
A Paradigmatic Case of Genetic Overlap Between Neurodevelopment Disorders and Schizophrenia Aligning with the Neurodevelopmental Continuum Hypothesis

 , , , , ,
, , , , ,  and
and
Abstract
:1. Introduction
2. Case Presentation
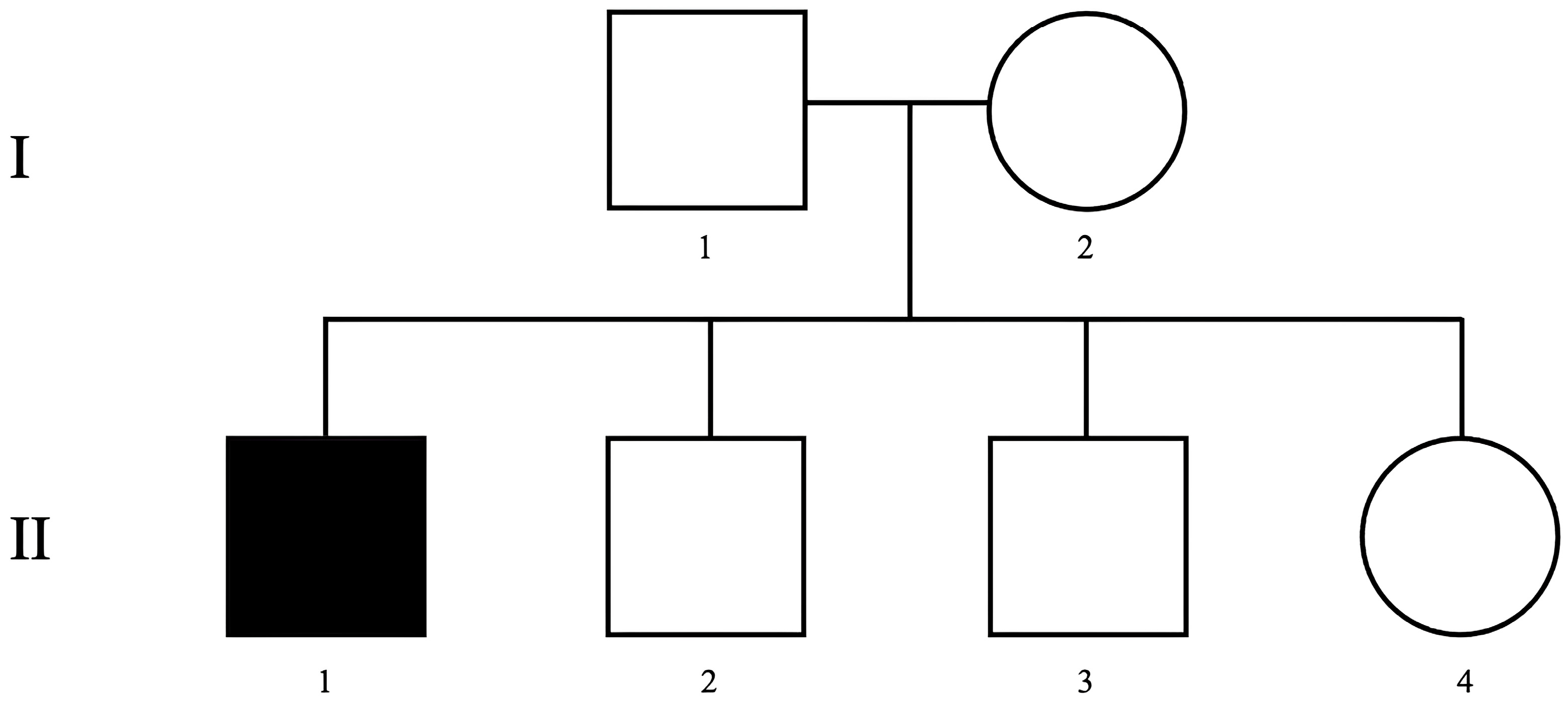
2.1. Patient Report
2.2. Molecular Analysis
2.3. Results of Genetic Testing
3. Discussion
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Kahn, R.S.; Sommer, I.E.; Murray, R.M.; Meyer-Lindenberg, A.; Weinberger, D.R.; Cannon, T.D.; O’Donovan, M.; Correll, C.U.; Kane, J.M.; van Os, J.; et al. Schizophrenia. Nat. Rev. Dis. Primers 2015, 1, 15067. [Google Scholar] [CrossRef] [PubMed]
- Schultz, S.H.; North, S.W.; Shields, C.G. Schizophrenia: A Review. Am. Fam. Physician 2007, 75, 1821–1829. [Google Scholar]
- Schizophrenia Working Group of the Psychiatric Genomics Consortium Biological Insights from 108 Schizophrenia-Associated Genetic Loci. Nature 2014, 511, 421–427. [CrossRef]
- Redon, R.; Ishikawa, S.; Fitch, K.R.; Feuk, L.; Perry, G.H.; Andrews, T.D.; Fiegler, H.; Shapero, M.H.; Carson, A.R.; Chen, W.; et al. Global Variation in Copy Number in the Human Genome. Nature 2006, 444, 444–454. [Google Scholar] [CrossRef]
- Rutkowski, T.P.; Schroeder, J.P.; Gafford, G.M.; Warren, S.T.; Weinshenker, D.; Caspary, T.; Mulle, J.G. Unraveling the Genetic Architecture of Copy Number Variants Associated with Schizophrenia and Other Neuropsychiatric Disorders. J. Neurosci. Res. 2017, 95, 1144–1160. [Google Scholar] [CrossRef] [PubMed]
- Lee, P.H.; Feng, Y.-C.A.; Smoller, J.W. Pleiotropy and Cross-Disorder Genetics Among Psychiatric Disorders. Biol. Psychiatry 2021, 89, 20–31. [Google Scholar] [CrossRef]
- Lombardo, B.; Esposito, D.; Iossa, S.; Vitale, A.; Verdesca, F.; Perrotta, C.; Di Leo, L.; Costa, V.; Pastore, L.; Franzé, A. Intragenic Deletion in MACROD2: A Family with Complex Phenotypes Including Microcephaly, Intellectual Disability, Polydactyly, Renal and Pancreatic Malformations. Cytogenet. Genome Res. 2019, 158, 25–31. [Google Scholar] [CrossRef]
- Ranieri, A.; La Monica, I.; Di Iorio, M.R.; Lombardo, B.; Pastore, L. Genetic Alterations in a Large Population of Italian Patients Affected by Neurodevelopmental Disorders. Genes 2024, 15, 427. [Google Scholar] [CrossRef] [PubMed]
- Lombardo, B.; Ceglia, C.; Tarsitano, M.; Pierucci, I.; Salvatore, F.; Pastore, L. Identification of a Deletion in the NDUFS4 Gene Using Array-Comparative Genomic Hybridization in a Patient with Suspected Mitochondrial Respiratory Disease. Gene 2014, 535, 376–379. [Google Scholar] [CrossRef]
- Sanna, V.; Ceglia, C.; Tarsitano, M.; Lombardo, B.; Coppola, A.; Zarrilli, F.; Castaldo, G.; Di Minno, G. Aberrant F8 Gene Intron 1 Inversion with Concomitant Duplication and Deletion in a Severe Hemophilia A Patient from Southern Italy. J. Thromb. Haemost. 2013, 11, 195–197. [Google Scholar] [CrossRef]
- Rees, E.; Creeth, H.D.J.; Hwu, H.-G.; Chen, W.J.; Tsuang, M.; Glatt, S.J.; Rey, R.; Kirov, G.; Walters, J.T.R.; Holmans, P.; et al. Schizophrenia, Autism Spectrum Disorders and Developmental Disorders Share Specific Disruptive Coding Mutations. Nat. Commun. 2021, 12, 5353. [Google Scholar] [CrossRef] [PubMed]
- Owen, M.J.; O’Donovan, M.C. Schizophrenia and the Neurodevelopmental Continuum: Evidence from Genomics. World Psychiatry 2017, 16, 227–235. [Google Scholar] [CrossRef] [PubMed]
- American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders; American Psychiatric Association Publishing: Washington, DC, USA, 2022; ISBN 0-89042-575-2. [Google Scholar]
- Cooper, S.-A.; Smiley, E.; Morrison, J.; Allan, L.; Williamson, A.; Finlayson, J.; Jackson, A.; Mantry, D. Psychosis and Adults with Intellectual Disabilities. Prevalence, Incidence, and Related Factors. Soc. Psychiatry Psychiatr. Epidemiol. 2007, 42, 530–536. [Google Scholar] [CrossRef] [PubMed]
- Ranieri, A.; Veneruso, I.; La Monica, I.; Pascale, M.; Pastore, L.; D’Argenio, V.; Lombardo, B. Combined ACGH and Exome Sequencing Analysis Improves Autism Spectrum Disorders Diagnosis: A Case Report. Medicina 2022, 58, 522. [Google Scholar] [CrossRef]
- Gillentine, M.A.; Schaaf, C.P. The Human Clinical Phenotypes of Altered CHRNA7 Copy Number. Biochem. Pharmacol. 2015, 97, 352–362. [Google Scholar] [CrossRef]
- Felix, R.A.; Chavez, V.A.; Novicio, D.M.; Morley, B.J.; Portfors, C.V. Nicotinic Acetylcholine Receptor Subunit α 7 -Knockout Mice Exhibit Degraded Auditory Temporal Processing. J. Neurophysiol. 2019, 122, 451–465. [Google Scholar] [CrossRef]
- Budisteanu, M.; Papuc, S.M.; Streata, I.; Cucu, M.; Pirvu, A.; Serban-Sosoi, S.; Erbescu, A.; Andrei, E.; Iliescu, C.; Ioana, D.; et al. The Phenotypic Spectrum of 15q13.3 Region Duplications: Report of 5 Patients. Genes 2021, 12, 1025. [Google Scholar] [CrossRef]
- Yin, J.; Chen, W.; Yang, H.; Xue, M.; Schaaf, C.P. Chrna7 Deficient Mice Manifest No Consistent Neuropsychiatric and Behavioral Phenotypes. Sci. Rep. 2017, 7, 39941. [Google Scholar] [CrossRef]
- Antony, I.; Narasimhan, M.; Shen, R.; Prakasam, R.; Kaushik, K.; Chapman, G.; Kroll, K.L. Duplication Versus Deletion Through the Lens of 15q13.3: Clinical and Research Implications of Studying Copy Number Variants Associated with Neuropsychiatric Disorders in Induced Pluripotent Stem Cell-Derived Neurons. Stem Cell Rev. Rep. 2023, 19, 639–650. [Google Scholar] [CrossRef]
- Botto, L.D.; May, K.; Fernhoff, P.M.; Correa, A.; Coleman, K.; Rasmussen, S.A.; Merritt, R.K.; O’Leary, L.A.; Wong, L.-Y.; Elixson, E.M.; et al. A Population-Based Study of the 22q11.2 Deletion: Phenotype, Incidence, and Contribution to Major Birth Defects in the Population. Pediatrics 2003, 112, 101–107. [Google Scholar] [CrossRef]
- Shprintzen, R.J. Velo-cardio-facial Syndrome: 30 Years of Study. Dev. Disabil. Res. Rev. 2008, 14, 3–10. [Google Scholar] [CrossRef] [PubMed]
- Leader, G.; Curtin, A.; Shprintzen, R.J.; Whelan, S.; Coyne, R.; Mannion, A. Adaptive Living Skills, Sleep Problems, and Mental Health Disorders in Adults with 22q11.21 Deletion Syndrome. Res. Dev. Disabil. 2023, 136, 104491. [Google Scholar] [CrossRef] [PubMed]
- Fung, W.L.A.; Butcher, N.J.; Costain, G.; Andrade, D.M.; Boot, E.; Chow, E.W.C.; Chung, B.; Cytrynbaum, C.; Faghfoury, H.; Fishman, L.; et al. Practical Guidelines for Managing Adults with 22q11.2 Deletion Syndrome. Genet. Med. 2015, 17, 599–609. [Google Scholar] [CrossRef]
- Fung, W.L.A.; McEvilly, R.; Fong, J.; Silversides, C.; Chow, E.; Bassett, A. Elevated Prevalence of Generalized Anxiety Disorder in Adults With 22q11.2 Deletion Syndrome. Am. J. Psychiatry 2010, 167, 998. [Google Scholar] [CrossRef]
- Gothelf, D.; Frisch, A.; Munitz, H.; Rockah, R.; Laufer, N.; Mozes, T.; Hermesh, H.; Weizman, A.; Frydman, M. Clinical Characteristics of Schizophrenia Associated with Velo-Cardio-Facial Syndrome. Schizophr. Res. 1999, 35, 105–112. [Google Scholar] [CrossRef] [PubMed]
- Tang, S.X.; Yi, J.J.; Calkins, M.E.; Whinna, D.A.; Kohler, C.G.; Souders, M.C.; McDonald-McGinn, D.M.; Zackai, E.H.; Emanuel, B.S.; Gur, R.C.; et al. Psychiatric Disorders in 22q11.2 Deletion Syndrome Are Prevalent but Undertreated. Psychol. Med. 2014, 44, 1267–1277. [Google Scholar] [CrossRef]
- Schneider, M.; Debbané, M.; Bassett, A.S.; Chow, E.W.C.; Fung, W.L.A.; van den Bree, M.B.M.; Owen, M.; Murphy, K.C.; Niarchou, M.; Kates, W.R.; et al. Psychiatric Disorders From Childhood to Adulthood in 22q11.2 Deletion Syndrome: Results From the International Consortium on Brain and Behavior in 22q11.2 Deletion Syndrome. Am. J. Psychiatry 2014, 171, 627–639. [Google Scholar] [CrossRef]
- Qin, X.; Chen, J.; Zhou, T. 22q11.2 Deletion Syndrome and Schizophrenia. Acta Biochim. Biophys. Sin. 2020, 52, 1181–1190. [Google Scholar] [CrossRef]
- Shifman, S.; Bronstein, M.; Sternfeld, M.; Pisanté-Shalom, A.; Lev-Lehman, E.; Weizman, A.; Reznik, I.; Spivak, B.; Grisaru, N.; Karp, L.; et al. A Highly Significant Association between a COMT Haplotype and Schizophrenia. Am. J. Hum. Genet. 2002, 71, 1296–1302. [Google Scholar] [CrossRef]
- Michaelovsky, E.; Gothelf, D.; Korostishevsky, M.; Frisch, A.; Burg, M.; Carmel, M.; Steinberg, T.; Inbar, D.; Apter, A.; Weizman, A. Association between a Common Haplotype in the COMT Gene Region and Psychiatric Disorders in Individuals with 22q11.2DS. Int. J. Neuropsychopharmacol. 2008, 11, 351–363. [Google Scholar] [CrossRef]
- Zinkstok, J.; Schmitz, N.; Van Amelsvoort, T.; Moeton, M.; Baas, F.; Linszen, D. Genetic Variation in COMT and PRODH Is Associated with Brain Anatomy in Patients with Schizophrenia. Genes. Brain Behav. 2008, 7, 61–69. [Google Scholar] [CrossRef]
- Liu, H.; Heath, S.C.; Sobin, C.; Roos, J.L.; Galke, B.L.; Blundell, M.L.; Lenane, M.; Robertson, B.; Wijsman, E.M.; Rapoport, J.L.; et al. Genetic Variation at the 22q11 PRODH2/DGCR6 Locus Presents an Unusual Pattern and Increases Susceptibility to Schizophrenia. Proc. Natl. Acad. Sci. USA 2002, 99, 3717–3722. [Google Scholar] [CrossRef] [PubMed]
- Merico, D.; Costain, G.; Butcher, N.J.; Warnica, W.; Ogura, L.; Alfred, S.E.; Brzustowicz, L.M.; Bassett, A.S. MicroRNA Dysregulation, Gene Networks, and Risk for Schizophrenia in 22q11.2 Deletion Syndrome. Front. Neurol. 2014, 5, 238. [Google Scholar] [CrossRef] [PubMed]
- van Beveren, N.J.M.; Krab, L.C.; Swagemakers, S.; Buitendijk, G.; Boot, E.; van der Spek, P.; Elgersma, Y.; van Amelsvoort, T.A.M.J. Functional Gene-Expression Analysis Shows Involvement of Schizophrenia-Relevant Pathways in Patients with 22q11 Deletion Syndrome. PLoS ONE 2012, 7, e33473. [Google Scholar] [CrossRef]
- Squarcione, C.; Torti, M.C.; Di Fabio, F.; Biondi, M. 22q11 Deletion Syndrome: A Review of the Neuropsychiatric Features and Their Neurobiological Basis. Neuropsychiatr. Dis. Treat. 2013, 9, 1873–1884. [Google Scholar] [CrossRef]
- Meechan, D.W.; Maynard, T.M.; Wu, Y.; Gopalakrishna, D.; Lieberman, J.A.; LaMantia, A.-S. Gene Dosage in the Developing and Adult Brain in a Mouse Model of 22q11 Deletion Syndrome. Mol. Cell. Neurosci. 2006, 33, 412–428. [Google Scholar] [CrossRef]
- Williams, H.J.; Owen, M.J.; O’Donovan, M.C. Is COMT a Susceptibility Gene for Schizophrenia? Schizophr. Bull. 2007, 33, 635–641. [Google Scholar] [CrossRef]
- Gogos, J.; Santha, M.; Takacs, Z.; Beck, K.D.; Luine, V.; Lucas, L.R.; Nadler, J.V.; Karayiorgou, M. The Gene Encoding Proline Dehydrogenase Modulates Sensorimotor Gating in Mice. Nat. Genet. 1999, 21, 434–439. [Google Scholar] [CrossRef]
- Lang, U.E.; Puls, I.; Müller, D.J.; Strutz-Seebohm, N.; Gallinat, J. Molecular Mechanisms of Schizophrenia. Cell. Physiol. Biochem. 2007, 20, 687–702. [Google Scholar] [CrossRef]
- Renick, S.E.; Kleven, D.T.; Chan, J.; Stenius, K.; Milner, T.A.; Pickel, V.M.; Fremeau, R.T. The Mammalian Brain High-Affinity l-Proline Transporter Is Enriched Preferentially in Synaptic Vesicles in a Subpopulation of Excitatory Nerve Terminals in Rat Forebrain. J. Neurosci. 1999, 19, 21–33. [Google Scholar] [CrossRef]
- Raux, G.; Bumsel, E.; Hecketsweiler, B.; van Amelsvoort, T.; Zinkstok, J.; Manouvrier-Hanu, S.; Fantini, C.; Brévière, G.-M.M.; Di Rosa, G.; Pustorino, G.; et al. Involvement of Hyperprolinemia in Cognitive and Psychiatric Features of the 22q11 Deletion Syndrome. Hum. Mol. Genet. 2007, 16, 83–91. [Google Scholar] [CrossRef]
- Zarchi, O.; Carmel, M.; Avni, C.; Attias, J.; Frisch, A.; Michaelovsky, E.; Patya, M.; Green, T.; Weinberger, R.; Weizman, A.; et al. Schizophrenia-like Neurophysiological Abnormalities in 22q11.2 Deletion Syndrome and Their Association to COMT and PRODH Genotypes. J. Psychiatr. Res. 2013, 47, 1623–1629. [Google Scholar] [CrossRef]
- Ren, D.; Luo, B.; Chen, P.; Yu, L.; Xiong, M.; Fu, Z.; Zhou, T.; Chen, W.-B.; Fei, E. DiGeorge Syndrome Critical Region Gene 2 (DGCR2), a Schizophrenia Risk Gene, Regulates Dendritic Spine Development through Cell Adhesion. Cell Biosci. 2023, 13, 134. [Google Scholar] [CrossRef] [PubMed]
- Shifman, S.; Levit, A.; Chen, M.-L.; Chen, C.-H.; Bronstein, M.; Weizman, A.; Yakir, B.; Navon, R.; Darvasi, A. A Complete Genetic Association Scan of the 22q11 Deletion Region and Functional Evidence Reveal an Association between DGCR2 and Schizophrenia. Hum. Genet. 2006, 120, 160–170. [Google Scholar] [CrossRef]
- Molinard-Chenu, A.; Dayer, A. The Candidate Schizophrenia Risk Gene DGCR2 Regulates Early Steps of Corticogenesis. Biol. Psychiatry 2018, 83, 692–706. [Google Scholar] [CrossRef]
- Forstner, A.J.; Degenhardt, F.; Schratt, G.; Nöthen, M.M. MicroRNAs as the Cause of Schizophrenia in 22q11.2 Deletion Carriers, and Possible Implications for Idiopathic Disease: A Mini-Review. Front. Mol. Neurosci. 2013, 6, 47. [Google Scholar] [CrossRef]
- Stark, K.L.; Xu, B.; Bagchi, A.; Lai, W.-S.; Liu, H.; Hsu, R.; Wan, X.; Pavlidis, P.; Mills, A.A.; Karayiorgou, M.; et al. Altered Brain MicroRNA Biogenesis Contributes to Phenotypic Deficits in a 22q11-Deletion Mouse Model. Nat. Genet. 2008, 40, 751–760. [Google Scholar] [CrossRef] [PubMed]
- Schofield, C.M.; Hsu, R.; Barker, A.J.; Gertz, C.C.; Blelloch, R.; Ullian, E.M. Monoallelic Deletion of the MicroRNA Biogenesis Gene Dgcr8 Produces Deficits in the Development of Excitatory Synaptic Transmission in the Prefrontal Cortex. Neural Dev. 2011, 6, 11. [Google Scholar] [CrossRef] [PubMed]
- Harrington, J.; Wheway, G.; Willaime-Morawek, S.; Gibson, J.; Walters, Z.S. Pathogenic KDM5B Variants in the Context of Developmental Disorders. Biochim. Biophys. Acta (BBA)-Gene Regul. Mech. 2022, 1865, 194848. [Google Scholar] [CrossRef]
- Vallianatos, C.N.; Iwase, S. Disrupted Intricacy of Histone H3K4 Methylation in Neurodevelopmental Disorders. Epigenomics 2015, 7, 503–519. [Google Scholar] [CrossRef]
- Chen, K.; Luan, X.; Liu, Q.; Wang, J.; Chang, X.; Snijders, A.M.; Mao, J.-H.; Secombe, J.; Dan, Z.; Chen, J.-H.; et al. Drosophila Histone Demethylase KDM5 Regulates Social Behavior through Immune Control and Gut Microbiota Maintenance. Cell Host Microbe 2019, 25, 537–552.e8. [Google Scholar] [CrossRef] [PubMed]
- Gilbert, J.; Man, H.-Y. Fundamental Elements in Autism: From Neurogenesis and Neurite Growth to Synaptic Plasticity. Front. Cell Neurosci. 2017, 11, 359. [Google Scholar] [CrossRef] [PubMed]
- Peykov, S.; Berkel, S.; Schoen, M.; Weiss, K.; Degenhardt, F.; Strohmaier, J.; Weiss, B.; Proepper, C.; Schratt, G.; Nöthen, M.M.; et al. Identification and Functional Characterization of Rare SHANK2 Variants in Schizophrenia. Mol. Psychiatry 2015, 20, 1489–1498. [Google Scholar] [CrossRef]
- Bergen, S.E.; Ploner, A.; Howrigan, D.; CNV Analysis Group and the Schizophrenia Working Group of the Psychiatric Genomics Consortium; O’Donovan, M.C.; Smoller, J.W.; Sullivan, P.F.; Sebat, J.; Neale, B.; Kendler, K.S. Joint Contributions of Rare Copy Number Variants and Common SNPs to Risk for Schizophrenia. Am. J. Psychiatry 2019, 176, 29–35. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
| Clinical Characteristics | Details |
|---|---|
| Age at evaluation | 32 |
| Sex | Male |
| Referred diagnosis | Treatment- resistant schizophrenia |
| Onset | Childhood |
| Pharmacological treatment at evaluation | Valproic acid 1000 mg/day; aripiprazole 400 mg/monthly; fluvoxamine 100 mg/day; delorazepam 6 mg/day; levomepromazine 25 mg/day |
| Physical characteristics | Narrow palpebral fissures; fourth fingers clinodactyly; flat feet; rearfoot valgus (bilaterally) |
| PANSS score total | 115 |
| PANSS Positive score | 22 |
| PANSS Negative score | 26 |
| PANSS General score | 67 |
| IQ assessment | 48 |
| Current pharmacological treatment | Paliperidone monthly, lithium carbonate |
| Patient | Number of Reads | Average Depth | Coverage Percentage (≥50×) | Number of Variants (Total) |
|---|---|---|---|---|
| Proband (II.1) | 31,477,808 | 193.36 | 91.2% | 14,869 |
| Chr | Gene | Nucleotide Variant * | Protein * | Status | ACMG # Classification |
|---|---|---|---|---|---|
| 1 | KDM5B | NM_006618.5: c.3007T>A (exon 20) | p.Tyr1003Asn | Het | VUS |
| 11 | SHANK2 | NM_012309.5: c.332G>A (exon 3) | p.Arg111His | Het | VUS |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Iannotta, F.; La Monica, I.; Di Iorio, M.R.; Freda, V.; Sica, A.; de Bartolomeis, A.; Pastore, L.; Iasevoli, F.; Lombardo, B. A Paradigmatic Case of Genetic Overlap Between Neurodevelopment Disorders and Schizophrenia Aligning with the Neurodevelopmental Continuum Hypothesis. Int. J. Mol. Sci. 2025, 26, 3970. https://doi.org/10.3390/ijms26093970
Iannotta F, La Monica I, Di Iorio MR, Freda V, Sica A, de Bartolomeis A, Pastore L, Iasevoli F, Lombardo B. A Paradigmatic Case of Genetic Overlap Between Neurodevelopment Disorders and Schizophrenia Aligning with the Neurodevelopmental Continuum Hypothesis. International Journal of Molecular Sciences. 2025; 26(9):3970. https://doi.org/10.3390/ijms26093970
Chicago/Turabian StyleIannotta, Federica, Ilaria La Monica, Maria Rosaria Di Iorio, Vittorio Freda, Antonia Sica, Andrea de Bartolomeis, Lucio Pastore, Felice Iasevoli, and Barbara Lombardo. 2025. "A Paradigmatic Case of Genetic Overlap Between Neurodevelopment Disorders and Schizophrenia Aligning with the Neurodevelopmental Continuum Hypothesis" International Journal of Molecular Sciences 26, no. 9: 3970. https://doi.org/10.3390/ijms26093970
APA StyleIannotta, F., La Monica, I., Di Iorio, M. R., Freda, V., Sica, A., de Bartolomeis, A., Pastore, L., Iasevoli, F., & Lombardo, B. (2025). A Paradigmatic Case of Genetic Overlap Between Neurodevelopment Disorders and Schizophrenia Aligning with the Neurodevelopmental Continuum Hypothesis. International Journal of Molecular Sciences, 26(9), 3970. https://doi.org/10.3390/ijms26093970