
Exploring Molecular and Genetic Differences in Angelica biserrata Roots Under Environmental Changes


Abstract
:1. Introduction
2. Results
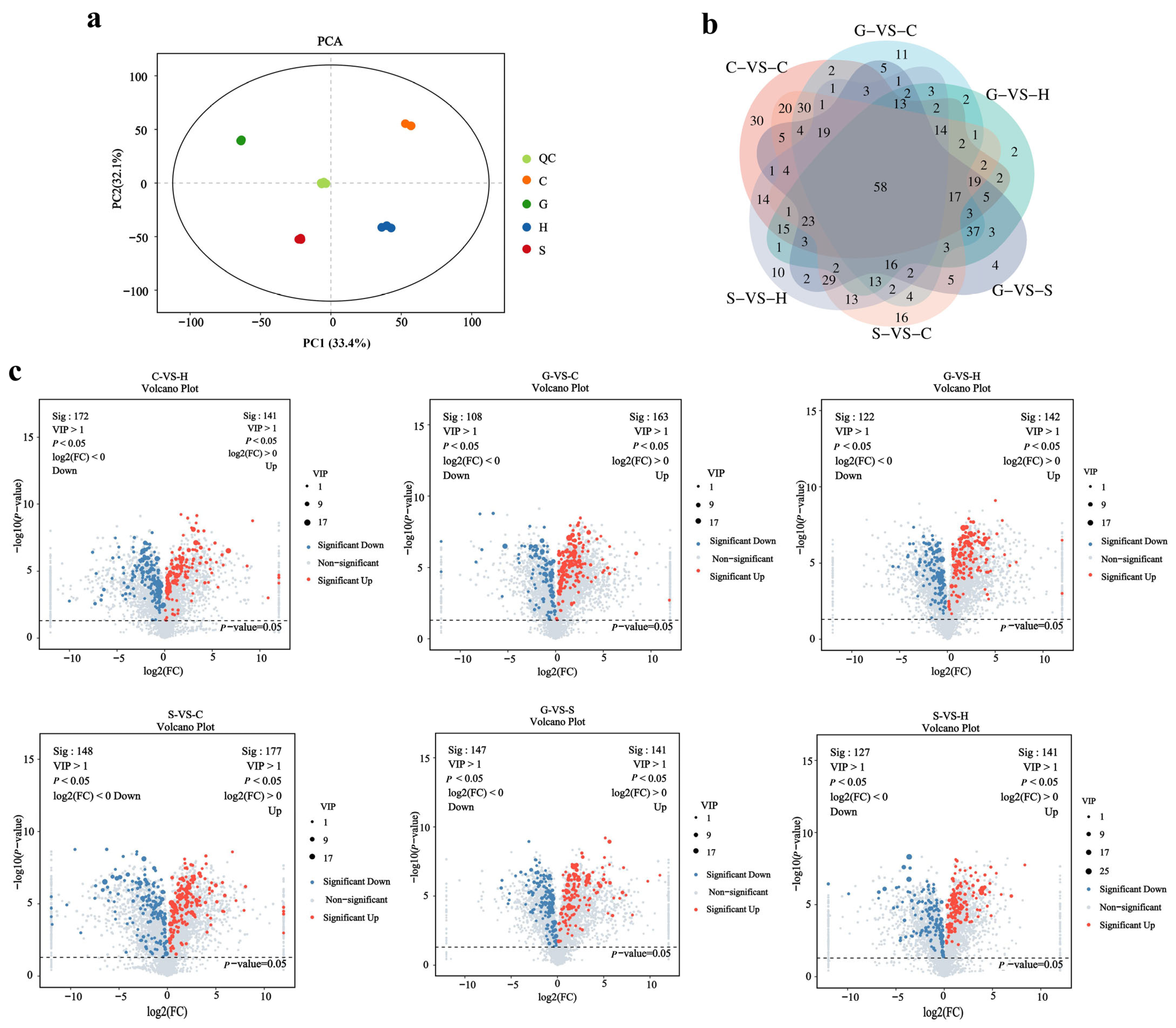
2.1. Metabolic Differences and Similarities of Ecotype Samples
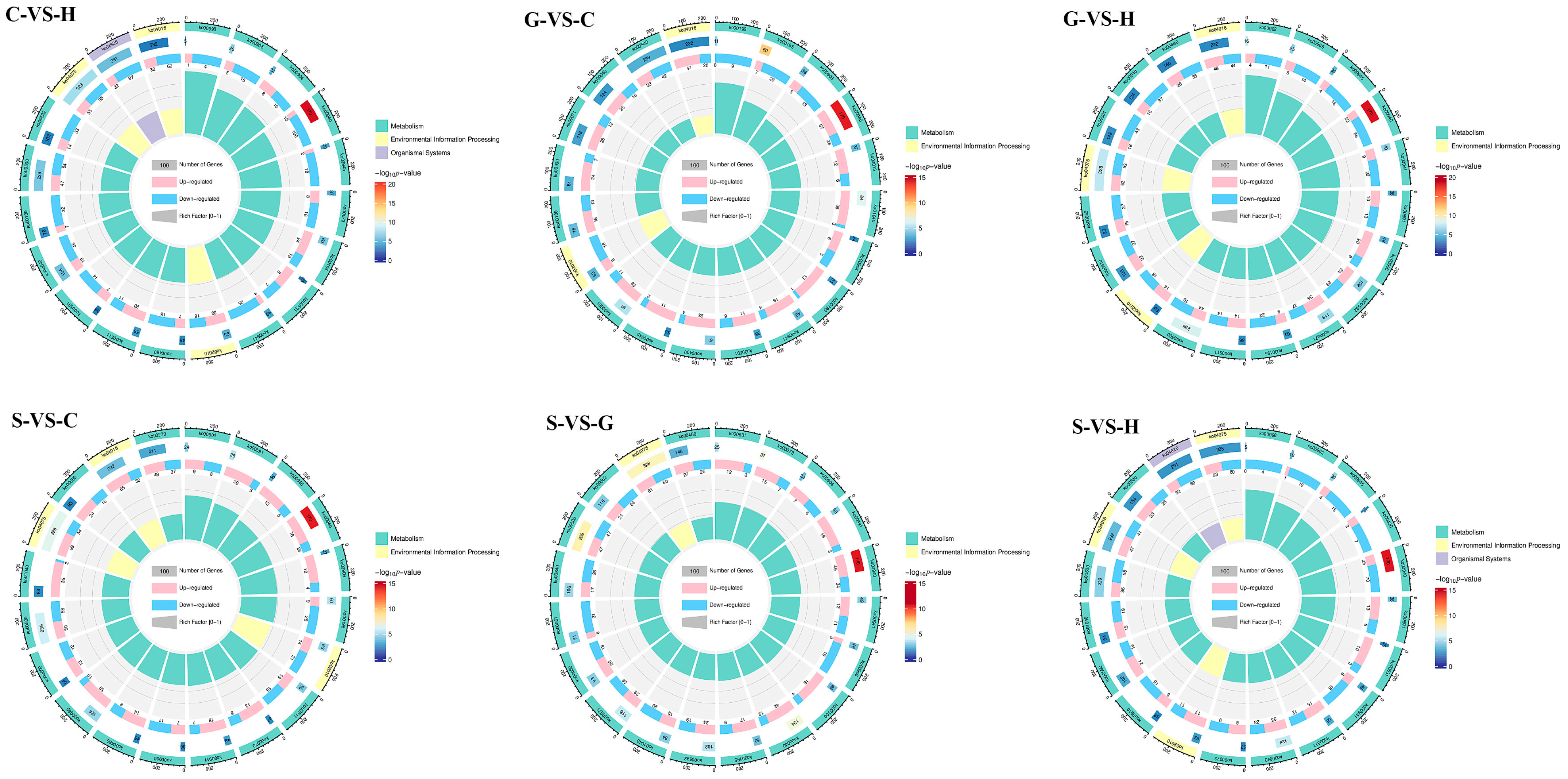
2.2. Genetic Information Differences and Similarities of Ecotype Samples
2.3. Comprehensive Analysis of Metabolomics and Transcriptomics
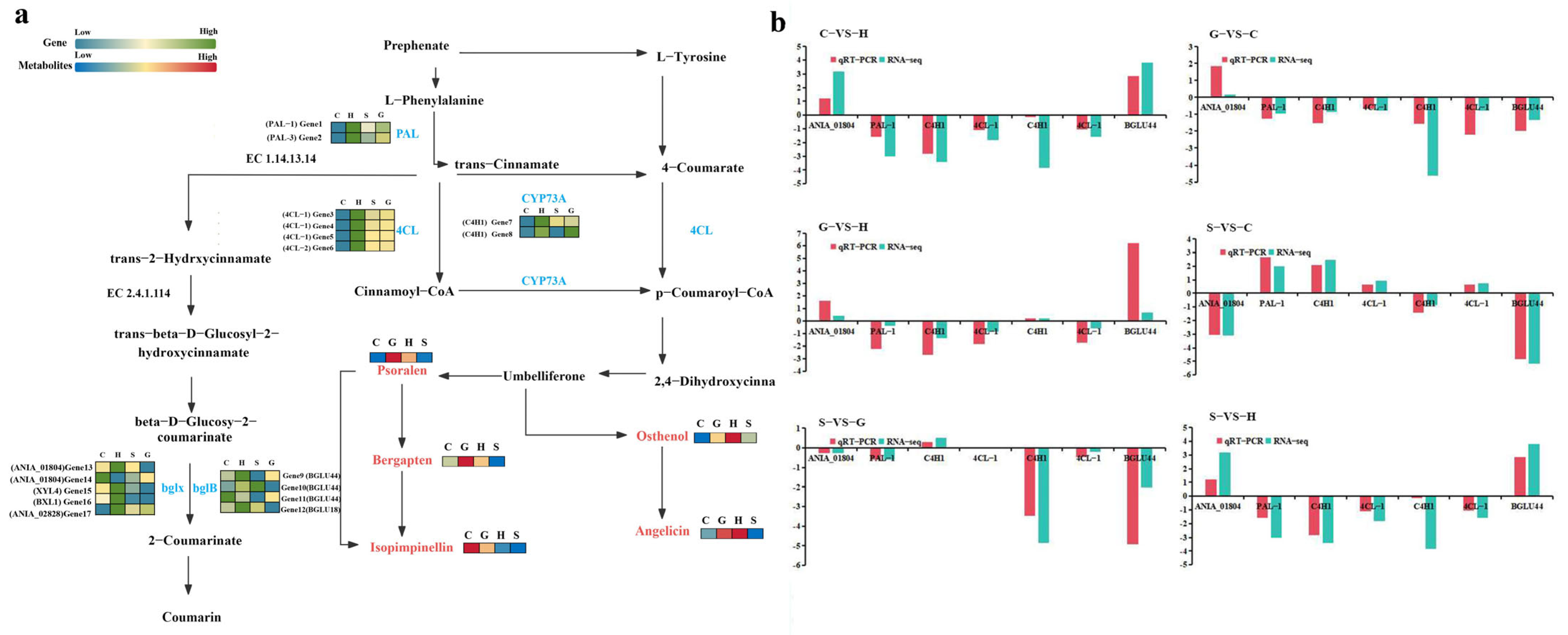
2.4. Analysis of the Synthetic Pathway of the Main Active Compound: Coumarins
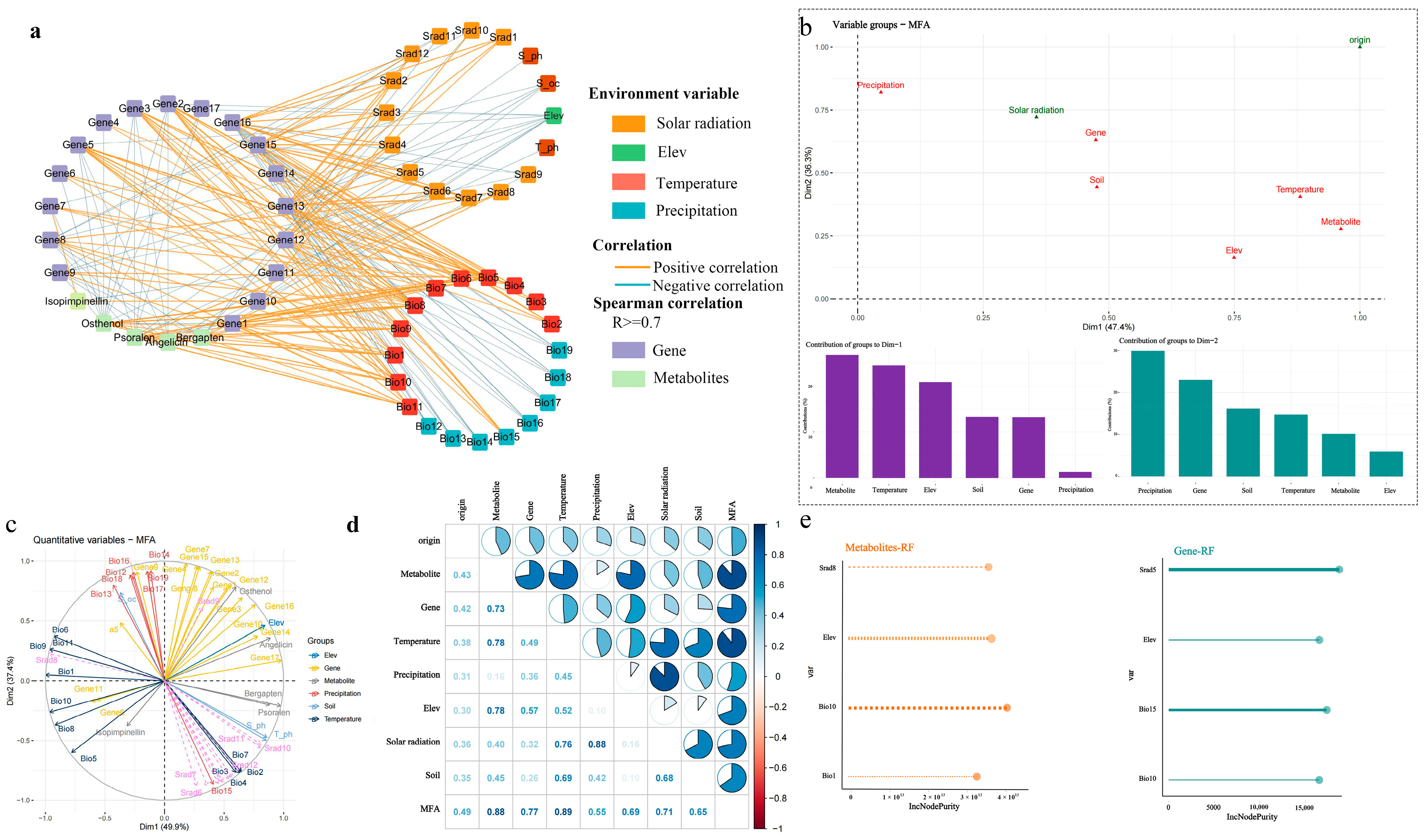
2.5. Interaction Between Environment and Global Transcriptome as Well as Metabolome
2.6. Interaction and Interaction Network Between Environment, Coumarin Metabolites, and Genes
2.7. Optimal Environment for Active Compound Accumulation
3. Discussion
3.1. Differences Between Metabolites and Heritage Information of Ecotype A. biserrata Roots
3.2. Key Environmental Variables Affecting A. biserrata Root Active Substance Accumulation and Gene Expression
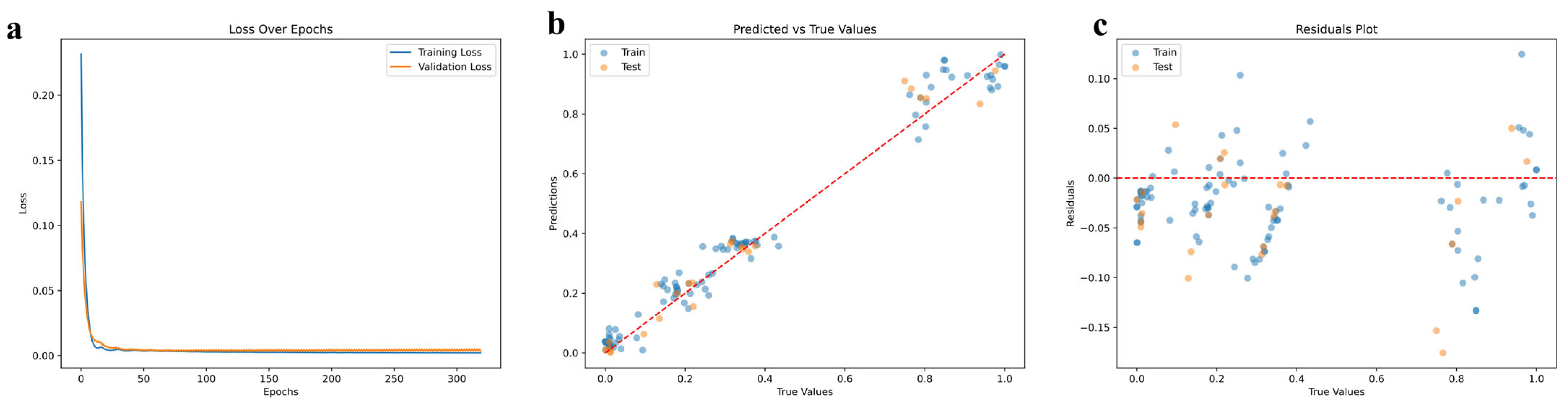
3.3. Deep Learning Predicts the Optimal Suitable Environment for Active Substances of A. biserrata Roots
4. Materials and Methods
4.1. Preparation of A. biserrata Root Materials
4.2. Transcriptome Analysis
4.3. Metabolite Determination and Analysis
4.4. Quantitative Real-Time Polymerase Chain Reaction (qRT-PCR)
4.5. Environment Variable
4.6. Deep Learning and Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Dessie, Y.; Amsalu, N.; Fassil, A.; Liyew, M. Antibacterial Potential of Selected Traditional Medicinal Plants for Wound Healing in Sekela District, Northwestern Ethiopia. J. Herbs Spices Med. Plants 2025, 31, 113–130. [Google Scholar] [CrossRef]
- Zhou, S.; Yan, X.; Yang, J.; Qian, C.; Yin, X.; Fan, X.; Fang, T.; Gao, Y.; Chang, Y.; Liu, W.; et al. Variations in Flavonoid Metabolites Along Altitudinal Gradient in a Desert Medicinal Plant Agriophyllum squarrosum. Front. Plant Sci. 2021, 12, 683265. [Google Scholar] [CrossRef] [PubMed]
- Hosseini, S.M.; Samsampour, D.; Ebrahimi, M.; Abadía, J.; Khanahmadi, M. Effect of drought stress on growth parameters, osmolyte contents, antioxidant enzymes and glycyrrhizin synthesis in licorice (Glycyrrhiza glabra L.) grown in the field. Phytochemistry 2018, 156, 124–134. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.F.; Zhang, L.; Yang, X.W. Distribution Assessments of Coumarins from Angelicae Pubescentis Radix in Rat Cerebrospinal Fluid and Brain by Liquid Chromatography Tandem Mass Spectrometry Analysis. Molecules 2018, 1, 225. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.Q.; Wu, H.W.; Yu, X.K.; Zhang, X.; Luo, H.Y.; Tang, L.Y.; Wang, Z.J. Traditional Chinese medicine of Angelicae pubescentis radix: A review of phytochemistry, pharmacology and pharmacokinetics. Front. Pharmacol. 2020, 11, 355. [Google Scholar] [CrossRef] [PubMed]
- Guo, Q.Q.; Du, G.C.; Li, Y.X.; Liang, C.Y.; Wang, C.; Zhang, Y.N.; Li, R.G. Nematotoxic coumarins from Angelica pubescens Maxim. f. biserrata Shan et Yuan roots and their physiological effects on Bursaphelenchus xylophilus. J. Nematol. 2018, 50, 559–568. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.L.; Zeng, J.G. Research Advances on Chemical Constituents and Pharmacological Effects of Angelica pubescen. Mod. Chin. Med. 2019, 21, 1739–1748. [Google Scholar]
- Zhu, Y.; Feng, J.; Liu, Q.Y.; Yang, S. Studies on Chemical Constituents of Radix Angelicae pubescentis. In Proceedings of the 2023 8th International Symposium on Energy Science and Chemical Engineering, Guangzhou, China, 24–26 March 2023; College of Commerce Liaoning Economic Management Cadre Institute: Dalian, China; Endocrinology Department Affiliated Hospital of Liaoning University of Traditional Chinese Medicine: Shenyang, China; College of Pharmacy Liaoning University of Traditional Chinese Medicine: Shenyang, China, 2025; pp. 205–209. [Google Scholar]
- Cao, L.D.; Hu, C.; Gu, J.; He, L.; Teng, X.F.; Tian, S.Y.; Li, Z. Studies on the chemical constituents from the roots of Angelicae pubescentis and their inhibition of osteoclastongenesis. Lishizhen Med. Mater. Medica Res. 2020, 33, 2918–2923. [Google Scholar]
- Wang, B.; Liu, X.; Zhou, A.; Meng, M.; Li, Q. Simultaneous analysis of coumarin derivatives in extracts of Radix Angelicae pubescentis (Duhuo) by HPLC-DAD-ESI-MSn technique. Anal. Methods 2014, 10, 39. [Google Scholar] [CrossRef]
- Zhen, M.D.; Song, M.M.; He, Z.H.; Li, H.Z. Molecular authentication of the medicinal species of Rhizoma et Radix Heraclei, Radix Angelicae Sinensis, Radix Angelicae Pubescentis and Rhizoma et Radix Notopterygii by integrating ITS2 and its secondary structure. Acta Pharm. Sin. 2021, 56, 2289–2294. [Google Scholar]
- Onda, Y.; Mochida, K. Exploring Genetic Diversity in Plants Using High-Throughput Sequencing Techniques. Curr. Genom. 2016, 17, 358–367. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Du, L.; Qu, Z.; Wang, H.; Dong, S.; Li, X.; Zhao, H. Integrated metabolomics and proteomics analysis to study the changes in Scutellaria baicalensis at different growth stages. Food Chem. 2023, 419, 136043. [Google Scholar] [CrossRef] [PubMed]
- Shen, S.Q.; Zhan, C.S.; Yang, C.K.; Fernie, A.R.; Luo, J. Metabolomics-centered mining of plant metabolic diversity and function: Past decade and future perspectives. Mol. Plant 2022, 16, 43–63. [Google Scholar] [CrossRef] [PubMed]
- Klupczyńska, E.A.; Pawłowski Tomasz, A. Regulation of Seed Dormancy and Germination Mechanisms in a Changing Environment. Int. J. Mol. Sci. 2021, 22, 1357. [Google Scholar] [CrossRef]
- Bao, K.; Jing, Z.H.; Wang, Q.; Huang, Z.; Han, D.; Dai, S.; Liu, C.; Wu, Q.; Xu, F. Quality analysis of Euryales Semen from different origins and varieties based on untargeted metabolomics. J. Chromatogr. B 2022, 1191, 123114. [Google Scholar] [CrossRef]
- Zhang, J.S.; Wang, N.; Chen, W.X.; Zhang, W.; Zhang, H.; Yu, H.; Yi, Y. Integrated metabolomics and transcriptomics reveal metabolites difference between wild and cultivated Ophiocordyceps sinensis. Food Res. Int. 2022, 163, 112275. [Google Scholar] [CrossRef]
- Lee, H.; Koo, J.H.; Lee, C.K.; Song, Y.; Joo, W.K.; Chae, C.J. Prediction and Classification of Phenol Contents in Cnidium officinale Makino Using a Stacking Ensemble Model in Climate Change Scenarios. Agronomy 2024, 14, 1766. [Google Scholar] [CrossRef]
- Nazarenko, D.V.; Kharyuk, P.V.; Oseledets, I.V.; Rodin, I.A.; Shpigun, O.A. Machine learning for LC-MS medicinal plants identification. Chemom. Intell. Lab. Syst. 2016, 156, e174–e180. [Google Scholar] [CrossRef]
- Meng, T.; Jing, X.; Yan, Z.; Pedrycz, W. A survey on machine learning for data fusion. Inf. Fusion 2020, 57, e115–e129. [Google Scholar] [CrossRef]
- Liu, C.; Zuo, Z.; Xu, F.; Wang, Y. Study of the suitable climate factors and geographical origins traceability of Panax notoginseng based on correlation analysis and spectral images combined with machine learning. Front. Plant Sci. 2023, 13, 1009727. [Google Scholar] [CrossRef]
- Gharghory, S.M. Deep Network based on Long Short-Term Memory for Time Series Prediction of Microclimate Data inside the Greenhouse. Int. J. Comput. Intell. Appl. 2020, 19, 2050013. [Google Scholar] [CrossRef]
- Shi, D.Y.; Yuan, P.; Liang, L.W.; Gao, L.; Li, M.; Diao, M. Integration of Deep Learning and Sparrow Search Algorithms to Optimize Greenhouse Microclimate Prediction for Seedling Environment Suitability. Agronomy 2024, 14, 254. [Google Scholar] [CrossRef]
- Maleki, M.; Wraith, D. Mixtures of multivariate restricted skew-normal factor analyzer models in a Bayesian framework. Comput. Stat. 2019, 34, 1039–1053. [Google Scholar] [CrossRef]
- Arsalan, G.; Ali, S.A.; Meisam, A.; Mohammadzadeh, A.; Jamali, S. Application of Artificial Neural Networks for Mangrove Mapping Using Multi-Temporal and Multi-Source Remote Sensing Imagery. Water 2020, 14, 244. [Google Scholar]
- Ben Said, L.; Basem, A.; Sultan, A.J.; Singh, P.K.; Jasim, D.J.; Anqi, A.E.; Rajab, H.; Ahmed, M.; Rajhi, W. Harnessing meta-heuristic, Bayesian, and search-based techniques in optimizing machine learning models for improved energy storage with microencapsulated PCMs. Int. Commun. Heat Mass Transf. 2025, 162, 108537. [Google Scholar] [CrossRef]
- Liu, M.; Zhuang, P.; Lai, F. A Bayesian optimization-genetic algorithm-based approach for automatic parameter calibration of soil models: Application to clay and sand model. Comput. Geotech. 2024, 176, 106717. [Google Scholar] [CrossRef]
- Mahani, M.R.; Nechepurenko, I.A.; Rahimof, Y.; Wicht, A. Optimizing data acquisition: A Bayesian approach for efficient machine learning model training. Mach. Learn. Sci. Technol. 2024, 5, 35013. [Google Scholar] [CrossRef]
- Cho, H.; Kim, Y.; Lee, E.; Choi, D.; Lee, Y.; Rhee, W. Basic Enhancement Strategies When Using Bayesian Optimization for Hyperparameter Tuning of Deep Neural Networks. IEEE Access 2020, 8, 52588–52608. [Google Scholar] [CrossRef]
- Jakubus, M.; Bakinowska, E. Varied macronutrient uptake by plants as an effect of different fertilisation schemes evaluated by PCA. Acta Agric. Scand. Sect. B—Soil Plant Sci. 2020, 70, 56–68. [Google Scholar] [CrossRef]
- Dong, N.; Lin, H. Contribution of phenylpropanoid metabolism to plant development and plant–environment interactions. J. Integr. Plant Biol. 2021, 63, 180–209. [Google Scholar] [CrossRef]
- Kumar, V.; Nautiyal, C.S. Plant Abiotic and Biotic Stress Alleviation: From an Endophytic Microbial Perspective. Curr. Microbiol. 2022, 79, 311. [Google Scholar] [CrossRef] [PubMed]
- Ranner, J.L.; Schalk, S.; Martyniak, C.; Parniske, M.; Gutjahr, C.; Stark, T.D.; Dawid, C. Primary and Secondary Metabolites in Lotus japonicus. J. Agric. Food Chem. 2023, 71, 11277–11303. [Google Scholar] [CrossRef]
- Du, J.L.; Lu, X.; Geng, Z.P.; Yuan, Y.; Liu, Y.; Li, J.; Wang, M.; Wang, J. Metabolites changes of Eucommia ulmoides Olive samaras from different regions and cultivars. Ind. Crops Prod. 2022, 189, 115824. [Google Scholar] [CrossRef]
- Zhang, C.; Jiang, Y.; Liu, C.; Shi, L.; Li, J.; Zeng, Y.; Guo, L.; Wang, S. Identification of Medicinal Compounds of Fagopyri Dibotryis Rhizome from Different Origins and Its Varieties Using UPLC-MS/MS-Based Metabolomics. Metabolites 2022, 12, 790. [Google Scholar] [CrossRef] [PubMed]
- Dong, H.; Li, M.; Jin, L.; Xie, X.; Li, M.; Wei, J. Cool Temperature Enhances Growth, Ferulic Acid and Flavonoid Biosynthesis While Inhibiting Polysaccharide Biosynthesis in Angelica sinensis. Molecules 2022, 27, 320. [Google Scholar] [CrossRef] [PubMed]
- Han, X.X.; Li, C.; Sun, S.; Ji, J.; Nie, B.; Maker, G.; Ren, Y.; Wang, L. The chromosome-level genome of female ginseng (Angelica sinensis) provides insights into molecular mechanisms and evolution of coumarin biosynthesis. Plant J. Cell Mol. Biol. 2022, 112, 1224–1237. [Google Scholar] [CrossRef]
- Han, F.; Ling, M.X.; Luo, C.; Xiao, Z.; Reng, X.Y.; Tan, Q.S.; Zhang, W.W. Analysis of quality differences in Angelica sinensis and its counterfeit products from different regions. J. Southwest China Norm. Univ. (Nat. Sci. Ed.) 2019, 44, 34–39. [Google Scholar]
- Leisner, C.P.; Potnis, N.; SanzSaez, A. Crosstalk and trade-offs: Plant responses to climate change-associated abiotic and biotic stresses. Plant Cell Environ. 2022, 10, 2946–2963. [Google Scholar] [CrossRef]
- Zandalinas, S.I.; Sengupta, S.; Fritschi, F.B.; Azad, R.K.; Nechushtai, R.; Mittler, R. The impact of multifactorial stress combination on plant growth and survival. New Phytol. 2021, 3, 1034–1048. [Google Scholar] [CrossRef]
- Chen, Z.H.; Soltis, D.E. Evolution of Environmental Stress Responses in Plants. Plant Cell Environ. 2020, 12, 2827–2831. [Google Scholar] [CrossRef]
- Mikkelsen, B.L.; Olsen, C.E.; Lyngkjær, M.F. Accumulation of secondary metabolites in healthy and diseased barley, grown under future climate levels of CO2, ozone and temperature. Phytochemistry 2014, 118, 162–173. [Google Scholar] [CrossRef] [PubMed]
- Paukov, A.; Teptina, A.; Morozova, M.; Kruglova, E.; Favero-Longo, S.E.; Bishop, C.; Rajakaruna, N. The Effects of Edaphic and Climatic Factors on Secondary Lichen Chemistry: A Case Study Using Saxicolous Lichens. Diversity 2019, 11, 94. [Google Scholar] [CrossRef]
- Gerganova, M.; Popova, A.V.; Stanoeva, D.; Velitchkova, M. Tomato plants acclimate better to elevated temperature and high light than to treatment with each factor separately. Plant Physiol. Biochem. 2016, 104, 234–241. [Google Scholar] [CrossRef]
- Li, M.F.; Liu, X.Z.; Wei, J.H. Selection of high altitude planting area of Angelica sinensis based on biomass, bioactive compounds accumulation and antioxidant capacity. Chin. Tradit. Herb. Drugs 2020, 51, 474–481. [Google Scholar]
- Xu, M.; Li, X.; Liu, M.; Shi, Y.; Zhou, H.; Zhang, B.; Yan, J. Spatial variation patterns of plant herbaceous community response to warming along latitudinal and altitudinal gradients in mountainous forests of the Loess Plateau, China. Environ. Exp. Bot. 2020, 172, 103983. [Google Scholar] [CrossRef]
- Chauhan, S.; Dhalaria, R.; Ghoshal, S.; Kanwal, K.; Verma, R. Altitudinal Impact on Phytochemical Composition and Mycorrhizal Diversity of Taxus Contorta Griff in the Temperate Forest of Shimla District. J. Basic Microbiol. 2024, 64, e2400016. [Google Scholar] [CrossRef]
- Večeřová, K.; Klem, K.; Veselá, B.; Holub, P.; Grace, J.; Urban, O. Combined Effect of Altitude, Season and Light on the Accumulation of Extractable Terpenes in Norway Spruce Needles. Forests 2021, 12, 1737. [Google Scholar] [CrossRef]
- Zou, K.; Liu, X.; Zhang, D.; Yang, Q.; Fu, S.; Meng, D.; Chang, W.; Li, R.; Yin, H.; Liang, Y. Flavonoid Biosynthesis Is Likely More Susceptible to Elevation and Tree Age Than Other Branch Pathways Involved in Phenylpropanoid Biosynthesis in Ginkgo Leaves. Front. Plant Sci. 2019, 10, 983. [Google Scholar] [CrossRef]
- Costa, S.D.; Gerschlauer, F.; Kiese, R.; Fischer, M.; Kleyer, M.; Hemp, A. Plant niche breadths along environmental gradients and their relationship to plant functional traits. Divers. Distrib. 2018, 24, 1869–1882. [Google Scholar] [CrossRef]
- Guo, X.L.; Lin, X.M.; Guo, J.; You, J.M. The Research Status and Prospect of Radix Angelicae Pubescentis. J. Anhui Agric. Sci. 2014, 42, 11673–11674. [Google Scholar]
- Dixon, R.A.; Paiva, N.L. Stress-Induced Phenylpropanoid Metabolism. Plant Cell Online 1995, 7, 1085–1097. [Google Scholar] [CrossRef]
- Escobar, L.A.; Silva, O.; Acevedo, P.; Nunes-Nesi, A.; Alberdi, M.; Reyes-Díaz, M. Different levels of UV-B resistance in Vaccinium corymbosum cultivars reveal distinct backgrounds of phenylpropanoid metabolites. Plant Physiol. Biochem. 2017, 118, 541–550. [Google Scholar] [CrossRef] [PubMed]
- Lei, L.; Yuan, X.; Fu, K.; Chen, Y.; Lu, Y.; Shou, N.; Wu, D.; Chen, X.; Shi, J.; Zhang, M.; et al. Pseudotargeted metabolomics revealed the adaptive mechanism of Draba oreades Schrenk at high altitude. Front. Plant Sci. 2022, 13, 1052640. [Google Scholar] [CrossRef]
- Silambarasan, T.S.; Mukesh, K.D. Eco-Technological Evaluation of Natural Phytochemicals Potential Drug Molecules Against Main Protease: A Machine Learning Algorithm. Cureus 2024, 16, e57151. [Google Scholar]
- Chinese Pharmacopoeia Commission. Pharmacopoeia of People’s Republic of China; China Medical Science and Technology Press: Beijing, China, 2020; Volume 1, pp. 133–134. [Google Scholar]
- Mihang, J.; Liu, X.J.; Liu, L.Y. Different Responses of Terrestrial Carbon Fluxes to Environmental Changes in Cold Temperate Forest Ecosystems. Forests 2024, 15, 1340. [Google Scholar] [CrossRef]
- Qaderi, M.M.; Martel, A.B.; Strugnell, C.A. Environmental Factors Regulate Plant Secondary Metabolites. Plants 2023, 3, 447. [Google Scholar] [CrossRef]
- Wang, R.X.; Zhang, T.J.; He, F.; Fu, R.; Jing, W.G.; Guo, X.; Wei, F. Identification Analysis of Angelicae sinensis radix and Angelicae pubescentis radix Based on Quantized “Digital Identity” and UHPLC-QTOF-MSE Analysis. J. Am. Soc. Mass Spectrom. 2024, 35, 2222–2229. [Google Scholar] [CrossRef] [PubMed]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 15, 2114–2120. [Google Scholar] [CrossRef]
- Thawng, C.N.; Smith, G.B. A transcriptome software comparison for the analyses of treatments expected to give subtle gene expression responses. BMC Genom. 2022, 23, 452. [Google Scholar] [CrossRef]
- García, C.J.; Alacid, V.; Tomás-Barberán, F.A.; García, C.; Palazón, P. Untargeted Metabolomics to Explore the Bacteria Exo-Metabolome Related to Plant Biostimulants. Agronomy 2022, 8, 1926. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
- He, P.; Guo, L.F.; Liu, Y.Z.; Meng, F.; Peng, C. Spatial dynamic simulation of important cash crops based on phenology with Scutellaria baicalensis Georgi as an example. Eur. J. Agron. 2023, 144, 126748. [Google Scholar] [CrossRef]
- Farcuh, M.; Li, B.; Rivero, R.M.; Shlizerman, L.; Sadka, A.; Blumwald, E. Sugar metabolism reprogramming in a non-climacteric bud mutant of a climacteric plum fruit during development on the tree. J. Exp. Bot. 2017, 68, 5813–5828. [Google Scholar] [CrossRef] [PubMed]
- Ghany, E.A.S.; Mahmood, A.M.; Aziz, E. Adaptive Dynamic Learning Rate Optimization Technique for Colorectal Cancer Diagnosis Based on Histopathological Image Using EfficientNet-B0 Deep Learning Model. Electronics 2024, 13, 3126. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Class | Count | Percent |
|---|---|---|
| Benzene and Substituted Derivatives | 314 | 6.04% |
| Carboxylic Acids and Derivatives | 544 | 10.46% |
| Coumarins and Derivatives | 108 | 2.08% |
| Fatty Acyls | 707 | 13.6% |
| Flavonoids | 291 | 5.6% |
| Glycerophospholipids | 95 | 1.83% |
| Organooxygen Compounds | 649 | 12.48% |
| Prenol Lipids | 462 | 8.88% |
| Steroids and Steroid Derivatives | 134 | 2.58% |
| Others | 1896 | 36.46% |
| C-VS-H | G-VS-C | S-VS-C | G-VS-H | G-VS-S | S-VS-H | |
|---|---|---|---|---|---|---|
| Organooxygen compounds | 18.8% | 14% | 15.3% | 16.6% | 13.8% | 14.9% |
| Prenol lipids | 8.6% | 8.8% | 11.3% | 8.7% | 11.1% | 12.6% |
| Coumarins and derivatives | 8.9% | 9.2% | 6.7% | 11.3% | 10.4% | 9.7% |
| Carboxylic acids and derivatives | 10.2% | 10.3% | 10.1% | 8.7% | 7.9% | 10% |
| Fatty acyls | 8.9% | 10.7% | 11.6% | 11.7% | 10.4% | 13% |
| Flavonoids | 8.9% | 5.9% | 7.3% | 7.1% | 8.3% | 6.7% |
| Enzyme | Numbering | Gene ID | Gene Name | Regulation | |||||
|---|---|---|---|---|---|---|---|---|---|
| C-VS-H | G-VS-C | G-VS-H | S-VS-C | S-VS-G | S-VS-H | ||||
| phenylalanine ammonia-lyase (PAL), K10775 | Gene2 | TRINITY_DN23454_c1_g2_i2_3 | PAL-3 | Down | UP | Down | UP | Down | Down |
| Gene1 | TRINITY_DN21310_c0_g2_i1_4 | PAL-1 | Down | UP | Down | UP | Down | Down | |
| 4-coumarate--CoA ligas (4CL), K01904 | Gene3 | TRINITY_DN18825_c0_g1_i2_4 | 4CL-1 | Down | UP | Down | UP | Down | Down |
| Gene4 | TRINITY_DN22443_c1_g2_i2_2 | 4CL-1 | Down | UP | Down | UP | Down | Down | |
| Gene6 | TRINITY_DN22443_c1_g4_i1_2 | 4CL-2 | Down | UP | Down | UP | UP | Down | |
| Gene5 | TRINITY_DN23319_c1_g4_i1_3 | 4CL-1 | Down | UP | Down | UP | UP | Down | |
| trans-cinnamate 4-monooxygenase (CYP73A), K00487 | Gene8 | TRINITY_DN19642_c0_g1_i1_3 | C4H1 | Down | UP | UP | Down | Down | Down |
| Gene7 | TRINITY_DN18621_c0_g1_i3_4 | C4H1 | Down | UP | Down | UP | UP | Down | |
| beta-glucosidase (bglB) K05350 | Gene9 | TRINITY_DN18641_c0_g1_i2_3 | BGLU44 | Down | UP | Down | Down | Down | Down |
| Gene10 | TRINITY_DN19407_c0_g1_i2_4 | BGLU44 | Down | Down | Down | UP | UP | UP | |
| Gene12 | TRINITY_DN21033_c0_g1_i8_4 | BGLU18 | Down | Down | Down | UP | UP | Down | |
| Gene11 | TRINITY_DN21908_c0_g2_i2_4 | BGLU44 | UP | Down | UP | Down | Down | Down | |
| beta-glucosidase (bglX) K05349 | Gene13 | TRINITY_DN19157_c0_g1_i1_4 | ANIA_01804 | Down | Down | Down | UP | UP | Down |
| Gene14 | TRINITY_DN20626_c2_g1_i5_1 | ANIA_01804 | UP | Down | UP | Down | Down | UP | |
| Gene15 | TRINITY_DN20683_c0_g1_i2_3 | XYL4 | Down | Down | Down | Down | UP | Down | |
| Gene16 | TRINITY_DN20767_c0_g1_i18_2 | BXL1 | Down | Down | Down | UP | UP | Down | |
| Gene17 | TRINITY_DN22550_c1_g4_i2_3 | ANIA_02828 | Down | UP | Down | UP | Down | Down | |
| Training Set Metrics | Test Set Metrics | |
|---|---|---|
| Mean Squared Error (MSE) | 0.0025 | 0.0043 |
| Root Mean Squared Error (RMSE) | 0.0495 | 0.0654 |
| Mean Absolute Error (MAE) | 0.0388 | 0.05 |
| Coefficient of Determination (R2) | 0.9772 | 0.9552 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hu, C.; Li, Q.; Ding, X.; Jiang, K.; Liang, W. Exploring Molecular and Genetic Differences in Angelica biserrata Roots Under Environmental Changes. Int. J. Mol. Sci. 2025, 26, 3894. https://doi.org/10.3390/ijms26083894
Hu C, Li Q, Ding X, Jiang K, Liang W. Exploring Molecular and Genetic Differences in Angelica biserrata Roots Under Environmental Changes. International Journal of Molecular Sciences. 2025; 26(8):3894. https://doi.org/10.3390/ijms26083894
Chicago/Turabian StyleHu, Chaogui, Qian Li, Xiaoqin Ding, Kan Jiang, and Wei Liang. 2025. "Exploring Molecular and Genetic Differences in Angelica biserrata Roots Under Environmental Changes" International Journal of Molecular Sciences 26, no. 8: 3894. https://doi.org/10.3390/ijms26083894
APA StyleHu, C., Li, Q., Ding, X., Jiang, K., & Liang, W. (2025). Exploring Molecular and Genetic Differences in Angelica biserrata Roots Under Environmental Changes. International Journal of Molecular Sciences, 26(8), 3894. https://doi.org/10.3390/ijms26083894