

In Vitro Study of Vitamin D Effects on Immune, Endothelial, and Vascular Smooth Muscle Cells in Chronic Kidney Disease
Abstract
1. Introduction
2. Results
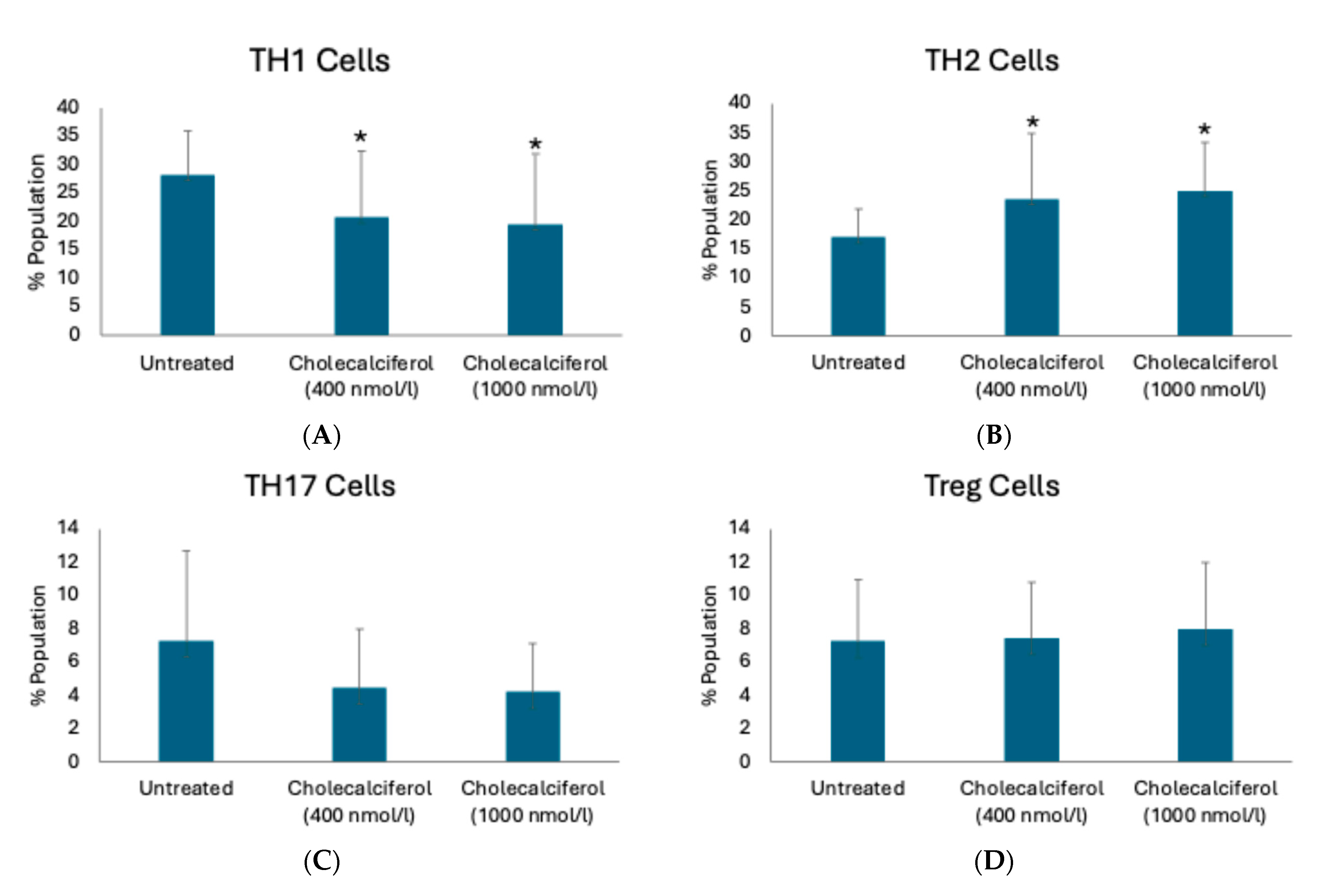
2.1. T-Cell Phenotype in CKD Subjects Is Altered upon Vitamin D Supplementation
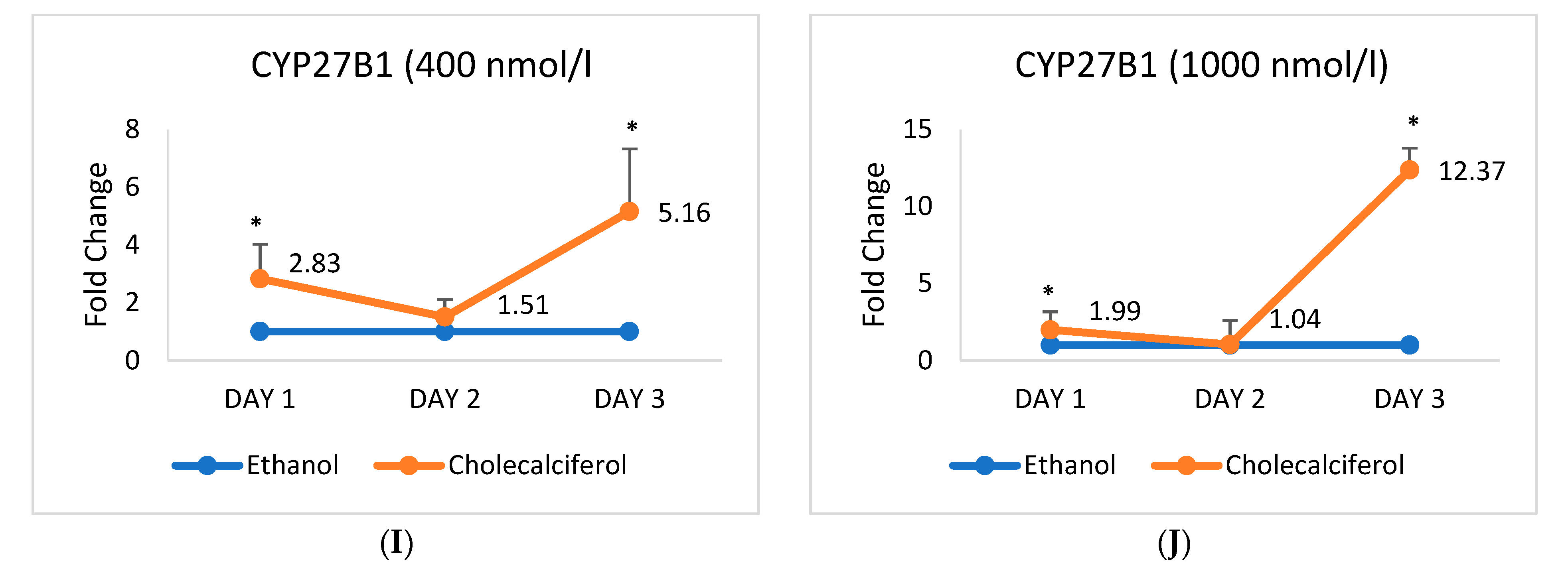
2.2. Expression of mRNA Encoding Vascular Smooth Muscle Cell Markers Alters upon the Cholecalciferol Treatment
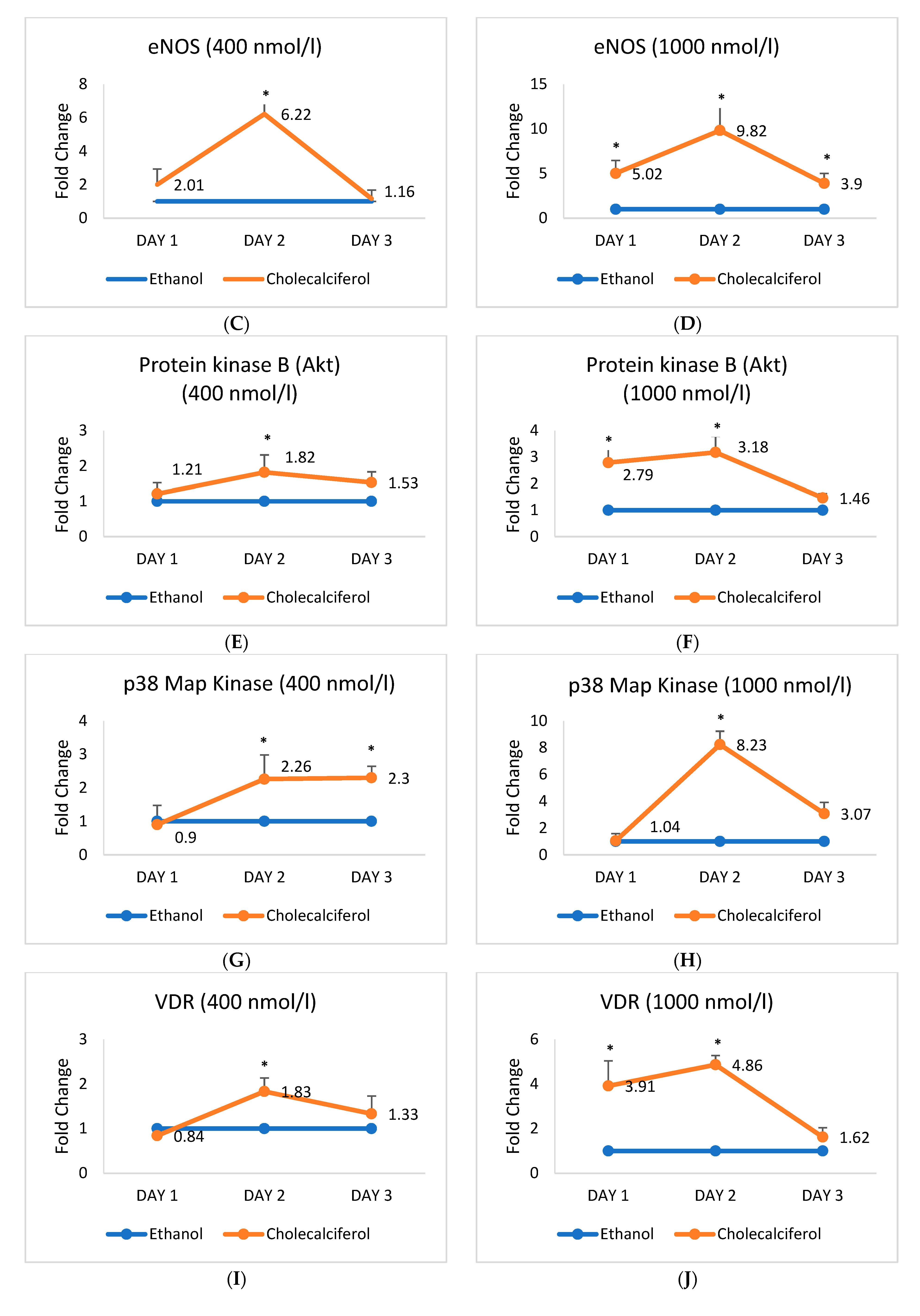
2.3. Expression of mRNA Encoding Endothelial Cell Markers Alters upon the Cholecalciferol Treatment
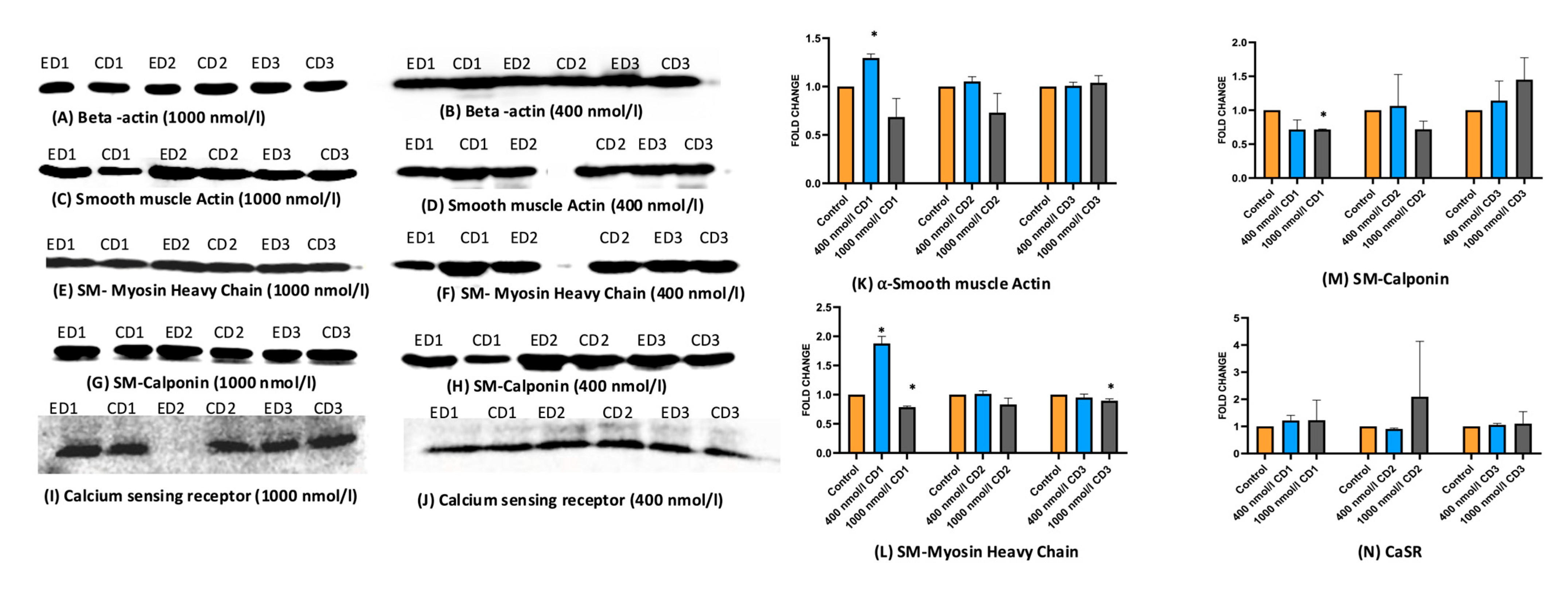
2.4. Expression of Protein Markers of HASMCs and HAECs Alters upon the Cholecalciferol Treatment
3. Discussion
4. Materials and Methods
4.1. Study Design
4.2. Study Population for T-Cell Phenotyping
4.3. PBMC Culture
4.3.1. Flow Cytometry Analysis
4.3.2. Endothelial and Vascular Cell Culture
4.4. mRNA Expression Analysis
4.5. Protein Expression Analysis
4.6. Measurement of Nitrite Concentration in HAEC Cell Supernatants
4.7. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Herzog, C.A.; Asinger, R.W.; Berger, A.K.; Charytan, D.M.; Diez, J.; Hart, R.G.; Eckardt, K.U.; Kasiske, B.L.; McCullough, P.A.; Passman, R.S.; et al. Cardiovascular disease in chronic kidney disease. A clinical update from Kidney Disease: Improving Global Outcomes (KDIGO). Kidney Int. 2011, 80, 572–586. [Google Scholar] [CrossRef] [PubMed]
- Said, S.; Hernandez, G.T. The link between chronic kidney disease and cardiovascular disease. J. Nephropathol. 2014, 3, 99–104. [Google Scholar] [PubMed]
- Saly, D.L.; Eswarappa, M.S.; Street, S.E.; Deshpande, P. Renal cell cancer and chronic kidney disease. Adv. Chronic Kidney. Dis. 2021, 28, 460–468.e461. [Google Scholar] [CrossRef] [PubMed]
- Syed-Ahmed, M.; Narayanan, M. Immune dysfunction and risk of infection in chronic kidney disease. Adv. Chronic Kidney. Dis. 2019, 26, 8–15. [Google Scholar] [CrossRef]
- Kreft, B.; Klouche, M.; Kreft, R.; Kirchner, H.; Sack, K. Low efficiency of active immunization against diphtheria in chronic hemodialysis patients. Kidney Int. 1997, 52, 212–216. [Google Scholar] [CrossRef]
- Matas, A.J.; Simmons, R.L.; Kjellstrand, C.M.; Buselmeier, T.J.; Najarian, J.S. Increased incidence of malignancy during chronic renal failure. Lancet 1975, 1, 883–886. [Google Scholar] [CrossRef]
- Espi, M.; Koppe, L.; Fouque, D.; Thaunat, O. Chronic Kidney Disease-Associated Immune Dysfunctions: Impact of Protein-Bound Uremic Retention Solutes on Immune Cells. Toxins 2020, 12, 300. [Google Scholar] [CrossRef]
- Mansouri, L.; Paulsson, J.M.; Moshfegh, A.; Jacobson, S.H.; Lundahl, J. Leukocyte proliferation and immune modulator production in patients with chronic kidney disease. PLoS ONE 2013, 8, e73141. [Google Scholar] [CrossRef]
- Moore, C.L.; Skolnik-David, H.; Sharp, P.A. Sedimentation analysis of polyadenylation-specific complexes. Mol. Cell. Biol. 1988, 8, 226–233. [Google Scholar]
- Stachowski, J.; Pollok, M.; Burrichter, H.; Spithaler, C.; Baldamus, C.A. Signalling via the TCR/CD3 antigen receptor complex in uremia is limited by the receptors number. Nephron 1993, 64, 369–375. [Google Scholar] [CrossRef]
- Lisowska, K.A.; Debska-Slizien, A.; Jasiulewicz, A.; Heleniak, Z.; Bryl, E.; Witkowski, J.M. Hemodialysis affects phenotype and proliferation of CD4-positive T lymphocytes. J. Clin. Immunol. 2012, 32, 189–200. [Google Scholar] [CrossRef] [PubMed]
- Vanhoutte, P.M.; Zhao, Y.; Xu, A.; Leung, S.W. Thirty Years of Saying NO: Sources, Fate, Actions, and Misfortunes of the Endothelium-Derived Vasodilator Mediator. Circ. Res. 2016, 119, 375–396. [Google Scholar] [CrossRef] [PubMed]
- Vila Cuenca, M.; Hordijk, P.L.; Vervloet, M.G. Most exposed: The endothelium in chronic kidney disease. Nephrol. Dial. Transplant. 2020, 35, 1478–1487. [Google Scholar] [CrossRef] [PubMed]
- Roumeliotis, S.; Mallamaci, F.; Zoccali, C. Endothelial Dysfunction in Chronic Kidney Disease, from Biology to Clinical Outcomes: A 2020 Update. J. Clin. Med. 2020, 9, 2359. [Google Scholar] [CrossRef]
- Bacchetta, J.; Pelletier, S. Vitamin D deficiency is associated with mortality in maintenance dialysis: Moving forward from epidemiology to clinical trials. Nephrol. Dial. Transplant. 2018, 33, 1679–1682. [Google Scholar] [CrossRef]
- Duranton, F.; Rodriguez-Ortiz, M.E.; Duny, Y.; Rodriguez, M.; Daures, J.P.; Argiles, A. Vitamin D treatment and mortality in chronic kidney disease: A systematic review and meta-analysis. Am. J. Nephrol. 2013, 37, 239–248. [Google Scholar] [CrossRef]
- Jayedi, A.; Soltani, S.; Shab-Bidar, S. Vitamin D status and all-cause mortality in patients with chronic kidney disease: A systematic review and dose-response meta-analysis. J. Clin. Endocrinol. Metab. 2017, 102, 2136–2145. [Google Scholar] [CrossRef]
- Pilz, S.; Iodice, S.; Zittermann, A.; Grant, W.B.; Gandini, S. Vitamin D status and mortality risk in CKD: A meta-analysis of prospective studies. Am. J. Kidney Dis. 2011, 58, 374–382. [Google Scholar] [CrossRef]
- Umar, M.; Sastry, K.S.; Chouchane, A.I. Role of Vitamin D Beyond the Skeletal Function: A Review of the Molecular and Clinical Studies. Int. J. Mol. Sci. 2018, 19, 1618. [Google Scholar] [CrossRef]
- Panichi, V.; Migliori, M.; Taccola, D.; Filippi, C.; De Nisco, L.; Giovannini, L.; Palla, R.; Tetta, C.; Camussi, G. Effects of 1,25(OH)2D3 in experimental mesangial proliferative nephritis in rats. Kidney Int. 2001, 60, 87–95. [Google Scholar] [CrossRef]
- Prietl, B.; Pilz, S.; Wolf, M.; Tomaschitz, A.; Obermayer-Pietsch, B.; Graninger, W.; Pieber, T.R. Vitamin D supplementation and regulatory T cells in apparently healthy subjects: Vitamin D treatment for autoimmune diseases? Isr. Med. Assoc. J. 2010, 12, 136–139. [Google Scholar] [PubMed]
- Ardalan, M.R.; Maljaei, H.; Shoja, M.M.; Piri, A.R.; Khosroshahi, H.T.; Noshad, H.; Argani, H. Calcitriol started in the donor, expands the population of CD4+CD25+ T cells in renal transplant recipients. Transplant. Proc. 2007, 39, 951–953. [Google Scholar] [CrossRef] [PubMed]
- Lang, C.L.; Wang, M.H.; Hung, K.Y.; Chiang, C.K.; Lu, K.C. Altered molecular repertoire of immune system by renal dysfunction in the elderly: Is prediction and targeted prevention in the horizon? EPMA J. 2013, 4, 17. [Google Scholar] [CrossRef] [PubMed]
- Hiemstra, T.F.; Lim, K.; Thadhani, R.; Manson, J.E. Vitamin D and Atherosclerotic Cardiovascular Disease. J. Clin. Endocrinol. Metab. 2019, 104, 4033–4050. [Google Scholar] [CrossRef]
- Kumar, V.; Yadav, A.K.; Lal, A.; Kumar, V.; Singhal, M.; Billot, L.; Gupta, K.L.; Banerjee, D.; Jha, V. A Randomized Trial of Vitamin D Supplementation on Vascular Function in CKD. J. Am. Soc. Nephrol. 2017, 28, 3100–3108. [Google Scholar] [CrossRef]
- Kumar, V.; Yadav, A.K.; Singhal, M.; Kumar, V.; Lal, A.; Banerjee, D.; Gupta, K.L.; Jha, V. Vascular function and cholecalciferol supplementation in CKD: A self-controlled case series. J. Steroid Biochem. Mol. Biol. 2018, 180, 19–22. [Google Scholar] [CrossRef]
- Ardalan, M.-R.; Sanadgol, H.; Nasri, H.; Baradaran, A.; Tamadon, M.-R.; Rafieian-Kopaei, M. Impact of vitamin D on the immune system in kidney disease. J. Parathyr. Dis. 2013, 1, 17–20. [Google Scholar]
- Brito, R.B.d.O.; Rebello, J.F.; Grabulosa, C.C.; Pinto, W.; Morales, A.; Elias, R.M.; Moyses, R.M.A.; Dalboni, M.A. 25-vitamin D reduces inflammation in uremic environment. Sci. Rep. 2020, 10, 128. [Google Scholar] [CrossRef]
- Rengarajan, J.; Szabo, S.J.; Glimcher, L.H. Transcriptional regulation of Th1/Th2 polarization. Immunol. Today 2000, 21, 479–483. [Google Scholar] [CrossRef]
- Staeva-Vieira, T.P.; Freedman, L.P. 1,25-dihydroxyvitamin D3 inhibits IFN-gamma and IL-4 levels during in vitro polarization of primary murine CD4+ T cells. J. Immunol. 2002, 168, 1181–1189. [Google Scholar] [CrossRef]
- Correale, J.; Ysrraelit, M.C.; Gaitan, M.I. Immunomodulatory effects of Vitamin D in multiple sclerosis. Brain 2009, 132, 1146–1160. [Google Scholar] [CrossRef] [PubMed]
- Maboshe, W.; Macdonald, H.M.; Wassall, H.; Fraser, W.D.; Tang, J.C.Y.; Fielding, S.; Barker, R.N.; Vickers, M.A.; Ormerod, A.; Thies, F. Low-Dose Vitamin D3 Supplementation Does Not Affect Natural Regulatory T Cell Population but Attenuates Seasonal Changes in T Cell-Produced IFN-gamma: Results From the D-SIRe2 Randomized Controlled Trial. Front. Immunol. 2021, 12, 623087. [Google Scholar] [CrossRef] [PubMed]
- Treiber, G.; Prietl, B.; Frohlich-Reiterer, E.; Lechner, E.; Ribitsch, A.; Fritsch, M.; Rami-Merhar, B.; Steigleder-Schweiger, C.; Graninger, W.; Borkenstein, M.; et al. Cholecalciferol supplementation improves suppressive capacity of regulatory T-cells in young patients with new-onset type 1 diabetes mellitus-A randomized clinical trial. Clin. Immunol. 2015, 161, 217–224. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; DeWispelaere, A.; Dastvan, F.; Osborne, W.R.; Blechner, C.; Windhorst, S.; Daum, G. Smooth Muscle-Alpha Actin Inhibits Vascular Smooth Muscle Cell Proliferation and Migration by Inhibiting Rac1 Activity. PLoS ONE 2016, 11, e0155726. [Google Scholar] [CrossRef]
- Jia, X.; Gu, Y.; Groome, L.J.; Al-Kofahi, M.; Alexander, J.S.; Li, W.; Wang, Y. 1,25(OH)2D3 Induces Placental Vascular Smooth Muscle Cell Relaxation by Phosphorylation of Myosin Phosphatase Target Subunit 1Ser507: Potential Beneficial Effects of Vitamin D on Placental Vasculature in Humans. Biol. Reprod. 2016, 94, 116. [Google Scholar] [CrossRef]
- Ma, R.; Gu, Y.; Zhao, S.; Sun, J.; Groome, L.J.; Wang, Y. Expressions of vitamin D metabolic components VDBP, CYP2R1, CYP27B1, CYP24A1, and VDR in placentas from normal and preeclamptic pregnancies. Am. J. Physiol. Endocrinol. Metab. 2012, 303, E928–E935. [Google Scholar] [CrossRef]
- Molinari, C.; Uberti, F.; Grossini, E.; Vacca, G.; Carda, S.; Invernizzi, M.; Cisari, C. 1α,25-dihydroxycholecalciferol induces nitric oxide production in cultured endothelial cells. Cell Physiol. Biochem. 2011, 27, 661–668. [Google Scholar] [CrossRef]
- Queen, L.R.; Ji, Y.; Xu, B.; Young, L.; Yao, K.; Wyatt, A.W.; Rowlands, D.J.; Siow, R.C.; Mann, G.E.; Ferro, A. Mechanisms underlying beta2-adrenoceptor-mediated nitric oxide generation by human umbilical vein endothelial cells. J. Physiol. 2006, 576, 585–594. [Google Scholar] [CrossRef]
- Janubova, M.; Zitnanova, I. The effects of vitamin D on different types of cells. Steroids 2024, 202, 109350. [Google Scholar] [CrossRef]
- Girgis, C.M.; Mokbel, N.; Cha, K.M.; Houweling, P.J.; Abboud, M.; Fraser, D.R.; Mason, R.S.; Clifton-Bligh, R.J.; Gunton, J.E. The vitamin D receptor (VDR) is expressed in skeletal muscle of male mice and modulates 25-hydroxyvitamin D (25OHD) uptake in myofibers. Endocrinology 2014, 155, 3227–3237. [Google Scholar] [CrossRef]
- Durham, A.L.; Speer, M.Y.; Scatena, M.; Giachelli, C.M.; Shanahan, C.M. Role of smooth muscle cells in vascular calcification: Implications in atherosclerosis and arterial stiffness. Cardiovasc. Res. 2018, 114, 590–600. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Cai, Y.; Yang, F.; Yang, Y.; Cui, Z.; Shi, D.; Bai, R. Vascular smooth muscle cell phenotypic switching in atherosclerosis. Heliyon 2024, 10, e37727. [Google Scholar] [CrossRef] [PubMed]
- Drueke, T.B.; Massy, Z.A. Role of vitamin D in vascular calcification: Bad guy or good guy? Nephrol. Dial. Transpl. 2012, 27, 1704–1707. [Google Scholar] [CrossRef] [PubMed]
- Torremade, N.; Bozic, M.; Panizo, S.; Barrio-Vazquez, S.; Fernandez-Martin, J.L.; Encinas, M.; Goltzman, D.; Arcidiacono, M.V.; Fernandez, E.; Valdivielso, J.M. Vascular Calcification Induced by Chronic Kidney Disease Is Mediated by an Increase of 1alpha-Hydroxylase Expression in Vascular Smooth Muscle Cells. J. Bone Min. Res. 2016, 31, 1865–1876. [Google Scholar] [CrossRef]
- Levey, A.S.; Stevens, L.A.; Schmid, C.H.; Zhang, Y.L.; Castro, A.F., 3rd; Feldman, H.I.; Kusek, J.W.; Eggers, P.; Van Lente, F.; Greene, T.; et al. A new equation to estimate glomerular filtration rate. Ann. Intern. Med. 2009, 150, 604–612. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| T-Cell Subpopulation | T-Cell Marker | Untreated (Cell Percentage) | 400 nmol/L Cholecalciferol (Cell Percentage) | p-Value | 1000 nmol/L Cholecalciferol (Cell Percentage) | p-Value |
|---|---|---|---|---|---|---|
| TH1 cells | CD3+CD4+CXCR3+ | 28.9 ± 18.8 | 23.1 ± 14.1 | 0.42 | 23.5 ± 15.9 | 0.55 |
| CD3+CD4+Tbet+ | 26.6 ± 8.6 | 23.8 ± 9.6 | 0.31 | 25.0 ± 14.2 | 0.72 | |
| CD3+CD4+IFNγ+ | 28.2 ± 7.7 | 20.7 ± 11.7 | 0.04 | 19.5 ± 12.5 | 0.02 | |
| TH2 cells | CD3+CD4+IL4+ | 31.8 ± 8.6 | 30.1 ± 11.9 | 0.38 | 31.9 ± 9.7 | 0.78 |
| CD3+CD4+STAT6+ | 17.1 ± 4.8 | 23.6 ± 11.3 | 0.02 | 25.0 ± 8.3 | 0.002 | |
| CD3+CD4+GATA3+ | 27.7 ± 9.9 | 22.3 ± 5.7 | 0.07 | 23.3 ± 6.4 | 0.18 | |
| CD3+CD4+CCR4+CCR6- | 18.3 ± 5.4 | 24.1 ± 7.0 | 0.02 | 26.0 ± 9.8 | 0.009 | |
| TH17 cells | CD3+CD4+IL17A+ | 7.3 ± 5.4 | 4.5 ± 3.5 | 0.06 | 4.2 ± 2.8 | 0.06 |
| CD3+CD4+RORγt+ | 7.9 ± 7.0 | 4.1 ± 4.1 | 0.13 | 3.9 ± 3.8 | 0.07 | |
| CD3+CD4+CCR4+CCR6+ | 15.4 ± 9.9 | 8.2 ± 8.3 | 0.02 | 10.0 ± 8.3 | 0.14 | |
| Treg cells | CD3+CD4+CD25+ | 14.8 ± 4.1 | 13.6 ± 6.2 | 0.53 | 14.0 ± 8.0 | 0.41 |
| CD3+CD4+CD25+CD127lowFOXP3+ | 3.00 ± 1.2 | 3.3 ± 2.4 | 0.88 | 3.1 ± 1.7 | 0.79 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kamboj, K.; Kumar, V.; Yadav, A.K. In Vitro Study of Vitamin D Effects on Immune, Endothelial, and Vascular Smooth Muscle Cells in Chronic Kidney Disease. Int. J. Mol. Sci. 2025, 26, 3967. https://doi.org/10.3390/ijms26093967
Kamboj K, Kumar V, Yadav AK. In Vitro Study of Vitamin D Effects on Immune, Endothelial, and Vascular Smooth Muscle Cells in Chronic Kidney Disease. International Journal of Molecular Sciences. 2025; 26(9):3967. https://doi.org/10.3390/ijms26093967
Chicago/Turabian StyleKamboj, Kajal, Vivek Kumar, and Ashok Kumar Yadav. 2025. "In Vitro Study of Vitamin D Effects on Immune, Endothelial, and Vascular Smooth Muscle Cells in Chronic Kidney Disease" International Journal of Molecular Sciences 26, no. 9: 3967. https://doi.org/10.3390/ijms26093967
APA StyleKamboj, K., Kumar, V., & Yadav, A. K. (2025). In Vitro Study of Vitamin D Effects on Immune, Endothelial, and Vascular Smooth Muscle Cells in Chronic Kidney Disease. International Journal of Molecular Sciences, 26(9), 3967. https://doi.org/10.3390/ijms26093967