
HCM-Associated MuRF1 Variants Compromise Ubiquitylation and Are Predicted to Alter Protein Structure
 , , , , , and
, , , , , and
Abstract
1. Introduction
2. Results
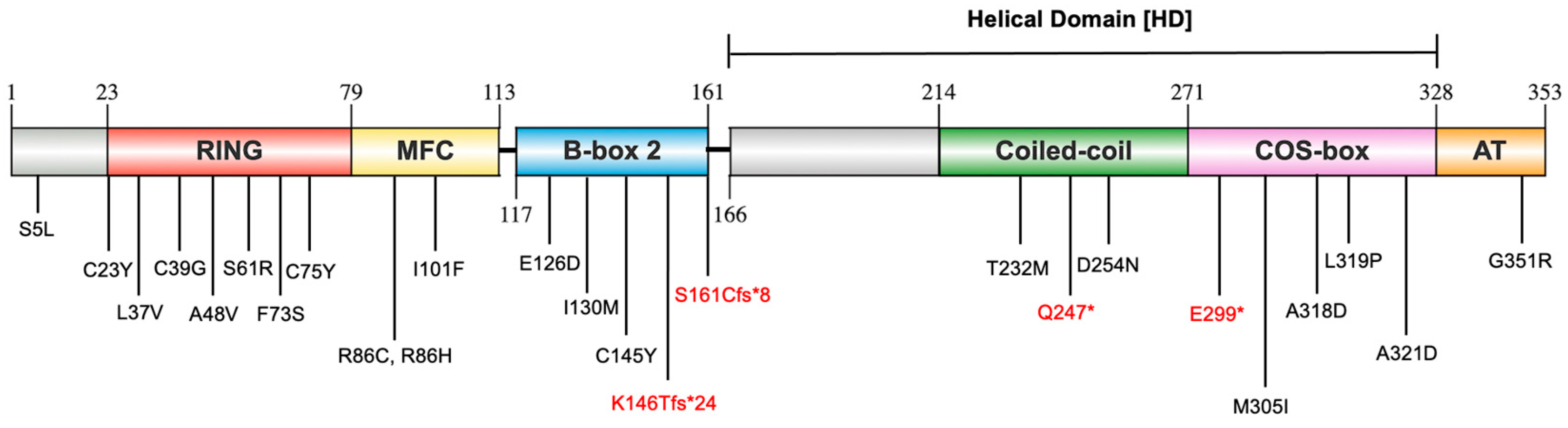
2.1. Distribution of Identified MuRF1 Variants Across MuRF1 Structural Domains
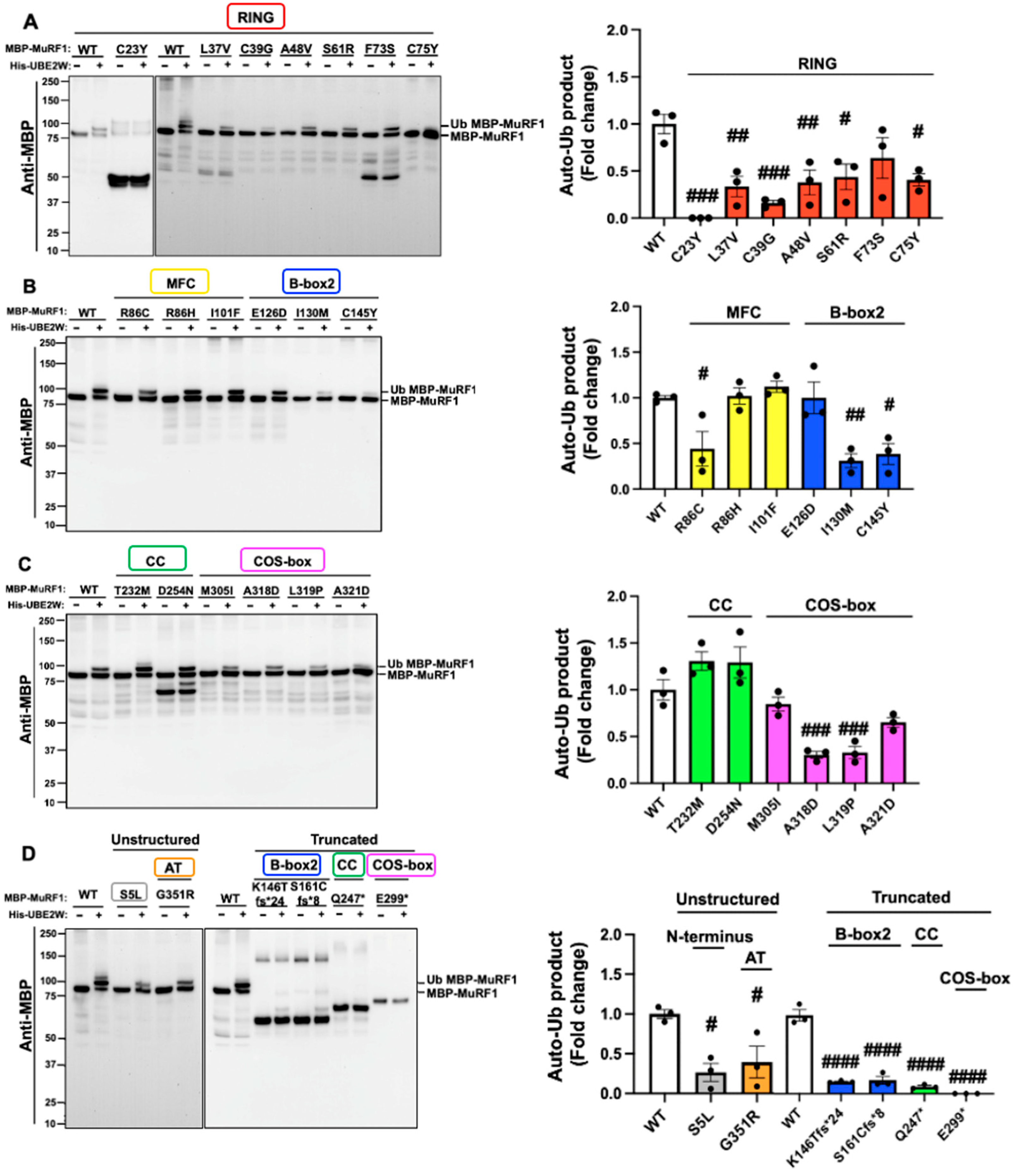
2.2. The Majority of MuRF1 Variants Impair MuRF1 Auto-Monoubiquitylation
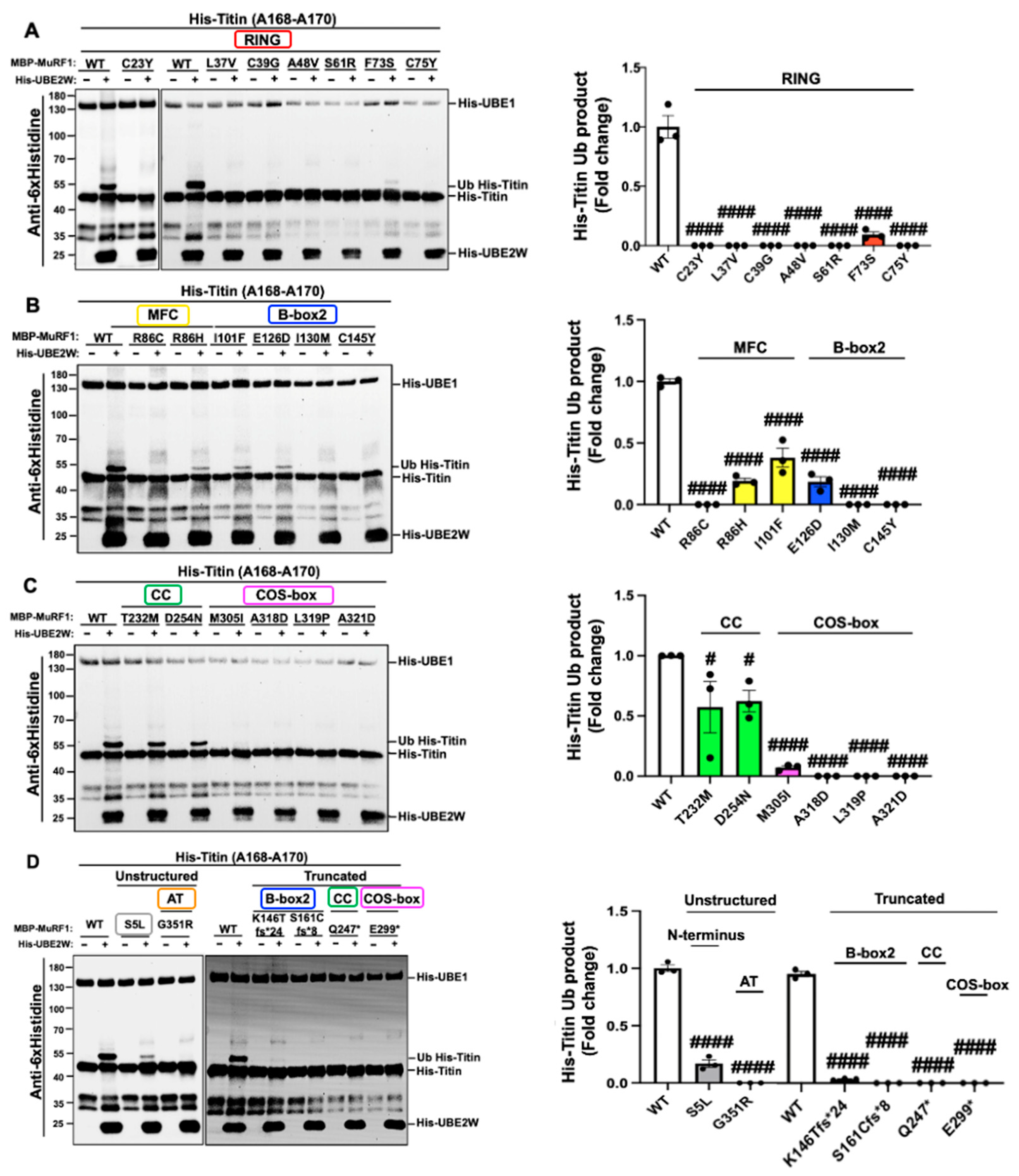
2.3. MuRF1 Variants Preferentially Hinder Monoubiquitylation of the Titin Fragment (A168-A170) Compared to Auto-Monoubiquitylation
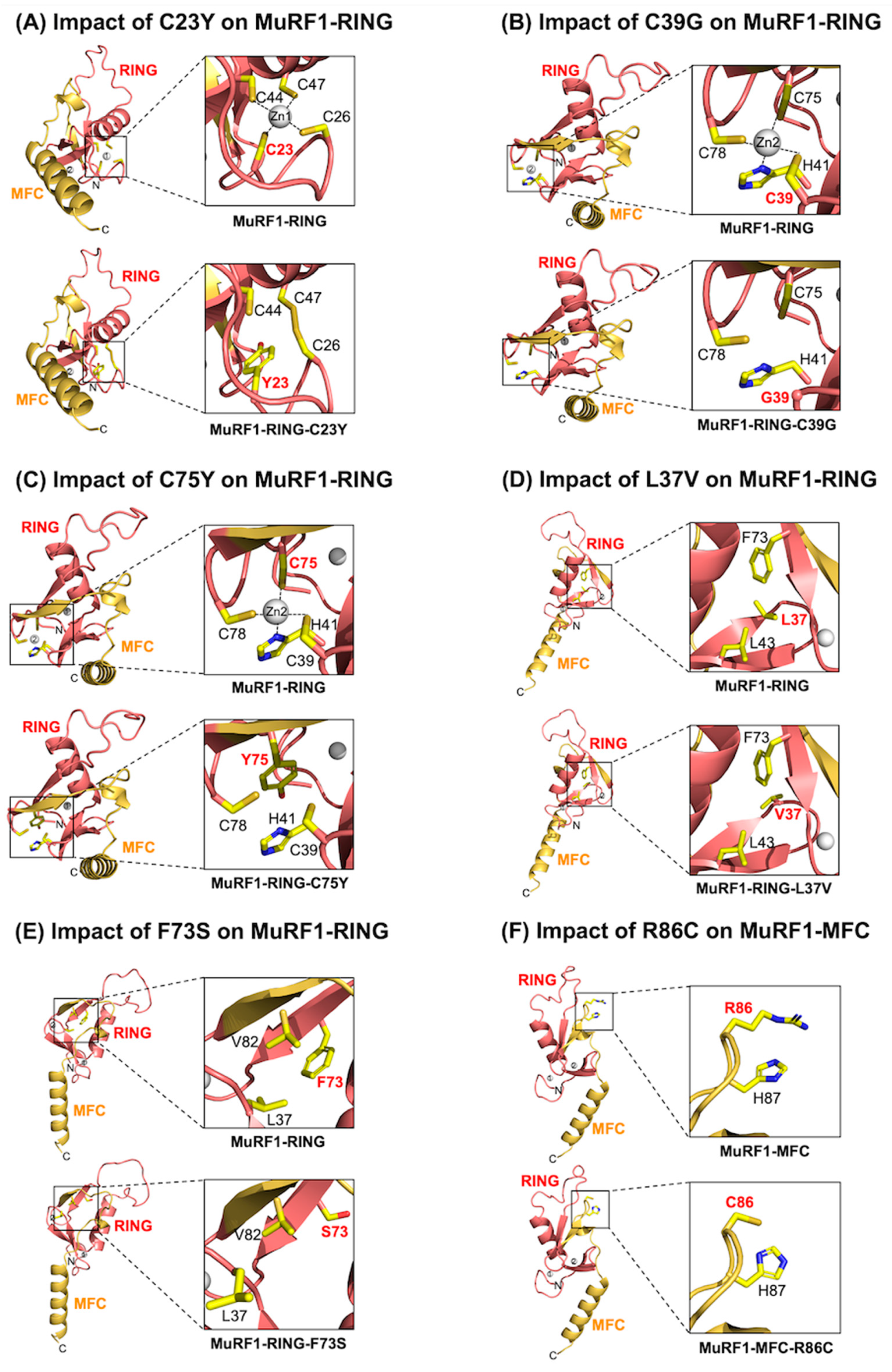
2.4. Assessment of the MuRF1-RING Domain: Variants Impair Zinc Binding and Compromise Core Interactions Critical for MuRF1’s Structural Integrity
2.5. Assessment of the MuRF1-MFC Domain: R86C May Disrupt the Structural Integrity of the MuRF1-MFC Core, Whereas R86H and I101F Are Not Predicted to Induce Structural Changes
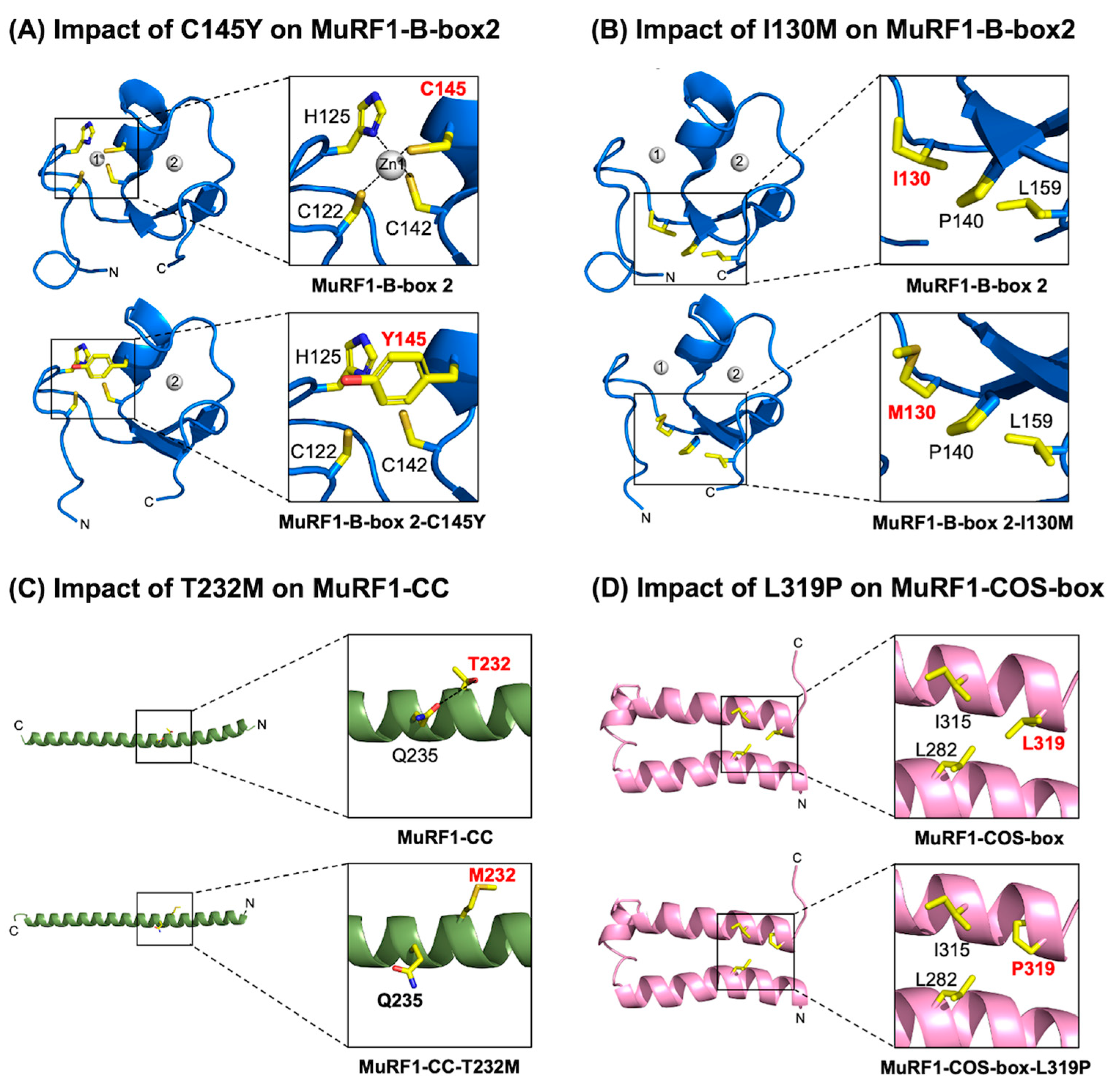
2.6. Assessment of the MuRF1-B-box2 Domain: Variant C145Y Disrupts Zinc Binding, While I130M Compromises Core Interactions
2.7. Assessment of the MuRF1-CC Domain: The T232M Variant Potentially Destabilises the α-Helical Structure
2.8. Assessment of the MuRF1-COS-Box Domain: Variant L319P May Compromise MuRF1’s Structural Integrity
3. Discussion
4. Materials and Methods
4.1. PCR Mutagenesis
4.2. Protein Expression and Purification
4.3. In Vitro Ubiquitylation
4.4. Immunoblotting
4.5. Statistical Analysis
4.6. Structure Modelling of MuRF1-RING-MFC, MuRF1-COS-Box, and the MuRF1/UBE2W Complex
4.7. Evaluating the Impact of MuRF1 Variants on MuRF1 Structure
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| ANP | Atrial Natriuretic Peptide |
| AT | Acidic tail |
| Anti-6xHis | Anti-6xHistidines Antibody |
| Anti-FK2 | Anti-Ubiquitinylated proteins Antibody, clone FK2 |
| Anti-MBP (HRP) | Anti-MBP Monoclonal Antibody, HRP conjugated |
| ATP | Adenosine triphosphate |
| Auto-mUb | Auto-monoubiquitylation |
| B-box2 | B-box type 2 |
| Bis-Tris | 2-[bis(2-hydroxyethyl)amino]-2-(hydroxymethyl)propane-1,3-diol |
| BNP | B-type Natriuretic Peptide |
| CC | Coiled-coil |
| COS-box | C-terminal subgroup One Signature-box |
| DNA | Deoxyribonucleic Acid |
| dNTP | Deoxynucleoside triphosphate |
| DTT | Dithiothreitol |
| E1, UBE1 | Ubiquitin-activating enzyme |
| E2, UBE2 | Ubiquitin-conjugating enzyme |
| E3, UBE3 | Ubiquitin-protein ligase |
| EPR | Electron paramagnetic resonance |
| FnIII | Fibronectin type III |
| FWD | Forward |
| HADDOCK | High Ambiguity Driven protein-protein Docking |
| HCM | Hypertrophic cardiomyopathy |
| HD | Helical domain |
| HECT | Homologous to E6AP C-terminus |
| HEPES | 4-(2-Hydroxyethyl)piperazine-1-ethane-sulfonic acid |
| His-, His-tag | Histidine tag |
| His-Titin-mUb | His-Titin (A168-A170) monoubiquitylation |
| HRP | Horseradish peroxidase |
| Ig | Immunoglobulin-like |
| IPTG | Isopropyl β-D-1-thiogalactopyranoside |
| MBP | Maltose-binding protein |
| MFC | MuRF family-specific motif |
| MgCl2 | Magnesium chloride |
| MID1 | Midline-1 protein |
| MOPS | 3-(N-morpholino)propanesulfonic acid |
| MuRF1 | Muscle RING-finger protein-1 protein |
| NaCl | Sodium chloride |
| NEB Tm Calculator | New England Biolabs Melting Temperature Calculator |
| NMD | Nonsense-mediated decay pathway |
| NuPAGE LDS | NuPAGE Lithium Dodecyl Sulphate |
| OD | Optical density |
| PCR | Polymerase chain reaction |
| PDB | Protein Data Bank |
| PELDOR | Pulsed electron-electron double resonance |
| Phyre2 | Protein Homology/analogY Recognition Engine V 2.2 |
| PMSF | Phenylmethylsulfonyl fluoride |
| PRALINE | PRofile ALIgNEment |
| PVDF | Polyvinylidene fluoride |
| PyMOL | PyMOL Molecular Graphics System version 2.6.0 |
| RBR | RING-between-RING |
| REV | Reverse |
| RING | Really Interesting New Gene |
| RMSD | Root Mean Square Deviation |
| RPM | Revolutions per minute |
| SDS-PAGE | Sodium dodecyl sulphate–polyacrylamide gel electrophoresis |
| SEM | Standard error of the mean |
| SMS-Ident and Sim | Sequence Manipulation Suite-Identity and Similarity |
| TBS-T | 1xTris-buffered saline-Tween 20 |
| TCEP | Tris(2-carboxyethyl)phosphine |
| TRIM18 | Tripartite motif-containing 18 |
| TRIM21 | Tripartite motif-containing 21 |
| TRIM25 | Tripartite motif-containing 25 |
| TRIM63 | Tripartite motif-containing 63 |
| Tris-HCl | Tris(hydroxymethyl)aminomethane hydrochloride |
| UBE2W | Ubiquitin-conjugating Enzyme E2W |
| UniProt | Universal Protein Resource |
| UPS | Ubiquitin–proteasome system |
| WT | Wild type |
| ZnSO4 | Zinc sulphate |
References
- Maron, B.J.; Rowin, E.J.; Maron, M.S. Global Burden of Hypertrophic Cardiomyopathy. JACC Heart Fail. 2018, 6, 376–378. [Google Scholar] [CrossRef] [PubMed]
- Lopes, L.R.; Ho, C.Y.; Elliott, P.M. Genetics of hypertrophic cardiomyopathy: Established and emerging implications for clinical practice. Eur. Heart J. 2024, 45, 2727–2734. [Google Scholar] [CrossRef] [PubMed]
- Maejima, Y.; Usui, S.; Zhai, P.; Takamura, M.; Kaneko, S.; Zablocki, D.; Yokota, M.; Isobe, M.; Sadoshima, J. Muscle-Specific RING Finger 1 Negatively Regulates Pathological Cardiac Hypertrophy Through Downregulation of Calcineurin A. Circ. Heart Fail. 2014, 7, 479–490. [Google Scholar] [CrossRef]
- Arya, R.; Kedar, V.; Hwang, J.R.; McDonough, H.; Li, H.-H.; Taylor, J.; Patterson, C. Muscle ring finger protein-1 inhibits PKCε activation and prevents cardiomyocyte hypertrophy. J. Cell Biol. 2004, 167, 1147–1159. [Google Scholar] [CrossRef]
- Mrosek, M.; Labeit, D.; Witt, S.; Heerklotz, H.; Castelmur, E.; Labeit, S.; Mayans, O. Molecular determinants for the recruitment of the ubiquitin-ligase MuRF-1 onto M-line titin. FASEB J. 2007, 21, 1383–1392. [Google Scholar] [CrossRef]
- Müller, E.; Salcan, S.; Bongardt, S.; Barbosa, D.M.; Krüger, M.; Kötter, S. E3-ligase knock down revealed differential titin degradation by autophagy and the ubiquitin proteasome system. Sci. Rep. 2021, 11, 21134. [Google Scholar] [CrossRef]
- Damgaard, R.B. The ubiquitin system: From cell signalling to disease biology and new therapeutic opportunities. Cell Death Differ. 2021, 28, 423–426. [Google Scholar] [CrossRef]
- Emmerich, C.H.; Cohen, P. Optimising methods for the preservation, capture and identification of ubiquitin chains and ubiquitylated proteins by immunoblotting. Biochem. Biophys. Res. Commun. 2015, 466, 1–14. [Google Scholar] [CrossRef]
- Su, M.; Wang, J.; Kang, L.; Wang, Y.; Zou, Y.; Feng, X.; Wang, D.; Ahmad, F.; Zhou, X.; Hui, R.; et al. Rare Variants in Genes Encoding MuRF1 and MuRF2 Are Modifiers of Hypertrophic Cardiomyopathy. Int. J. Mol. Sci. 2014, 15, 9302–9313. [Google Scholar] [CrossRef]
- Schlossarek, S.; Frey, N.; Carrier, L. Ubiquitin-proteasome system and hereditary cardiomyopathies. J. Mol. Cell. Cardiol. 2014, 71, 25–31. [Google Scholar] [CrossRef]
- Kedar, V.; McDonough, H.; Arya, R.; Li, H.; Rockman, H.A.; Patterson, C. Muscle-specific RING finger 1 is a bona fide ubiquitin ligase that degrades cardiac troponin I. Proc. Natl. Acad. Sci. USA 2004, 101, 18135–18140. [Google Scholar] [CrossRef] [PubMed]
- Willis, M.S.; Schisler, J.C.; Li, L.; Rodríguez, J.E.; Hilliard, E.G.; Charles, P.C.; Patterson, C. Cardiac muscle ring finger-1 increases susceptibility to heart failure in vivo. Circ. Res. 2009, 105, 80–88. [Google Scholar] [CrossRef] [PubMed]
- Willis, M.S.; Ike, C.; Li, L.; Wang, D.-Z.; Glass, D.J.; Patterson, C. Muscle Ring Finger 1, but not Muscle Ring Finger 2, Regulates Cardiac Hypertrophy In Vivo. Circ. Res. 2007, 100, 456–459. [Google Scholar] [CrossRef] [PubMed]
- Lord, S.O.; Dawson, P.W.J.; Chunthorng-Orn, J.; Ng, J.; Baehr, L.M.; Hughes, D.C.; Sridhar, P.; Knowles, T.; Bodine, S.C.; Lai, Y.-C. Uncovering the mechanisms of MuRF1-induced ubiquitylation and revealing similarities with MuRF2 and MuRF3. Biochem. Biophys. Rep. 2024, 37, 101636. [Google Scholar] [CrossRef]
- Bogomolovas, J.; Gasch, A.; Simkovic, F.; Rigden, D.J.; Labeit, S.; Mayans, O. Titin kinase is an inactive pseudokinase scaffold that supports MuRF1 recruitment to the sarcomeric M-line. Open Biol. 2014, 4, 140041. [Google Scholar] [CrossRef]
- Mrosek, M.; Meier, S.; Ucurum-Fotiadis, Z.; Von Castelmur, E.; Hedbom, E.; Lustig, A.; Grzesiek, S.; Labeit, D.; Labeit, S.; Mayans, O. Structural Analysis of B-Box 2 from MuRF1: Identification of a Novel Self-Association Pattern in a RING-like Fold. Biochemistry 2008, 47, 10722–10730. [Google Scholar] [CrossRef]
- Franke, B.; Gasch, A.; Rodriguez, D.; Chami, M.; Khan, M.M.; Rudolf, R.; Bibby, J.; Hanashima, A.; Bogomolovas, J.; Von Castelmur, E.; et al. Molecular basis for the fold organization and sarcomeric targeting of the muscle atrogin MuRF1. Open Biol. 2014, 4, 130172. [Google Scholar] [CrossRef]
- Peris-Moreno, D.; Taillandier, D.; Polge, C. MuRF1/TRIM63, Master Regulator of Muscle Mass. Int. J. Mol. Sci. 2020, 21, 6663. [Google Scholar] [CrossRef]
- Chen, S.N.; Czernuszewicz, G.; Tan, Y.; Lombardi, R.; Jin, J.; Willerson, J.T.; Marian, A.J. Human Molecular Genetic and Functional Studies Identify TRIM63, Encoding Muscle RING Finger Protein 1, as a Novel Gene for Human Hypertrophic Cardiomyopathy. Circ. Res. 2012, 111, 907–919. [Google Scholar] [CrossRef]
- Płoski, R.; Pollak, A.; Müller, S.; Franaszczyk, M.; Michalak, E.; Kosinska, J.; Stawinski, P.; Spiewak, M.; Seggewiss, H.; Bilinska, Z.T. Does p.Q247X in TRIM63 Cause Human Hypertrophic Cardiomyopathy? Circ. Res. 2014, 114, e2–e5. [Google Scholar] [CrossRef]
- Olivé, M.; Abdul-Hussein, S.; Oldfors, A.; González-Costello, J.; Van Der Ven, P.F.M.; Fürst, D.O.; González, L.; Moreno, D.; Torrejón-Escribano, B.; Alió, J.; et al. New cardiac and skeletal protein aggregate myopathy associated with combined MuRF1 and MuRF3 mutations. Hum. Mol. Genet. 2015, 24, 3638–3650. [Google Scholar] [CrossRef] [PubMed]
- Jokela, M.; Baumann, P.; Huovinen, S.; Penttilä, S.; Udd, B. Homozygous Nonsense Mutation p.Q274X in TRIM63 (MuRF1) in a Patient with Mild Skeletal Myopathy and Cardiac Hypertrophy. J. Neuromuscul. Dis. 2019, 6, 143–146. [Google Scholar] [CrossRef] [PubMed]
- Salazar-Mendiguchía, J.; Ochoa, J.P.; Palomino-Doza, J.; Domínguez, F.; Díez-López, C.; Akhtar, M.; Ramiro-León, S.; Clemente, M.M.; Pérez-Cejas, A.; Robledo, M.; et al. Mutations in TRIM63 cause an autosomal-recessive form of hypertrophic cardiomyopathy. Heart 2020, 106, 1342–1348. [Google Scholar] [CrossRef]
- Andreeva, S.; Chumakova, O.; Karelkina, E.; Lebedeva, V.; Lubimtseva, T.; Semenov, A.; Nikitin, A.; Speshilov, G.; Kozyreva, A.; Sokolnikova, P.; et al. Case Report: Two New Cases of Autosomal-Recessive Hypertrophic Cardiomyopathy Associated With TRIM63-Compound Heterozygous Variant. Front. Genet. 2022, 13, 743472. [Google Scholar] [CrossRef]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef]
- Kiss, L.; Rhinesmith, T.; Luptak, J.; Dickson, C.F.; Weidenhausen, J.; Smyly, S.; Yang, J.-C.; Maslen, S.L.; Sinning, I.; Neuhaus, D.; et al. Trim-Away ubiquitinates and degrades lysine-less and N-terminally acetylated substrates. Nat. Commun. 2023, 14, 2160. [Google Scholar] [CrossRef]
- Van Zundert, G.C.P.; Rodrigues, J.P.G.L.M.; Trellet, M.; Schmitz, C.; Kastritis, P.L.; Karaca, E.; Melquiond, A.S.J.; Van Dijk, M.; De Vries, S.J.; Bonvin, A.M.J.J. The HADDOCK2.2 Web Server: User-Friendly Integrative Modeling of Biomolecular Complexes. J. Mol. Biol. 2016, 428, 720–725. [Google Scholar] [CrossRef]
- Deshaies, R.J.; Joazeiro, C.A.P. RING Domain E3 Ubiquitin Ligases. Annu. Rev. Biochem. 2009, 78, 399–434. [Google Scholar] [CrossRef]
- Stevens, M.; Franke, B.; Skorupka, K.A.; Cafiso, D.S.; Pornillos, O.; Mayans, O.; Norman, D.G. Exploration of the TRIM Fold of MuRF1 Using EPR Reveals a Canonical Antiparallel Structure and Extended COS-Box. J. Mol. Biol. 2019, 431, 2900–2909. [Google Scholar] [CrossRef]
- Sanchez, J.G.; Okreglicka, K.; Chandrasekaran, V.; Welker, J.M.; Sundquist, W.I.; Pornillos, O. The tripartite motif coiled-coil is an elongated antiparallel hairpin dimer. Proc. Natl. Acad. Sci. USA 2014, 111, 2494–2499. [Google Scholar] [CrossRef]
- Kelley, L.A.; Mezulis, S.; Yates, C.M.; Wass, M.N.; Sternberg, M.J.E. The Phyre2 web portal for protein modeling, prediction and analysis. Nat. Protoc. 2015, 10, 845–858. [Google Scholar] [CrossRef] [PubMed]
- Wright, K.M.; Du, H.; Dagnachew, M.; Massiah, M.A. Solution structure of the microtubule-targeting COS domain of MID1. FEBS J. 2016, 283, 3089–3102. [Google Scholar] [CrossRef] [PubMed]
- Ruhrman Shahar, N.; Marek-Yagel, D.; Greenberg, R.; Isakov, O.; Naftali, M.; Friedman, E.; Bazak, L.; Monakier, D.; Veber, A.; Shalva, N.; et al. Mono and Biallelic Variants in TRIM63 Are Frequently Associated With a Unique Form of Hypertrophic Cardiomyopathy. Circ. Genomic Precis. Med. 2025, 18, e004864. [Google Scholar] [CrossRef] [PubMed]
- Genomics England PanelApp. Hypertrophic Cardiomyopathy (Version 4.21). Available online: https://panelapp.genomicsengland.co.uk/panels/49/ (accessed on 31 March 2025).
- Mayo Clinic Laboratories HCMGG. Overview: Hypertrophic Cardiomyopathy Gene Panel, Varies. Available online: https://www.mayocliniclabs.com/test-catalog/overview/617281 (accessed on 31 March 2025).
- Centner, T.; Yano, J.; Kimura, E.; McElhinny, A.S.; Pelin, K.; Witt, C.C.; Bang, M.-L.; Trombitas, K.; Granzier, H.; Gregorio, C.C.; et al. Identification of muscle specific ring finger proteins as potential regulators of the titin kinase domain. J. Mol. Biol. 2001, 306, 717–726. [Google Scholar] [CrossRef]
- Wang, B.; Merillat, S.A.; Vincent, M.; Huber, A.K.; Basrur, V.; Mangelberger, D.; Zeng, L.; Elenitoba-Johnson, K.; Miller, R.A.; Irani, D.N.; et al. Loss of the Ubiquitin-conjugating Enzyme UBE2W Results in Susceptibility to Early Postnatal Lethality and Defects in Skin, Immune, and Male Reproductive Systems. J. Biol. Chem. 2016, 291, 3030–3042. [Google Scholar] [CrossRef]
- Magnati, S.; Bracco, E. Never Fold to Fold Continuously: A Conundrum in Ubiquitin–Proteasome System (UPS)-Mediated Protein Quality Control (PQC). Biophysica 2024, 4, 158–167. [Google Scholar] [CrossRef]
- DeBoever, C.; Tanigawa, Y.; Lindholm, M.E.; McInnes, G.; Lavertu, A.; Ingelsson, E.; Chang, C.; Ashley, E.A.; Bustamante, C.D.; Daly, M.J.; et al. Medical relevance of protein-truncating variants across 337,205 individuals in the UK Biobank study. Nat. Commun. 2018, 9, 1612. [Google Scholar] [CrossRef]
- Austin, E.D.; Phillips, J.A.; Cogan, J.D.; Hamid, R.; Yu, C.; Stanton, K.C.; Phillips, C.A.; Wheeler, L.A.; Robbins, I.M.; Newman, J.H.; et al. Truncating and missense BMPR2 mutations differentially affect the severity of heritable pulmonary arterial hypertension. Respir. Res. 2009, 10, 87. [Google Scholar] [CrossRef]
- Chen, D.; Geis-Asteggiante, L.; Gomes, F.P.; Ostrand-Rosenberg, S.; Fenselau, C. Top-Down Proteomic Characterization of Truncated Proteoforms. J. Proteome Res. 2019, 18, 4013–4019. [Google Scholar] [CrossRef]
- Baehr, L.M.; Hughes, D.C.; Lynch, S.A.; Van Haver, D.; Maia, T.M.; Marshall, A.G.; Radoshevich, L.; Impens, F.; Waddell, D.S.; Bodine, S.C. Identification of the MuRF1 Skeletal Muscle Ubiquitylome Through Quantitative Proteomics. Function 2021, 2, zqab029. [Google Scholar] [CrossRef]
- Blake, T.J.; Heath, K.G.; Langdon, W.Y. The truncation that generated the v-cbl oncogene reveals an ability for nuclear transport, DNA binding and acute transformation. EMBO J. 1993, 12, 2017–2026. [Google Scholar] [CrossRef] [PubMed]
- Saurin, A.J.; Borden, K.L.B.; Boddy, M.N.; Freemont, P.S. Does this have a familiar RING? Trends Biochem. Sci. 1996, 21, 208–214. [Google Scholar] [CrossRef] [PubMed]
- Huber, R. Flexibility and rigidity, requirements for the function of proteins and protein pigment complexes. Biochem. Soc. Trans. 1987, 15, 1009–1020. [Google Scholar] [CrossRef] [PubMed]
- Sljoka, A. Structural and Functional Analysis of Proteins Using Rigidity Theory. In Sublinear Computation Paradigm; Katoh, N., Higashikawa, Y., Ito, H., Nagao, A., Shibuya, T., Sljoka, A., Tanaka, K., Uno, Y., Eds.; Springer: Singapore, 2022; pp. 337–367. [Google Scholar]
- Fletcher, A.J.; Vaysburd, M.; Maslen, S.; Zeng, J.; Skehel, J.M.; Towers, G.J.; James, L.C. Trivalent RING Assembly on Retroviral Capsids Activates TRIM5 Ubiquitination and Innate Immune Signaling. Cell Host Microbe 2018, 24, 761–775.e6. [Google Scholar] [CrossRef]
- Vučković, S.; Dinani, R.; Nollet, E.E.; Kuster, D.W.D.; Buikema, J.W.; Houtkooper, R.H.; Nabben, M.; Van Der Velden, J.; Goversen, B. Characterization of cardiac metabolism in iPSC-derived cardiomyocytes: Lessons from maturation and disease modeling. Stem Cell Res. Ther. 2022, 13, 332. [Google Scholar] [CrossRef]
- Maass, A.; Leinwand, L.A. Animal models of hypertrophic cardiomyopathy. Curr. Opin. Cardiol. 2000, 15, 189–196. [Google Scholar] [CrossRef]
- Dvornikov, A.V.; De Tombe, P.P.; Xu, X. Phenotyping cardiomyopathy in adult zebrafish. Prog. Biophys. Mol. Biol. 2018, 138, 116–125. [Google Scholar] [CrossRef]
- Liu, H.; Naismith, J.H. An efficient one-step site-directed deletion, insertion, single and multiple-site plasmid mutagenesis protocol. BMC Biotechnol. 2008, 8, 91. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| MuRF1 Variants | Domain | Result Summary of This Study | ||
|---|---|---|---|---|
| Auto-mUb | His-Titin-mUb | Structural Modelling Predicted Impact on MuRF1 Structure | ||
| S5L [9] | N-terminal | Decreased # | Decreased #### | No prediction |
| C23Y [23] | RING | Abolished ### | Abolished #### | Impair zinc binding (site 1) |
| L37V [23] | RING | Decreased ## | Abolished #### | Compromise RING domain core region |
| C39G [24] | RING | Decreased ### | Abolished #### | Impair zinc binding (site 1) |
| A48V [19] | RING | Decreased ## | Abolished #### | Strengthen RING domain core region |
| S61R [9] | RING | Decreased # | Abolished #### | Strengthen RING domain core region |
| F73S [9] | RING | No change | Decreased #### | Compromise RING domain core region |
| C75Y [23,24] | RING | Decreased # | Abolished #### | Impair zinc binding (site 2) |
| R86C [9] | MFC | Decreased # | Abolished #### | Compromise MFC domain stability |
| R86H [9] | MFC | No change | Decreased #### | No change; structurally neutral |
| I101F [9] | MFC | No change | Decreased #### | No change; structurally neutral |
| E126D [9] | B-box2 | No change | Decreased #### | No change; structurally neutral |
| I130M [19] | B-box2 | Decreased ## | Abolished #### | Compromise B-box2 domain core region |
| C145Y [23] | B-box2 | Decreased # | Abolished #### | Impair zinc binding (site 1) |
| K146Tfs*24 [23] | B-box2 | Decreased #### | Decreased #### | No prediction |
| S161Cfs*8 [23,24] | B-box2 | Decreased #### | Abolished #### | No prediction |
| T232M [9] | CC | No change | Decreased # | Destabilise CC domain |
| Q247* [19,20,21,22,23,24] | CC | Decreased #### | Abolished #### | No prediction |
| D254N [9] | CC | No change | Decreased # | No change; structurally neutral |
| E299* [9] | COS-box | Abolished #### | Abolished #### | No prediction |
| M305I [9] | COS-box | No change | Decreased #### | No change; structurally neutral |
| A318D [9] | COS-box | Decreased ### | Abolished #### | Strengthen COS-box domain stability |
| L319P [23] | COS-box | Decrease ### | Abolished #### | Compromise COS-box core region |
| A321D [9] | COS-box | No change | Abolished #### | No change; structurally neutral |
| G351R [9] | AT | Decreased # | Abolished #### | No prediction |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chunthorng-Orn, J.; Noureddine, M.; Dawson, P.W.J.; Lord, S.O.; Ng, J.; Boyton, L.; Gehmlich, K.; Mohammed, F.; Lai, Y.-C. HCM-Associated MuRF1 Variants Compromise Ubiquitylation and Are Predicted to Alter Protein Structure. Int. J. Mol. Sci. 2025, 26, 3921. https://doi.org/10.3390/ijms26083921
Chunthorng-Orn J, Noureddine M, Dawson PWJ, Lord SO, Ng J, Boyton L, Gehmlich K, Mohammed F, Lai Y-C. HCM-Associated MuRF1 Variants Compromise Ubiquitylation and Are Predicted to Alter Protein Structure. International Journal of Molecular Sciences. 2025; 26(8):3921. https://doi.org/10.3390/ijms26083921
Chicago/Turabian StyleChunthorng-Orn, Jitpisute, Maya Noureddine, Peter W. J. Dawson, Samuel O. Lord, Jimi Ng, Luke Boyton, Katja Gehmlich, Fiyaz Mohammed, and Yu-Chiang Lai. 2025. "HCM-Associated MuRF1 Variants Compromise Ubiquitylation and Are Predicted to Alter Protein Structure" International Journal of Molecular Sciences 26, no. 8: 3921. https://doi.org/10.3390/ijms26083921
APA StyleChunthorng-Orn, J., Noureddine, M., Dawson, P. W. J., Lord, S. O., Ng, J., Boyton, L., Gehmlich, K., Mohammed, F., & Lai, Y.-C. (2025). HCM-Associated MuRF1 Variants Compromise Ubiquitylation and Are Predicted to Alter Protein Structure. International Journal of Molecular Sciences, 26(8), 3921. https://doi.org/10.3390/ijms26083921