.2. Reprogramming ML II Fibroblasts into iPSCs
4.3. Culture and Expansion of iPSCs
4.4. Detection of Episomal Vectors
4.5. Immunostaining of the Pluripotency and Differentiation Markers
4.6. Reverse Transcription and Quantitative Real-Time PCR (RT-qPCR)
4.7. Genetic Characterization
4.8. Measurement of Enzymatic Activity
4.9. Western Blot for Alpha-Galactosidase
4.10. Filipin Staining
4.11. Cell Proliferation Assay with Alamar Blue
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Dogterom, E.J.; Wagenmakers, M.A.E.M.; Wilke, M.; Demirdas, S.; Muschol, N.M.; Pohl, S.; Meijden, J.C.v.d.; Rizopoulos, D.; Ploeg, A.T.v.d.; Oussoren, E. Mucolipidosis Type II and Type III: A Systematic Review of 843 Published Cases. Genet. Med. 2021, 23, 2047–2056. [Google Scholar] [CrossRef] [PubMed]
- Velho, R.V.; Harms, F.L.; Danyukova, T.; Ludwig, N.F.; Friez, M.J.; Cathey, S.S.; Filocamo, M.; Tappino, B.; Güneş, N.; Tüysüz, B.; et al. The Lysosomal Storage Disorders Mucolipidosis Type II, Type III Alpha/Beta, and Type III Gamma: Update on GNPTAB and GNPTG Mutations. Hum. Mutat. 2019, 40, 842–864. [Google Scholar] [CrossRef]
- Matos, L.; Vilela, R.; Rocha, M.; Santos, J.I.; Coutinho, M.F.; Gaspar, P.; Prata, M.J.; Alves, S. Development of an Antisense Oligonucleotide-Mediated Exon Skipping Therapeutic Strategy for Mucolipidosis II: Validation at RNA Level. Hum. Gene Ther. 2020, 31, 775–783. [Google Scholar] [CrossRef] [PubMed]
- Coutinho, M.; Encarnação, M.; Gomes, R.; da Silva Santos, L.; Martins, S.; Sirois-Gagnon, D.; Bargal, R.; Filocamo, M.; Raas-Rothschild, A.; Tappino, B.; et al. Origin and Spread of a Common Deletion Causing Mucolipidosis Type II: Insights from Patterns of Haplotypic Diversity. Clin. Genet. 2011, 80, 273–280. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, K.; Tanabe, K.; Ohnuki, M.; Narita, M.; Ichisaka, T.; Tomoda, K.; Yamanaka, S. Induction of Pluripotent Stem Cells from Adult Human Fibroblasts by Defined Factors. Cell 2007, 131, 861–872. [Google Scholar] [CrossRef] [PubMed]
- Cerneckis, J.; Cai, H.; Shi, Y. Induced Pluripotent Stem Cells (IPSCs): Molecular Mechanisms of Induction and Applications. Signal Transduct. Target. Ther. 2024, 9, 112. [Google Scholar] [CrossRef]
- Kobolák, J.; Molnár, K.; Varga, E.; Bock, I.; Jezsó, B.; Téglási, A.; Zhou, S.; Lo Giudice, M.; Hoogeveen-Westerveld, M.; Pijnappel, W.P.; et al. Modelling the Neuropathology of Lysosomal Storage Disorders through Disease-Specific Human Induced Pluripotent Stem Cells. Exp. Cell Res. 2019, 380, 216–233. [Google Scholar] [CrossRef]
- Luciani, M.; Gritti, A.; Meneghini, V. Human IPSC-Based Models for the Development of Therapeutics Targeting Neurodegenerative Lysosomal Storage Diseases. Front. Mol. Biosci. 2020, 7, 224. [Google Scholar] [CrossRef]
- Morsy, A.; Carmona, A.V.; Trippier, P.C. Patient-Derived Induced Pluripotent Stem Cell Models for Phenotypic Screening in the Neuronal Ceroid Lipofuscinoses. Molecules 2021, 26, 6235. [Google Scholar] [CrossRef]
- Huang, W.; Zhou, Y.; Wang, J.; Jiang, C.; Zhang, Y.; Zhou, R. Generation of Two Heterozygous GAA Mutation-Carrying Human Induced Pluripotent Stem Cell Lines (XACHi005-A, XACHi006-A) from Parents of an Infant with Pompe Disease. Stem Cell Res. 2022, 64, 102934. [Google Scholar] [CrossRef]
- Gao, L.; Lu, Z.; Zhang, Y.; Liu, L.; Sun, J.; Fu, H.; Mao, J.; Hu, L. Clinical Characteristics and Induced Pluripotent Stem Cells (IPSCs) Disease Model of Fabry Disease Caused by a Novel GLA Mutation. QJM 2024, 117, 566–573. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.; Chen, S.; Jiang, Z.; Chen, D.; Chen, H.; Bai, G.; Yu, L.; Chen, W. Diminished GALNS Activity in Induced Pluripotent Stem Cells of Mucopolysaccharidosis IVA Caused by Compound p. S162Y and p. C165F Mutation. QJM 2024, 118, 17–23. [Google Scholar] [CrossRef]
- Pornsukjantra, T.; Saikachain, N.; Sutjarit, N.; Khongkrapan, A.; Tubsuwan, A.; Bhukhai, K.; Tim-Aroon, T.; Anurathapan, U.; Hongeng, S.; Asavapanumas, N. An Increase in ER Stress and Unfolded Protein Response in IPSCs-Derived Neuronal Cells from Neuronopathic Gaucher Disease Patients. Sci. Rep. 2024, 14, 9177. [Google Scholar] [CrossRef] [PubMed]
- Evans, L.M.P.; Gawron, J.; Sim, F.J.; Feltri, M.L.; Marziali, L.N. Human IPSC-Derived Myelinating Organoids and Globoid Cells to Study Krabbe Disease. PLoS ONE 2024, 19, e0314858. [Google Scholar] [CrossRef]
- Adewumi, O.; Aflatoonian, B.; Ahrlund-Richter, L.; Amit, M.; Andrews, P.W.; Beighton, G.; Bello, P.A.; Benvenisty, N.; Berry, L.S.; Bevan, S.; et al. Characterization of Human Embryonic Stem Cell Lines by the International Stem Cell Initiative. Nat. Biotechnol. 2007, 25, 803–816. [Google Scholar] [CrossRef]
- Duarte, A.J.; Ribeiro, D.; Santos, R.; Moreira, L.; Bragança, J.; Amaral, O. Induced Pluripotent Stem Cell Line (INSAi002-A) from a Fabry Disease Patient Hemizygote for the Rare p.W287X Mutation. Stem Cell Res. 2020, 45, 101794. [Google Scholar] [CrossRef]
- Vaz, I.M.; Borgonovo, T.; Kasai-Brunswick, T.H.; Dos Santos, D.S.; Mesquita, F.C.P.; Vasques, J.F.; Gubert, F.; Rebelatto, C.L.K.; Senegaglia, A.C.; Brofman, P.R.S. Chromosomal Aberrations after Induced Pluripotent Stem Cells Reprogramming. Genet. Mol. Biol. 2021, 44, e20200147. [Google Scholar] [CrossRef] [PubMed]
- Yu, C.; Sun, Q.; Zhou, H. Enzymatic Screening and Diagnosis of Lysosomal Storage Diseases. N. Am. J. Med. Sci. 2013, 6, 186–193. [Google Scholar] [CrossRef]
- Vanier, M.T.; Latour, P. Laboratory Diagnosis of Niemann-Pick Disease Type C: The Filipin Staining Test. Methods Cell Biol. 2015, 126, 357–375. [Google Scholar] [CrossRef]
- Otomo, T.; Higaki, K.; Nanba, E.; Ozono, K.; Sakai, N. Lysosomal Storage Causes Cellular Dysfunction in Mucolipidosis II Skin Fibroblasts. J. Biol. Chem. 2011, 286, 35283–35290. [Google Scholar] [CrossRef]
- Rampersad, S.N. Multiple Applications of Alamar Blue as an Indicator of Metabolic Function and Cellular Health in Cell Viability Bioassays. Sensors 2012, 12, 12347–12360. [Google Scholar] [CrossRef] [PubMed]
- Steichen, C.; Hannoun, Z.; Luce, E.; Hauet, T.; Dubart-Kupperschmitt, A. Genomic Integrity of Human Induced Pluripotent Stem Cells: Reprogramming, Differentiation and Applications. World J. Stem Cells 2019, 11, 729–747. [Google Scholar] [CrossRef]
- Gaudioso, Á.; Silva, T.P.; Dolores Ledesma, M. Models to Study Basic and Applied Aspects of Lysosomal Storage Disorders. Adv. Drug Deliv. Rev. 2022, 190, 114532. [Google Scholar] [CrossRef]
- Köse, S.; Aerts Kaya, F.; Kuşkonmaz, B.; Uçkan Çetınkaya, D. Characterization of Mesenchymal Stem Cells in Mucolipidosis Type II (I-Cell Disease). Turk. J. Biol. 2019, 43, 171–178. [Google Scholar] [CrossRef] [PubMed]
- Escribá, R.; Ferrer-Lorente, R.; Raya, Á. Inborn Errors of Metabolism: Lessons from IPSC Models. Rev. Endocr. Metab. Disord. 2021, 22, 1189–1200. [Google Scholar] [CrossRef] [PubMed]
- Alfadhel, M.; AlShehhi, W.; Alshaalan, H.; Al Balwi, M.; Eyaida, W. Mucolipidosis II: First Report from Saudi Arabia. Ann. Saudi Med. 2013, 33, 382–386. [Google Scholar] [CrossRef]
- Home·HPSCreg. Available online: https://hpscreg.eu/ (accessed on 24 January 2025).
- Ludwig, N.F.; Velho, R.V.; Sperb-Ludwig, F.; Acosta, A.X.; Ribeiro, E.M.; Kim, C.A.; Gandelman Horovitz, D.D.; Boy, R.; Rodovalho-Doriqui, M.J.; Lourenço, C.M.; et al. GNPTAB Missense Mutations Cause Loss of GlcNAc-1-Phosphotransferase Activity in Mucolipidosis Type II through Distinct Mechanisms. Int. J. Biochem. Cell Biol. 2017, 92, 90–94. [Google Scholar] [CrossRef]
- De Pace, R.; Coutinho, M.F.; Koch-Nolte, F.; Haag, F.; Prata, M.J.; Alves, S.; Braulke, T.; Pohl, S. Mucolipidosis II-Related Mutations Inhibit the Exit from the Endoplasmic Reticulum and Proteolytic Cleavage of GlcNAc-1-Phosphotransferase Precursor Protein (GNPTAB). Hum. Mutat. 2014, 35, 368–376. [Google Scholar] [CrossRef]
- Boonen, M.; Van Meel, E.; Oorschot, V.; Klumperman, J.; Kornfeld, S. Vacuolization of Mucolipidosis Type II Mouse Exocrine Gland Cells Represents Accumulation of Autolysosomes. Mol. Biol. Cell 2011, 22, 1135–1147. [Google Scholar] [CrossRef]
- Willenborg, M.; Schmidt, C.K.; Braun, P.; Landgrebe, J.; Von Figura, K.; Saftig, P.; Eskelinen, E.L. Mannose 6-Phosphate Receptors, Niemann-Pick C2 Protein, and Lysosomal Cholesterol Accumulation. J. Lipid Res. 2005, 46, 2559–2569. [Google Scholar] [CrossRef]
- Encarnação, M.; Ribeiro, I.; David, H.; Coutinho, M.F.; Quelhas, D.; Alves, S. Challenges in the Definitive Diagnosis of Niemann–Pick Type C—Leaky Variants and Alternative Transcripts. Genes 2023, 14, 1990. [Google Scholar] [CrossRef]
- Sitarska, D.; Ługowska, A. Laboratory Diagnosis of the Niemann-Pick Type C Disease: An Inherited Neurodegenerative Disorder of Cholesterol Metabolism. Metab. Brain Dis. 2019, 34, 1253–1260. [Google Scholar] [CrossRef]
- Khan, S.A.; Tomatsu, S.C. Mucolipidoses Overview: Past, Present, and Future. Int. J. Mol. Sci. 2020, 21, 6812. [Google Scholar] [CrossRef] [PubMed]
- Yokoi, A.; Niida, Y.; Kuroda, M.; Imi-Hashida, Y.; Toma, T.; Yachie, A. B-Cell-Specific Accumulation of Inclusion Bodies Loaded with HLA Class II Molecules in Patients with Mucolipidosis II (I-Cell Disease). Pediatr. Res. 2018 861 2018, 86, 85–91. [Google Scholar] [CrossRef]
- Martin, J.J.; Leroy, J.G.; van Eygen, M.; Ceuterick, C. I-Cell Disease—A Further Report on Its Pathology. Acta Neuropathol. 1984, 64, 234–242. [Google Scholar] [CrossRef] [PubMed]
- Swaroop, M.; Brooks, M.J.; Gieser, L.; Swaroop, A.; Zheng, W. Patient IPSC-Derived Neural Stem Cells Exhibit Phenotypes in Concordance with the Clinical Severity of Mucopolysaccharidosis I. Hum. Mol. Genet. 2018, 27, 3612–3626. [Google Scholar] [CrossRef] [PubMed]
- Otero, M.G.; Kim, J.; Kushwaha, Y.K.; Rajewski, A.; Nonis, F.D.; Santiskulvong, C.; Bannykh, S.I.; Oza, H.; Farooqi, H.M.U.; Babros, M.; et al. Cellular Modeling of CLN6 with IPSC-Derived Neurons and Glia. bioRxiv 2024. [Google Scholar] [CrossRef]
- Pham, V.; Sertori Finoti, L.; Cassidy, M.M.; Maguire, J.A.; Gagne, A.L.; Waxman, E.A.; French, D.L.; King, K.; Zhou, Z.; Gelb, M.H.; et al. A Novel IPSC Model Reveals Selective Vulnerability of Neurons in Multiple Sulfatase Deficiency. Mol. Genet. Metab. 2024, 141, 108116. [Google Scholar] [CrossRef]
- Broeders, M.; van Rooij, J.; Oussoren, E.; van Gestel, T.; Smith, C.; Kimber, S.; Verdijk, R.; Wagenmakers, M.; van den Hout, J.; van der Ploeg, A.; et al. Modeling Cartilage Pathology in Mucopolysaccharidosis VI Using IPSCs Reveals Early Dysregulation of Chondrogenic and Metabolic Gene Expression. Front. Bioeng. Biotechnol. 2022, 10, 949063. [Google Scholar] [CrossRef]
- ter Huurne, M.; Parker, B.L.; Liu, N.Q.; Qian, E.L.; Vivien, C.; Karavendzas, K.; Mills, R.J.; Saville, J.T.; Abu-Bonsrah, D.; Wise, A.F.; et al. GLA-Modified RNA Treatment Lowers GB3 Levels in IPSC-Derived Cardiomyocytes from Fabry-Affected Individuals. Am. J. Hum. Genet. 2023, 110, 1600–1605. [Google Scholar] [CrossRef]
- Goolab, S.; Terburgh, K.; du Plessis, C.; Scholefield, J.; Louw, R. CRISPR-Cas9 Mediated Knockout of NDUFS4 in Human IPSCs: A Model for Mitochondrial Complex I Deficiency. Biochim. Biophys. Acta Mol. Basis Dis. 2025, 1871, 167569. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, V.; Kravitz, J.; Gao, C.; Hochman, M.L.; Meng, D.; Chen, D.; Wang, Y.; Jegga, A.G.; Nelson, J.S.; Tan, W. Perturbations of Glutathione and Sphingosine Metabolites in Port Wine Birthmark Patient-Derived Induced Pluripotent Stem Cells. Metabolites 2023, 13, 983. [Google Scholar] [CrossRef]
- Lejri, I.; Cader, Z.; Grimm, A.; Eckert, A. Human IPSCs from Aged Donors Retain Their Mitochondrial Aging Signature. Int. J. Mol. Sci. 2024, 25, 11199. [Google Scholar] [CrossRef]
- Imamura, K.; Sakurai, Y.; Enami, T.; Shibukawa, R.; Nishi, Y.; Ohta, A.; Shu, T.; Kawaguchi, J.; Okada, S.; Hoenen, T.; et al. IPSC Screening for Drug Repurposing Identifies Anti-RNA Virus Agents Modulating Host Cell Susceptibility. FEBS Open Bio 2021, 11, 1452–1464. [Google Scholar] [CrossRef] [PubMed]
- Ebert, A.D.; Liang, P.; Wu, J.C. Induced Pluripotent Stem Cells as a Disease Modeling and Drug Screening Platform. J. Cardiovasc. Pharmacol. 2012, 60, 408. [Google Scholar] [CrossRef]
- Kido, J.; Nakamura, K.; Era, T. Role of Induced Pluripotent Stem Cells in Lysosomal Storage Diseases. Mol. Cell. Neurosci. 2020, 108, 103540. [Google Scholar] [CrossRef] [PubMed]
- Filocamo, M.; Mazzotti, R.; Corsolini, F.; Stroppiano, M.; Stroppiana, G.; Grossi, S.; Lualdi, S.; Tappino, B.; Lanza, F.; Galotto, S.; et al. Cell Line and DNA Biobank From Patients Affected by Genetic Diseases. Open J. Bioresour. 2014, 1, e2. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of Relative Gene Expression Data Using Real-Time Quantitative PCR and the 2−ΔΔCT Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Aerts, J.M.F.G.; Donker-Koopman, W.E.; Van Der Vliet, M.K.; Jonsson, L.M.V.; Ginns, E.I.; Murray, G.J.; Barranger, J.A.; Tager, J.M.; Schram, A.W. The Occurrence of Two Immunologically Distinguishable Beta-Glucocerebrosidases in Human Spleen. Eur. J. Biochem. 1985, 150, 565–574. [Google Scholar] [CrossRef]
- Gaspar, P.; Herrera, J.; Rodrigues, D.; Cerezo, S.; Delgado, R.; Andrade, C.F.; Forascepi, R.; Macias, J.; del Pino, M.D.; Prados, M.D.; et al. Frequency of Fabry Disease in Male and Female Haemodialysis Patients in Spain. BMC Med. Genet. 2010, 11, 19. [Google Scholar] [CrossRef]
- Crippa, S.; Alberti, G.; Passerini, L.; Savoia, E.O.; Mancino, M.; De Ponti, G.; Santi, L.; Berti, M.; Testa, M.; Hernandez, R.J.; et al. A GLB1 Transgene with Enhanced Therapeutic Potential for the Preclinical Development of Ex-Vivo Gene Therapy to Treat Mucopolysaccharidosis Type IVB. Mol. Ther. Methods Clin. Dev. 2024, 32, 101313. [Google Scholar] [CrossRef] [PubMed]
- Ribeiro, M.G.; Pinto, R.A.; Dos Santos, M.R.; Maia, M.; Sá Miranda, M.C. Biochemical Characterization of Beta-Hexosaminidase in Different Biological Specimens from Eleven Patients with GM2-Gangliosidosis B1 Variant. J. Inherit. Metab. Dis. 1991, 14, 715–720. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Test Sample ML II iPSCs | Reference Sample ML II Fibroblasts | ||
|---|---|---|---|
| Passage Number | 16 | 5 | |
| STR Markers | D8S1179 | 13, 14 | 13, 14 |
| D21S11 | 28, 30 | 28, 30 | |
| D7S820 | 10, 13 | 10, 13 | |
| CSF1PO | 12 | 12 | |
| D3S1358 | 16, 18 | 16, 18 | |
| TH01 | 6, 8 | 6, 8 | |
| D13S317 | 11 | 11 | |
| D16S539 | 9, 11 | 9, 11 | |
| D2S1338 | 22, 25 | 22, 25 | |
| D19S433 | 12, 13 | 12, 13 | |
| vWA | 17 | 17 | |
| TPOX | 8 | 8 | |
| D18S51 | 14, 16 | 14, 16 | |
| AMEL | X | X | |
| D5S818 | 9, 12 | 9, 12 | |
| FGA | 19, 24 | 19, 24 |
| Ratio Activity in ML II/Activity in WT | ||
|---|---|---|
| Fibroblasts | iPSCs | |
| Beta-glucosidase | 1.9 ± 0.3 | 1.3 ± 0.1 |
| Alpha-galactosidase | 0.16 ± 0.07 | 0.27 ± 0.07 |
| Beta-galactosidase | 0.02 ± 0.01 | 0.14 ± 0.07 |
| Hexosaminidase A | 0.18 ± 0.07 | 0.16 ± 0.05 |
| Total hexosaminidase | 0.18 ± 0.10 | 0.21 ± 0.16 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Moutinho, M.E.; Gonçalves, M.; Duarte, A.J.; Encarnação, M.; Coutinho, M.F.; Matos, L.; Santos, J.I.; Ribeiro, D.; Amaral, O.; Gaspar, P.; et al. Establishment of a Human iPSC Line from Mucolipidosis Type II That Expresses the Key Markers of the Disease. Int. J. Mol. Sci. 2025, 26, 3871. https://doi.org/10.3390/ijms26083871
Moutinho ME, Gonçalves M, Duarte AJ, Encarnação M, Coutinho MF, Matos L, Santos JI, Ribeiro D, Amaral O, Gaspar P, et al. Establishment of a Human iPSC Line from Mucolipidosis Type II That Expresses the Key Markers of the Disease. International Journal of Molecular Sciences. 2025; 26(8):3871. https://doi.org/10.3390/ijms26083871
Chicago/Turabian StyleMoutinho, Maria Eduarda, Mariana Gonçalves, Ana Joana Duarte, Marisa Encarnação, Maria Francisca Coutinho, Liliana Matos, Juliana Inês Santos, Diogo Ribeiro, Olga Amaral, Paulo Gaspar, and et al. 2025. "Establishment of a Human iPSC Line from Mucolipidosis Type II That Expresses the Key Markers of the Disease" International Journal of Molecular Sciences 26, no. 8: 3871. https://doi.org/10.3390/ijms26083871
APA StyleMoutinho, M. E., Gonçalves, M., Duarte, A. J., Encarnação, M., Coutinho, M. F., Matos, L., Santos, J. I., Ribeiro, D., Amaral, O., Gaspar, P., Alves, S., & Moreira, L. V. (2025). Establishment of a Human iPSC Line from Mucolipidosis Type II That Expresses the Key Markers of the Disease. International Journal of Molecular Sciences, 26(8), 3871. https://doi.org/10.3390/ijms26083871