
Molecular and Pathophysiological Mechanisms Leading to Ischemic Heart Disease in Patients with Diabetes Mellitus

 , , , ,
, , , ,  , , ,
, , , {kind=link}
{kind=link}
Abstract
1. Introduction
Methodology of the Review
2. The Connection Between Atherosclerosis and Diabetes Mellitus
3. Hyperglycemia in Diabetic Patients
3.1. Acute Effect of Hyperglycemia
3.2. Prolonged Effect of Hyperglycemia
3.3. The Impact of Hyperglycemia on Platelet Function
- In a hyperglycemic state, there is increased expression of glycoprotein Ib (GpIb) on the surface of platelets, which has an affinity for binding to von Willebrand factor (vWF) and GpIIb/IIIa, thereby promoting platelet interaction with fibrin [35].
3.4. The Impact of Hyperglycemia on the Coagulation Process
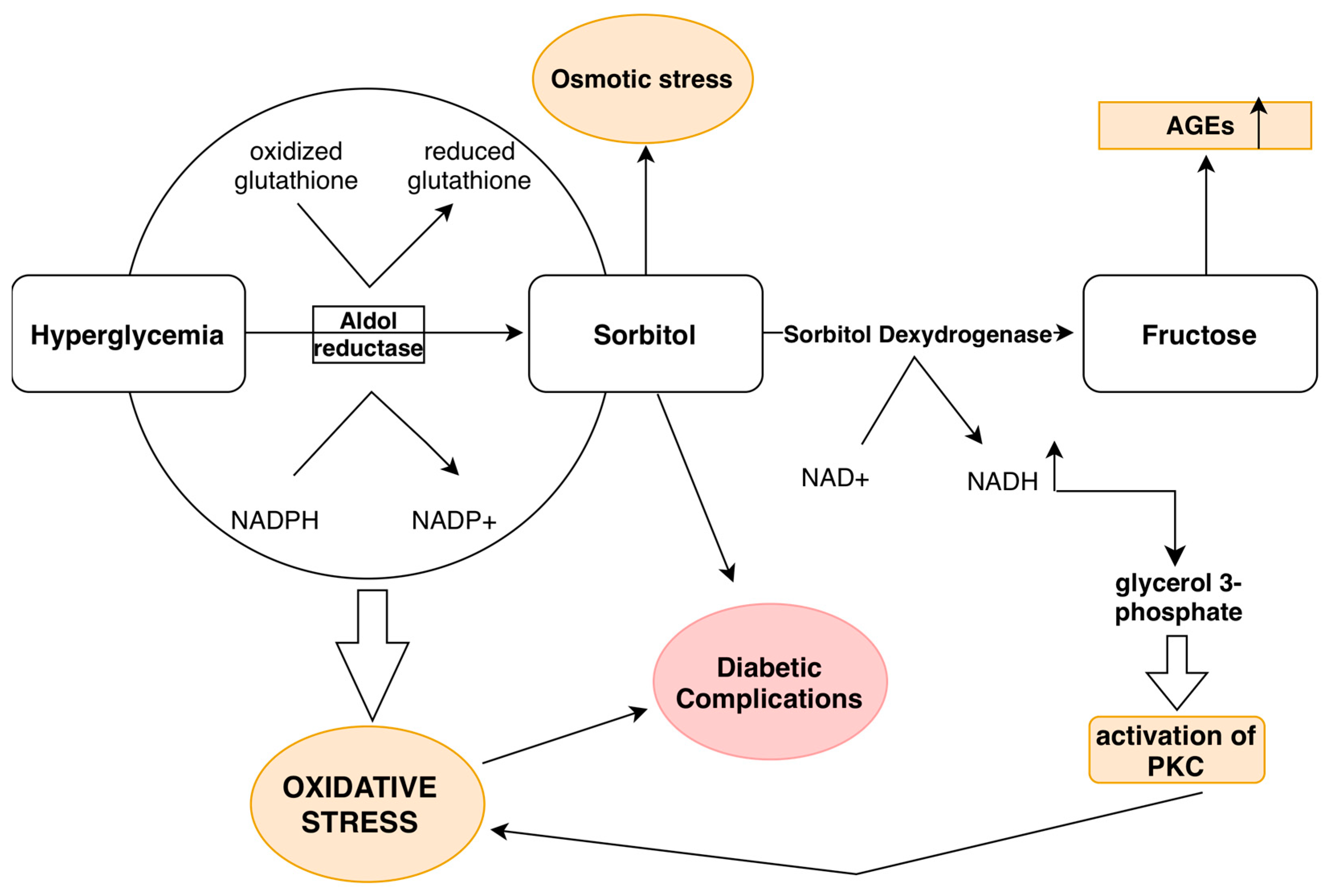
3.5. Hyperglycemia and the Polyol Pathway
4. Insulin Resistance
5. Dyslipidemia in Diabetic Patients
New Therapeutic Approaches in the Treatment of Dyslipidemia
6. Oxidative Stress
Non-Coding RNAs and Oxidative Stress
7. Chronic Inflammation
7.1. Chronic Inflammation, Therapeutic Dilemmas
7.2. NETosis and Chronic Inflammation
- Inhibition of NET formation by developing therapies that target key enzymes in the NETosis process, such as NADPH oxidase and peptidyl-arginine deiminase 4 (PAD4).
- Increasing the elimination of NETs through the use of DNases or other enzymes that degrade DNA.
- Neutralization of the procoagulant components of NETs, such as histones and proteases.
- An immunological approach using antibodies that bind to NET components, aiding in their elimination from circulation and reducing their impact on plaque destabilization and thrombosis.
8. The Significance of the Connection Between Diabetes Mellitus and Liver Fibrosis in the Development of Atherosclerosis
9. The Interrelationship Between Metabolic and Inflammatory Pathways in the Formation of an Atherosclerotic Plaque
10. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Jebari-Benslaiman, S.; Galicia-García, U.; Larrea-Sebal, A.; Olaetxea, J.R.; Alloza, I.; Vandenbroeck, K.; Benito-Vicente, A.; Martín, C. Pathophysiology of Atherosclerosis. Int. J. Mol. Sci. 2022, 23, 3346. [Google Scholar] [CrossRef]
- Rubanyi, G.M. The Role of Endothelium in Cardiovascular Homeostasis and Diseases. J. Cardiovasc. Pharmacol. 1993, 22, S1–S14. [Google Scholar] [CrossRef] [PubMed]
- Cheng, F.; Torzewski, M.; Degreif, A.; Rossmann, H.; Canisius, A.; Lackner, K.J. Impact of Glutathione Peroxidase-1 Deficiency on Macrophage Foam Cell Formation and Proliferation: Implications for Atherogenesis. PLoS ONE 2013, 8, e72063. [Google Scholar] [CrossRef] [PubMed]
- Poznyak, A.; Grechko, A.V.; Poggio, P.; Myasoedova, V.A.; Alfieri, V.; Orekhov, A.N. The Diabetes Mellitus–Atherosclerosis Connection: The Role of Lipid and Glucose Metabolism and Chronic Inflammation. Int. J. Mol. Sci. 2020, 21, 1835. [Google Scholar] [CrossRef] [PubMed]
- Liao, J.K. Linking endothelial dysfunction with endothelial cell activation. J. Clin. Investig. 2013, 123, 540–541. [Google Scholar] [CrossRef]
- Mofidi, R.; Crotty, T.B.; McCarthy, P.; Sheehan, S.J.; Mehigan, D.; Keaveny, T.V. Association between plaque instability, angiogenesis and symptomatic carotid occlusive disease. Br. J. Surg. 2001, 88, 945–950. [Google Scholar] [CrossRef]
- Stary, H.C.; Chandler, A.B.; Glagov, S.; Guyton, J.R.; Insull, W.; Rosenfeld, M.E.; Schaffer, S.A.; Schwartz, C.J.; Wagner, W.D.; Wissler, R.W. A definition of initial, fatty streak, and intermediate lesions of atherosclerosis. A report from the Committee on Vascular Lesions of the Council on Arteriosclerosis, American Heart Association. Circulation 1994, 89, 2462–2478. [Google Scholar] [CrossRef]
- Jiang, H.; Zhou, Y.; Nabavi, S.M.; Sahebkar, A.; Little, P.J.; Xu, S.; Weng, J.; Ge, J. Mechanisms of Oxidized LDL-Mediated Endothelial Dysfunction and Its Consequences for the Development of Atherosclerosis. Front. Cardiovasc. Med. 2022, 9, 925923. [Google Scholar] [CrossRef]
- Levitan, I.; Volkov, S.; Subbaiah, P.V. Oxidized LDL: Diversity, patterns of recognition, and pathophysiology. Antioxid. Redox Signal. 2010, 13, 39–75. [Google Scholar] [CrossRef]
- Poznyak, A.V.; Nikiforov, N.G.; Markin, A.M.; Kashirskikh, D.A.; Myasoedova, V.A.; Gerasimova, E.V.; Orekhov, A.N. Overview of OxLDL and Its Impact on Cardiovascular Health: Focus on Atherosclerosis. Front. Pharmacol. 2021, 11, 613780. [Google Scholar] [CrossRef]
- Choi, H.Y.; Rahmani, M.; Wong, B.W.; Allahverdian, S.; McManus, B.M.; Pickering, J.G.; Chan, T.; Francis, G.A. ATP-binding cassette transporter A1 expression and apolipoprotein A-I binding are impaired in intima-type arterial smooth muscle cells. Circulation 2009, 119, 3223–3231. [Google Scholar] [CrossRef] [PubMed]
- Xu, S.; Ilyas, I.; Little, P.J.; Li, H.; Kamato, D.; Zheng, X.; Luo, S.; Li, Z.; Liu, P.; Han, J.; et al. Endothelial Dysfunction in Atherosclerotic Cardiovascular Diseases and Beyond: From Mechanism to Pharmacotherapies. Pharmacol. Rev. 2021, 73, 924–967. [Google Scholar] [CrossRef]
- Kraiss, L.W.; Geary, R.L.; Mattsson, E.J.; Vergel, S.; Au, Y.T.; Clowes, A.W. Acute Reductions in Blood Flow and Shear Stress Induce Platelet-Derived Growth Factor-A Expression in Baboon Prosthetic Grafts. Circ. Res. 1996, 79, 45–53. [Google Scholar] [CrossRef]
- Watson, M.G.; Byrne, H.M.; Macaskill, C.; Myerscough, M.R. A two-phase model of early fibrous cap formation in atherosclerosis. J. Theor. Biol. 2018, 456, 123–136. [Google Scholar] [CrossRef] [PubMed]
- Jankauskas, S.S.; Kansakar, U.; Varzideh, F.; Wilson, S.; Mone, P.; Lombardi, A.; Gambardella, J.; Santulli, G. Heart failure in diabetes. Metabolism 2021, 125, 154910. [Google Scholar] [CrossRef]
- Wilson, S.; Mone, P.; Kansakar, U.; Jankauskas, S.S.; Donkor, K.; Adebayo, A.; Varzideh, F.; Eacobacci, M.; Gambardella, J.; Lombardi, A.; et al. Diabetes and restenosis. Cardiovasc. Diabetol. 2022, 21, 23. [Google Scholar] [CrossRef]
- Puri, R.; Nissen, S.E.; Ballantyne, C.M.; Barter, P.J.; Chapman, M.J.; Erbel, R.; Libby, P.; Raichlen, J.S.; John, J.S.; Wolski, K.; et al. Factors underlying regression of coronary atheroma with potent statin therapy. Eur. Heart J. 2013, 34, 1818–1825. [Google Scholar] [CrossRef] [PubMed]
- Bentzon, J.F.; Otsuka, F.; Virmani, R.; Falk, E. Mechanisms of Plaque Formation and Rupture. Circ. Res. 2014, 114, 1852–1866. [Google Scholar] [CrossRef]
- Osaki, T.; Ichinose, A. Current views of activating and regulatory mechanisms of blood coagulation. Nihon Rinsho 2014, 72, 1206–1211. [Google Scholar]
- Juricic, S.A.; Stojkovic, S.M.; Galassi, A.R.; Stankovic, G.R.; Orlic, D.N.; Vukcevic, V.D.; Milasinovic, D.G.; Aleksandric, S.B.; Tomasevic, M.V.; Dobric, M.R.; et al. Long-term follow-up of patients with chronic total coronary artery occlusion previously randomized to treatment with optimal drug therapy or percutaneous revascularization of chronic total occlusion (COMET-CTO). Front. Cardiovasc. Med. 2023, 9, 1014664. [Google Scholar] [CrossRef]
- An, Y.; Xu, B.-T.; Wan, S.-R.; Ma, X.-M.; Long, Y.; Xu, Y.; Jiang, Z.-Z. The role of oxidative stress in diabetes mellitus-induced vascular endothelial dysfunction. Cardiovasc. Diabetol. 2023, 22, 237. [Google Scholar] [CrossRef]
- Lin, X.; Xu, Y.; Pan, X.; Xu, J.; Ding, Y.; Sun, X.; Song, X.; Ren, Y.; Shan, P.F. Global, regional, and national burden and trend of diabetes in 195 countries and territories: An analysis from 1990 to 2025. Sci. Rep. 2020, 10, 14790. [Google Scholar] [CrossRef] [PubMed]
- Boden, G.; Rao, A.K. Effects of hyperglycemia and hyperinsulinemia on the tissue factor pathway of blood coagulation. Curr. Diabetes Rep. 2007, 7, 223–227. [Google Scholar] [CrossRef]
- Crisafulli, A.; Pagliaro, P.; Roberto, S.; Cugusi, L.; Mercuro, G.; Lazou, A.; Beauloye, C.; Bertrand, L.; Hausenloy, D.J.; Aragno, M.; et al. Diabetic Cardiomyopathy and Ischemic Heart Disease: Prevention and Therapy by Exercise and Conditioning. Int. J. Mol. Sci. 2020, 21, 2896. [Google Scholar] [CrossRef] [PubMed]
- Lien, C.-F.; Chen, S.-J.; Tsai, M.-C.; Lin, C.-S. Potential Role of Protein Kinase C in the Pathophysiology of Diabetes-Associated Atherosclerosis. Front. Pharmacol. 2021, 12, 716332. [Google Scholar] [CrossRef] [PubMed]
- Durpès, M.-C.; Morin, C.; Paquin-Veillet, J.; Beland, R.; Paré, M.; Guimond, M.-O.; Rekhter, M.; King, G.L.; Geraldes, P. PKC-β activation inhibits IL-18-binding protein causing endothelial dysfunction and diabetic atherosclerosis. Cardiovasc. Res. 2015, 106, 303–313. [Google Scholar] [CrossRef]
- Lachin, J.M.; Nathan, D.M. Understanding Metabolic Memory: The Prolonged Influence of Glycemia During the Diabetes Control and Complications Trial (DCCT) on Future Risks of Complications During the Study of the Epidemiology of Diabetes Interventions and Complications (EDIC). Diabetes Care 2021, 44, 2216–2224. [Google Scholar] [CrossRef]
- Bornfeldt, K.E. Does Elevated Glucose Promote Atherosclerosis? Pros and Cons. Circ. Res. 2016, 119, 190–193. [Google Scholar] [CrossRef]
- Katakami, N. Mechanism of Development of Atherosclerosis and Cardiovascular Disease in Diabetes Mellitus. J. Atheroscler. Thromb. 2018, 25, 27–39. [Google Scholar] [CrossRef]
- Zeadin, M.G.; Petlura, C.I.; Werstuck, G.H. Molecular Mechanisms Linking Diabetes to the Accelerated Development of Atherosclerosis. Can. J. Diabetes 2013, 37, 345–350. [Google Scholar] [CrossRef]
- Kaur, R.; Kaur, M.; Singh, J. Endothelial dysfunction and platelet hyperactivity in type 2 diabetes mellitus: Molecular insights and therapeutic strategies. Cardiovasc. Diabetol. 2018, 17, 121. [Google Scholar] [CrossRef] [PubMed]
- Ye, J.; Li, L.; Wang, M.; Ma, Q.; Tian, Y.; Zhang, Q.; Liu, J.; Li, B.; Zhang, B.; Liu, H.; et al. Diabetes Mellitus Promotes the Development of Atherosclerosis: The Role of NLRP3. Front. Immunol. 2022, 13, 900254. [Google Scholar] [CrossRef]
- Hegab, Z.; Mohamed, T.M.; Stafford, N.; Mamas, M.; Cartwright, E.J.; Oceandy, D. Advanced glycation end products reduce the calcium transient in cardiomyocytes by increasing production of reactive oxygen species and nitric oxide. FEBS Open Bio 2017, 7, 1672–1685. [Google Scholar] [CrossRef] [PubMed]
- Idris-Khodja, N.; Ouerd, S.; Mian, M.O.R.; Gornitsky, J.; Barhoumi, T.; Paradis, P.; Schiffrin, E.L. Endothelin-1 Overexpression Exaggerates Diabetes-Induced Endothelial Dysfunction by Altering Oxidative Stress. Am. J. Hypertens. 2016, 29, 1245–1251. [Google Scholar] [CrossRef]
- Beckman, J.A.; Creager, M.A.; Libby, P. Diabetes and Atherosclerosis. JAMA 2002, 287, 2570–2581. [Google Scholar] [CrossRef] [PubMed]
- Madonna, R.; Balistreri, C.R.; De Rosa, S.; Muscoli, S.; Selvaggio, S.; Selvaggio, G.; Ferdinandy, P.; De Caterina, R. Impact of Sex Differences and Diabetes on Coronary Atherosclerosis and Ischemic Heart Disease. J. Clin. Med. 2019, 8, 98. [Google Scholar] [CrossRef]
- Balteau, M.; Tajeddine, N.; de Meester, C.; Ginion, A.; Rosiers, C.D.; Brady, N.R.; Sommereyns, C.; Horman, S.; Vanoverschelde, J.-L.; Gailly, P.; et al. NADPH oxidase activation by hyperglycaemia in cardiomyocytes is independent of glucose metabolism but requires SGLT1. Cardiovasc. Res. 2011, 92, 237–246. [Google Scholar] [CrossRef]
- Schmidt, A.M.; Du Yan, S.; Wautier, J.-L.; Stern, D. Activation of receptor for advanced glycation end products: A mechanism for chronic vascular dysfunction in diabetic vasculopathy and atherosclerosis. Circ. Res. 1999, 84, 489–497. [Google Scholar] [CrossRef]
- Grainger, D.J.; Wakefield, L.; Bethell, H.W.; Farndale, R.W.; Metcalfe, J.C. Release and activation of platelet latent TGF–β in blood clots during dissolution with plasmin. Nat. Med. 1995, 1, 932–937. [Google Scholar] [CrossRef]
- Mone, P.; Lombardi, A.; Salemme, L.; Cioppa, A.; Popusoi, G.; Varzideh, F.; Pansini, A.; Jankauskas, S.S.; Forzano, I.; Avvisato, R.; et al. Stress Hyperglycemia Drives the Risk of Hospitalization for Chest Pain in Patients With Ischemia and Nonobstructive Coronary Arteries (INOCA). Diabetes Care 2023, 46, 450–454. [Google Scholar] [CrossRef]
- Razani, B.; Chakravarthy, M.V.; Semenkovich, C.F. Insulin Resistance and Atherosclerosis. Endocrinol. Metab. Clin. N. Am. 2008, 37, 603–621. [Google Scholar] [CrossRef] [PubMed]
- Sun, D.; Man, W.; Zhang, L. Roles of Insulin Resistance, Endothelial Dysfunction and Lifestyle Changes in the Development of Cardiovascular Disease in Diabetic Patients. Curr. Drug Targets 2017, 18, 1792–1799. [Google Scholar] [CrossRef]
- Jyotsna, F.; Ahmed, A.; Kumar, K.; Kaur, P.; Chaudhary, M.H.; Kumar, S.; Khan, E.; Khanam, B.; Shah, S.U.; Varrassi, G.; et al. Exploring the Complex Connection Between Diabetes and Cardiovascular Disease: Analyzing Approaches to Mitigate Cardiovascular Risk in Patients with Diabetes. Cureus 2023, 15, e43882. [Google Scholar] [CrossRef] [PubMed]
- Robinson, J.G.; Farnier, M.; Krempf, M.; Bergeron, J.; Luc, G.; Averna, M.; Stroes, E.S.; Langslet, G.; Raal, F.J.; El Shahawy, M.; et al. Efficacy and Safety of Alirocumab in Reducing Lipids and Cardiovascular Events. N. Engl. J. Med. 2015, 372, 1489–1499. [Google Scholar] [CrossRef] [PubMed]
- Tsukushi, S.; Katsuzaki, T.; Aoyama, I.; Takayama, F.; Miyazaki, T.; Shimokata, K.; Niwa, T. Increased erythrocyte 3-DG and AGEs in diabetic hemodialysis patients: Role of the polyol pathway. Kidney Int. 1999, 55, 1970–1976. [Google Scholar] [CrossRef] [PubMed]
- The Emerging Risk Factors Collaboration. Diabetes mellitus, fasting blood glucose concentration, and risk of vascular disease: A collaborative meta-analysis of 102 prospective studies. Lancet 2010, 375, 2215–2222. [Google Scholar] [CrossRef]
- Xiang, Y.; Cheng, J.; Wang, D.; Hu, X.; Xie, Y.; Stitham, J.; Atteya, G.; Du, J.; Tang, W.H.; Lee, S.H.; et al. Hyperglycemia repression of miR-24 coordinately upregulates endothelial cell expression and secretion of von Willebrand factor. Blood 2015, 125, 3377–3387. [Google Scholar] [CrossRef]
- Martín-Timón, I.; Sevillano-Collantes, C.; Segura-Galindo, A.; Del Canizo-Gomez, F.J. Type 2 diabetes and cardiovascular disease: Have all risk factors the same strength? World J. Diabetes 2014, 5, 444–470. [Google Scholar] [CrossRef]
- Giacco, F.; Brownlee, M. Oxidative stress and diabetic complications. Circ. Res. 2010, 107, 1058–1070. [Google Scholar] [CrossRef]
- Inoguchi, T.; Li, P.; Umeda, F.; Yu, H.Y.; Kakimoto, M.; Imamura, M.; Aoki, T.; Etoh, T.; Hashimoto, T.; Naruse, M.; et al. High glucose level and free fatty acid stimulate reactive oxygen species production through protein kinase C--dependent activation of NAD(P)H oxidase in cultured vascular cells. Diabetes 2000, 49, 1939–1945. [Google Scholar] [CrossRef]
- Wen, X.; Ji, Y.; Tang, H.; Jin, Z.; Su, W.; Zhou, L.; Xia, Z.-Y.; Li, L.; Lei, S. Caveolin-3: Therapeutic target for diabetic myocardial ischemia/reperfusion injury. Mol. Med. 2025, 31, 80. [Google Scholar] [CrossRef] [PubMed]
- Potenza, M.A.; Addabbo, F.; Montagnani, M. Vascular actions of insulin with implications for endothelial dysfunction. Am. J. Physiol. Endocrinol. Metab. 2009, 297, E568–E577. [Google Scholar] [CrossRef]
- Møller, S.; Kimer, N.; Hove, J.D.; Barløse, M.; Gluud, L.L. Cardiovascular disease and metabolic dysfunction-associated steatotic liver disease: Pathophysiology and diagnostic aspects. Eur. J. Prev. Cardiol. 2025, zwae306. [Google Scholar] [CrossRef] [PubMed]
- DeFronzo, R.A. Insulin resistance, lipotoxicity, type 2 diabetes and atherosclerosis: The missing links. The Claude Bernard Lecture 2009. Diabetologia 2010, 53, 1270–1287. [Google Scholar] [CrossRef] [PubMed]
- Albers, J.J.; Marcovina, S.M.; Imperatore, G.; Snively, B.M.; Stafford, J.; Fujimoto, W.Y.; Mayer-Davis, E.J.; Petitti, D.B.; Pihoker, C.; Dolan, L.; et al. Prevalence and Determinants of Elevated Apolipoprotein B and Dense Low-Density Lipoprotein in Youths with Type 1 and Type 2 Diabetes. J. Clin. Endocrinol. Metab. 2008, 93, 735–742. [Google Scholar] [CrossRef] [PubMed]
- Summerhill, V.I.; Grechko, A.V.; Yet, S.-F.; Sobenin, I.A.; Orekhov, A.N. The Atherogenic Role of Circulating Modified Lipids in Atherosclerosis. Int. J. Mol. Sci. 2019, 20, 3561. [Google Scholar] [CrossRef]
- Pitanga, T.N.; de Aragão França, L.; Rocha, V.C.J.; Meirelles, T.; Borges, V.M.; Gonçalves, M.S.; Pontes-De-Carvalho, L.C.; Noronha-Dutra, A.A.; Dos-Santos, W.L.C. Neutrophil-derived microparticles induce myeloperoxidase-mediated damage of vascular endothelial cells. BMC Cell Biol. 2014, 15, 21. [Google Scholar] [CrossRef] [PubMed]
- Bazzi, S.; Frangie, C.; Azar, E.; Daher, J. The effect of myeloperoxidase-oxidized LDL on THP-1 macrophage polarization and repolarization. Innate Immun. 2022, 28, 91–103. [Google Scholar] [CrossRef] [PubMed]
- Maguire, E.M.; Pearce, S.W.; Xiao, Q. Foam cell formation: A new target for fighting atherosclerosis and cardiovascular disease. Vasc. Pharmacol. 2019, 112, 54–71. [Google Scholar] [CrossRef]
- Berneis, K.K.; Krauss, R.M. Metabolic origins and clinical significance of LDL heterogeneity. J. Lipid Res. 2002, 43, 1363–1379. [Google Scholar] [CrossRef]
- Olufadi, R.; Byrne, C.D. Effects of VLDL and remnant particles on platelets. Pathophysiol. Haemost. Thromb. 2006, 35, 281–291. [Google Scholar] [CrossRef] [PubMed]
- Le Goff, W.; Bourron, O.; Materne, C.; Galier, S.; Phan, F.; Tan-Chen, S.; Guillas, I.; Hartemann, A.; Salem, J.-E.; Redheuil, A.; et al. Inverse relationship between circulating sphingosine-1-phosphate and precursor species and coronary artery calcification score in type 2 diabetes. Cardiovasc. Diabetol. 2025, 24, 85. [Google Scholar] [CrossRef]
- Li, Y.; Li, D.; Lin, J.; Zhou, L.; Yang, W.; Yin, X.; Xu, C.; Cao, Z.; Wang, Y. Proteomic signatures of type 2 diabetes predict the incidence of coronary heart disease. Cardiovasc. Diabetol. 2025, 24, 120. [Google Scholar] [CrossRef] [PubMed]
- Pizzino, G.; Irrera, N.; Cucinotta, M.; Pallio, G.; Mannino, F.; Arcoraci, V.; Squadrito, F.; Altavilla, D.; Bitto, A. Oxidative Stress: Harms and Benefits for Human Health. Oxid. Med. Cell. Longev. 2017, 2017, 8416763. [Google Scholar] [CrossRef]
- Förstermann, U.; Xia, N.; Li, H. Roles of Vascular Oxidative Stress and Nitric Oxide in the Pathogenesis of Atherosclerosis. Circ. Res. 2017, 120, 713–735. [Google Scholar] [CrossRef] [PubMed]
- Bohula, E.A.; Katz, J.N.; van Diepen, S.; Alviar, C.L.; Baird-Zars, V.M.; Park, J.-G.; Barnett, C.F.; Bhattal, G.; Barsness, G.W.; Burke, J.A.; et al. Demographics, Care Patterns, and Outcomes of Patients Admitted to Cardiac Intensive Care Units. JAMA Cardiol. 2019, 4, 928–935. [Google Scholar] [CrossRef]
- Esper, R.J.; Nordaby, R.A.; Vilariño, J.O.; Paragano, A.; Cacharrón, J.L.; Machado, R.A. Endothelial dysfunction: A comprehensive appraisal. Cardiovasc. Diabetol. 2006, 5, 4. [Google Scholar] [CrossRef]
- Vasu, S.; Kumano, K.; Darden, C.M.; Rahman, I.; Lawrence, M.C.; Naziruddin, B. MicroRNA Signatures as Future Biomarkers for Diagnosis of Diabetes States. Cells 2019, 8, 1533. [Google Scholar] [CrossRef]
- Duan, Y.; Zhang, S.; Xia, Y.; Li, H.; Liu, D.; Du, Y. Identification of novel target genes in exaggerated cardiac remodeling following myocardial infarction in diabetes. Front. Endocrinol. 2025, 16, 1536639. [Google Scholar] [CrossRef]
- Ghafouri-Fard, S.; Shoorei, H.; Taheri, M. Non-coding RNAs are involved in the response to oxidative stress. Biomed. Pharmacother. 2020, 127, 110228. [Google Scholar] [CrossRef]
- Barutta, F.; Bellini, S.; Mastrocola, R.; Bruno, G.; Gruden, G. MicroRNA and Microvascular Complications of Diabetes. Int. J. Endocrinol. 2018, 2018, 6890501. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.-J.; Huang, Y.-L.; Shih, Y.-Y.; Wu, H.-Y.; Peng, C.-T.; Lo, W.-Y. MicroRNA-146a decreases high glucose/thrombin-induced endothelial inflammation by inhibiting NAPDH oxidase 4 expression. Mediat. Inflamm. 2014, 2014, 379537. [Google Scholar] [CrossRef]
- Clempus, R.E.; Griendling, K.K. Reactive oxygen species signaling in vascular smooth muscle cells. Cardiovasc. Res. 2006, 71, 216–225. [Google Scholar] [CrossRef] [PubMed]
- Fish, J.E.; Santoro, M.M.; Morton, S.U.; Yu, S.; Yeh, R.-F.; Wythe, J.D.; Ivey, K.N.; Bruneau, B.G.; Stainier, D.Y.R.; Srivastava, D. miR-126 regulates angiogenic signaling and vascular integrity. Dev. Cell 2008, 15, 272–284. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Icli, B.; Wara, A.K.; Belkin, N.; He, S.; Kobzik, L.; Hunninghake, G.M.; Vera, M.P.; Blackwell, T.S.; Baron, R.M.; et al. MicroRNA-181b regulates NF-κB–mediated vascular inflammation. J. Clin. Investig. 2012, 122, 1973–1990. [Google Scholar] [CrossRef] [PubMed]
- Wu, W.; Xiao, H.; Laguna-Fernandez, A.; Villarreal, G., Jr.; Wang, K.-C.; Geary, G.G.; Zhang, Y.; Wang, W.-C.; Huang, H.-D.; Zhou, J.; et al. Flow-Dependent Regulation of Krüppel-Like Factor 2 Is Mediated by MicroRNA-92a. Circulation 2011, 124, 633–641. [Google Scholar] [CrossRef]
- Wojciechowska, A.; Braniewska, A.; Kozar-Kamińska, K. MicroRNA in cardiovascular biology and disease. Adv. Clin. Exp. Med. 2017, 26, 868–874. [Google Scholar] [CrossRef]
- Grebe, A.; Hoss, F.; Latz, E. NLRP3 Inflammasome and the IL-1 Pathway in Atherosclerosis. Circ. Res. 2018, 122, 1722–1740. [Google Scholar] [CrossRef]
- Rubartelli, A.; Lotze, M.T. Inside, outside, upside down: Damage-associated molecular-pattern molecules (DAMPs) and redox. Trends Immunol. 2007, 28, 429–436. [Google Scholar] [CrossRef]
- Kita, T.; Kume, N.; Minami, M.; Hayashida, K.; Murayama, T.; Sano, H.; Moriwaki, H.; Kataoka, H.; Nishi, E.; Horiuchi, H.; et al. Role of Oxidized LDL in Atherosclerosis. Ann. N. Y. Acad. Sci. 2001, 947, 199–206. [Google Scholar] [CrossRef]
- Wan, Z.; Fan, Y.; Liu, X.; Xue, J.; Han, Z.; Zhu, C.; Wang, X. NLRP3 inflammasome promotes diabetes-induced endothelial inflammation and atherosclerosis. Diabetes Metab. Syndr. Obes. 2019, 12, 1931–1942. [Google Scholar] [CrossRef]
- Verma, S.; Bain, S.C.; Buse, J.B.; Idorn, T.; Rasmussen, S.; Ørsted, D.D.; Nauck, M.A. Occurence of First and Recurrent Major Adverse Cardiovascular Events with Liraglutide Treatment Among Patients with Type 2 Diabetes and High Risk of Cardiovascular Events. JAMA Cardiol. 2019, 4, 1214–1220. [Google Scholar] [CrossRef]
- Karstoft, K.; Pedersen, B.K. Exercise and type 2 diabetes: Focus on metabolism and inflammation. Immunol. Cell Biol. 2016, 94, 146–150. [Google Scholar] [CrossRef] [PubMed]
- Monzo, L.; Kobayashi, M.; Ferreira, J.P.; Lamiral, Z.; Delles, C.; Clark, A.L.; Edelmann, F.; González, A.; Heymans, S.; Pellicori, P.; et al. Echocardiographic and biomarker characteristics in diabetes, coronary artery disease or both: Insights from HOMAGE trial. Cardiovasc. Diabetol. 2025, 24, 111. [Google Scholar] [CrossRef] [PubMed]
- Libby, P. Inflammation in atherosclerosis. Nature 2002, 420, 868–874. [Google Scholar] [CrossRef] [PubMed]
- Badimon, L.; Padró, T.; Vilahur, G. Atherosclerosis, platelets and thrombosis in acute ischaemic heart disease. Eur. Heart J. Acute Cardiovasc. Care 2012, 1, 60–74. [Google Scholar] [CrossRef]
- Brownlee, M. The pathobiology of diabetic complications: A unifying mechanism. Diabetes 2005, 54, 1615–1625. [Google Scholar] [CrossRef]
- Yang, G.; Chen, L. An Update of Microsomal Prostaglandin E Synthase-1 and PGE2 Receptors in Cardiovascular Health and Diseases. Oxidative Med. Cell. Longev. 2016, 2016, 5249086. [Google Scholar] [CrossRef]
- Overgaard, K.S.; Andersen, T.R.; Heinsen, L.J.; Pararajasingam, G.; Mohamed, R.A.; Madsen, F.S.; Biesenbach, I.I.A.; Højlund, K.; Lambrechtsen, J.; Auscher, S.; et al. Pericoronary adipose tissue attenuation predicts compositional plaque changes: A 12-month longitudinal study in individuals with type 2 diabetes without symptoms or known coronary artery disease. Cardiovasc. Diabetol. 2025, 24, 143. [Google Scholar] [CrossRef]
- Drucker, D.J. Mechanisms of Action and Therapeutic Application of Glucagon-like Peptide-1. Cell Metab. 2018, 27, 740–756. [Google Scholar] [CrossRef]
- Chen, S.; Coronel, R.; Hollmann, M.W.; Weber, N.C.; Zuurbier, C.J. Direct cardiac effects of SGLT2 inhibitors. Cardiovasc. Diabetol. 2022, 21, 45. [Google Scholar] [CrossRef]
- Luna-Marco, C.; Iannantuoni, F.; Hermo-Argibay, A.; Devos, D.; Salazar, J.D.; Víctor, V.M.; Rovira-Llopis, S. Cardiovascular benefits of SGLT2 inhibitors and GLP-1 receptor agonists through effects on mitochondrial function and oxidative stress. Free Radic. Biol. Med. 2024, 213, 19–35. [Google Scholar] [CrossRef]
- Deroissart, J.; Porsch, F.; Koller, T.; Binder, C.J. Anti-inflammatory and Immunomodulatory Therapies in Atherosclerosis. In Prevention and Treatment of Atherosclerosis: Improving State-of-the-Art Management and Search for Novel Targets; Springer: Cham, Switzerland, 2022. [Google Scholar]
- Steinberg, B.E.; Grinstein, S. Unconventional Roles of the NADPH Oxidase: Signaling, Ion Homeostasis, and Cell Death. Sci. STKE 2007, 2007, pe11. [Google Scholar] [CrossRef]
- Massberg, S.; Grahl, L.; von Bruehl, M.-L.; Manukyan, D.; Pfeiler, S.; Goosmann, C.; Brinkmann, V.; Lorenz, M.; Bidzhekov, K.; Khandagale, A.B.; et al. Reciprocal coupling of coagulation and innate immunity via neutrophil serine proteases. Nat. Med. 2010, 16, 887–896. [Google Scholar] [CrossRef] [PubMed]
- Adamidis, P.S.; Pantazi, D.; Moschonas, I.C.; Liberopoulos, E.; Tselepis, A.D. Neutrophil Extracellular Traps (NETs) and Atherosclerosis: Does Hypolipidemic Treatment Have an Effect? J. Cardiovasc. Dev. Dis. 2024, 11, 72. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Hong, W.; Wan, M.; Zheng, L. Molecular mechanisms and therapeutic target of NETosis in diseases. MedComm 2022, 3, e162. [Google Scholar] [CrossRef] [PubMed]
- Ridker, P.M.; Howard, C.P.; Walter, V.; Everett, B.; Libby, P.; Hensen, J.; Thuren, T. Effects of Interleukin-1β Inhibition with Canakinumab on Hemoglobin A1c, Lipids, C-Reactive Protein, Interleukin-6, and Fibrinogen. Circulation 2012, 126, 2739–2748. [Google Scholar] [CrossRef]
- Sun, Z.; Zhang, J.; Duan, J.; Wang, Q.; Yun, Z.; Lin, J.; Yang, Y.; Zuo, W.; Wang, Z.; Xiong, X.; et al. Cross-sectional study on the association between the fibrosis-4 index and co-occurring myocardial infarction in Chinese patients with type 2 diabetes mellitus. Front. Endocrinol. 2025, 16, 1551472. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Juricic, S.; Klac, J.; Stojkovic, S.; Tesic, M.; Jovanovic, I.; Aleksandric, S.; Dobric, M.; Zivkovic, S.; Maricic, B.; Simeunovic, D.; et al. Molecular and Pathophysiological Mechanisms Leading to Ischemic Heart Disease in Patients with Diabetes Mellitus. Int. J. Mol. Sci. 2025, 26, 3924. https://doi.org/10.3390/ijms26093924
Juricic S, Klac J, Stojkovic S, Tesic M, Jovanovic I, Aleksandric S, Dobric M, Zivkovic S, Maricic B, Simeunovic D, et al. Molecular and Pathophysiological Mechanisms Leading to Ischemic Heart Disease in Patients with Diabetes Mellitus. International Journal of Molecular Sciences. 2025; 26(9):3924. https://doi.org/10.3390/ijms26093924
Chicago/Turabian StyleJuricic, Stefan, Jovana Klac, Sinisa Stojkovic, Milorad Tesic, Ivana Jovanovic, Srdjan Aleksandric, Milan Dobric, Stefan Zivkovic, Bojan Maricic, Dejan Simeunovic, and et al. 2025. "Molecular and Pathophysiological Mechanisms Leading to Ischemic Heart Disease in Patients with Diabetes Mellitus" International Journal of Molecular Sciences 26, no. 9: 3924. https://doi.org/10.3390/ijms26093924
APA StyleJuricic, S., Klac, J., Stojkovic, S., Tesic, M., Jovanovic, I., Aleksandric, S., Dobric, M., Zivkovic, S., Maricic, B., Simeunovic, D., Lasica, R., Dikic, M., Banovic, M., & Beleslin, B. (2025). Molecular and Pathophysiological Mechanisms Leading to Ischemic Heart Disease in Patients with Diabetes Mellitus. International Journal of Molecular Sciences, 26(9), 3924. https://doi.org/10.3390/ijms26093924