
From Chemotherapy to Targeted Therapy: Unraveling Resistance in Acute Myeloid Leukemia Through Genetic and Non-Genetic Insights


Abstract
:1. Introduction
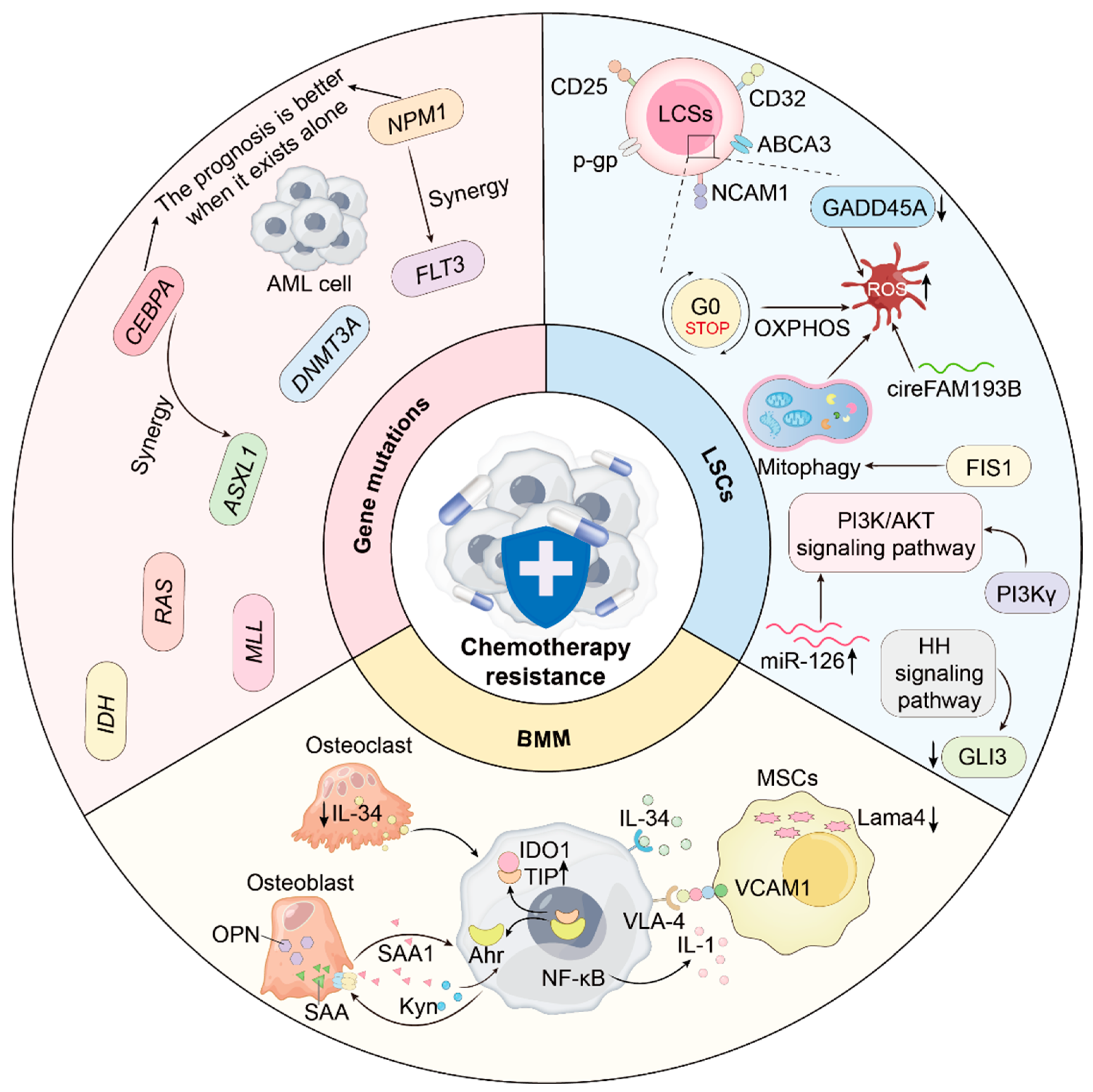
2. Chemotherapy Resistance Mechanisms
2.1. Gene Mutations and Clonal Heterogeneity
2.2. Leukemia Stem Cells (LSCs) and Multidimensional Networks
2.3. Bone Marrow Niche and Stromal Components
2.3.1. Mesenchymal Stem Cells (MSCs)
2.3.2. Osteoblasts and Osteoclasts
2.4. Optimized Chemotherapy Strategies
3. Targeted Therapy and Resistance Mechanisms
3.1. Mutation-Specific Inhibitors
3.1.1. FMS-like Tyrosine Kinase 3 (FLT3) Inhibitors
3.1.2. Isocitrate Dehydrogenase (IDH) Inhibitors
3.1.3. Menin Inhibitor
3.2. Hedgehog (HH) Signaling Pathway Inhibitor—Glasdegib
3.3. Apoptosis-Targeted Therapy—Venetoclax
3.4. Epigenetic Inhibitors—HMAs (Azacitidine and Decitabine)
3.5. Immunotherapy
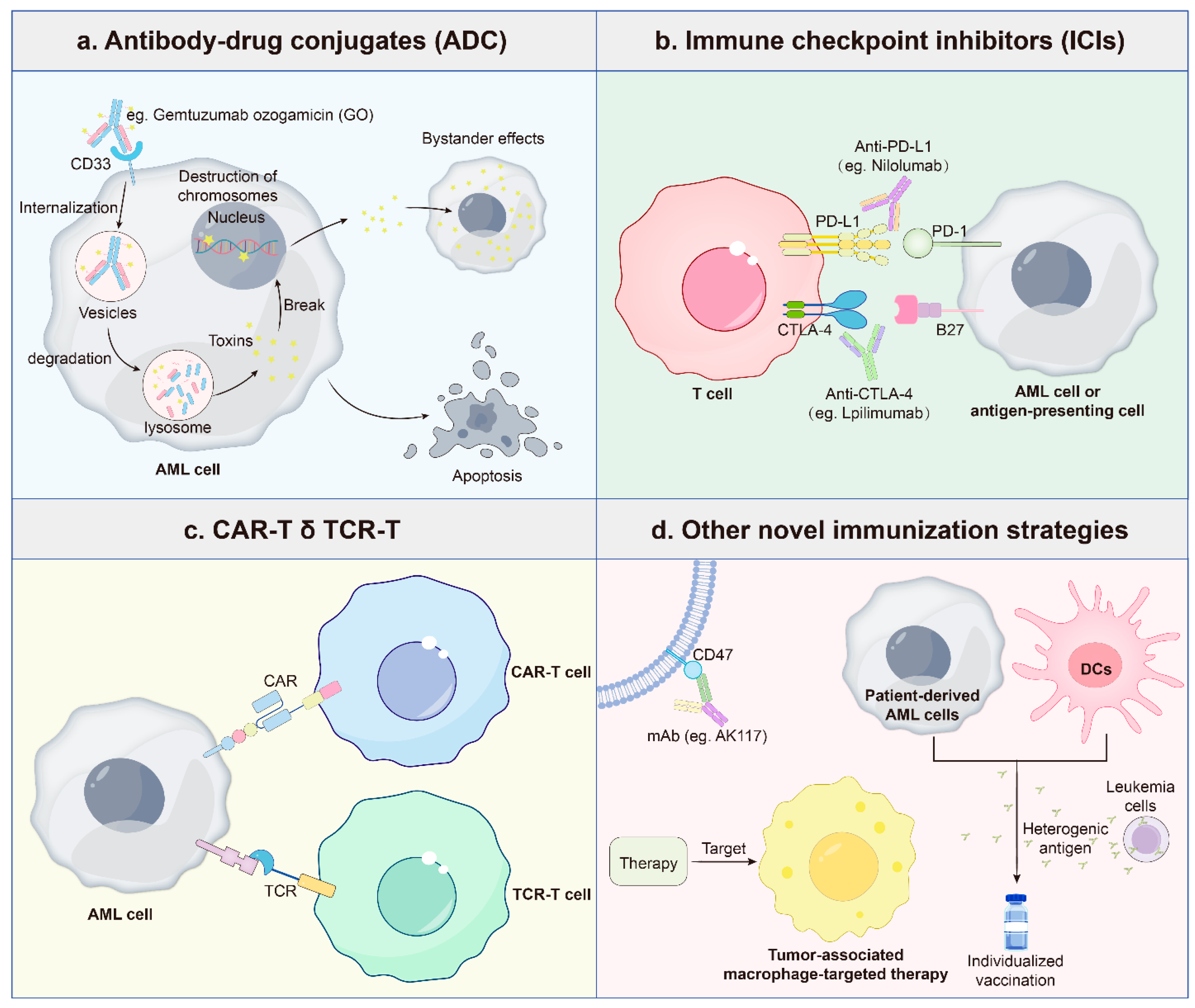
4. Integrative Perspectives and Future Directions
Author Contributions
Funding
Conflicts of Interest
References
- Döhner, H.; Estey, E.; Grimwade, D.; Amadori, S.; Appelbaum, F.R.; Büchner, T.; Dombret, H.; Ebert, B.L.; Fenaux, P.; Larson, R.A.; et al. Diagnosis and Management of AML in Adults: 2017 ELN Recommendations from an International Expert Panel. Blood 2017, 129, 424–447. [Google Scholar] [CrossRef]
- Döhner, H.; Estey, E.H.; Amadori, S.; Appelbaum, F.R.; Büchner, T.; Burnett, A.K.; Dombret, H.; Fenaux, P.; Grimwade, D.; Larson, R.A.; et al. Diagnosis and Management of Acute Myeloid Leukemia in Adults: Recommendations from an International Expert Panel, on Behalf of the European LeukemiaNet. Blood 2010, 115, 453–474. [Google Scholar] [CrossRef] [PubMed]
- DiNardo, C.D.; Erba, H.P.; Freeman, S.D.; Wei, A.H. Acute Myeloid Leukaemia. Lancet 2023, 401, 2073–2086. [Google Scholar] [CrossRef] [PubMed]
- Kantarjian, H.; Kadia, T.; DiNardo, C.; Daver, N.; Borthakur, G.; Jabbour, E.; Garcia-Manero, G.; Konopleva, M.; Ravandi, F. Acute Myeloid Leukemia: Current Progress and Future Directions. Blood Cancer J. 2021, 11, 41. [Google Scholar] [CrossRef]
- Bhansali, R.S.; Pratz, K.W.; Lai, C. Recent Advances in Targeted Therapies in Acute Myeloid Leukemia. J. Hematol. Oncol. 2023, 16, 29. [Google Scholar] [CrossRef] [PubMed]
- Krauss, A.C.; Gao, X.; Li, L.; Manning, M.L.; Patel, P.; Fu, W.; Janoria, K.G.; Gieser, G.; Bateman, D.A.; Przepiorka, D.; et al. FDA Approval Summary: (Daunorubicin and Cytarabine) Liposome for Injection for the Treatment of Adults with High-Risk Acute Myeloid Leukemia. Clin. Cancer Res. 2019, 25, 2685–2690. [Google Scholar] [CrossRef]
- Appelbaum, F.R.; Gundacker, H.; Head, D.R.; Slovak, M.L.; Willman, C.L.; Godwin, J.E.; Anderson, J.E.; Petersdorf, S.H. Age and Acute Myeloid Leukemia. Blood 2006, 107, 3481–3485. [Google Scholar] [CrossRef]
- Tardi, P.; Johnstone, S.; Harasym, N.; Xie, S.; Harasym, T.; Zisman, N.; Harvie, P.; Bermudes, D.; Mayer, L. In Vivo Maintenance of Synergistic Cytarabine:Daunorubicin Ratios Greatly Enhances Therapeutic Efficacy. Leuk. Res. 2009, 33, 129–139. [Google Scholar] [CrossRef]
- Lancet, J.E.; Uy, G.L.; Cortes, J.E.; Newell, L.F.; Lin, T.L.; Ritchie, E.K.; Stuart, R.K.; Strickland, S.A.; Hogge, D.; Solomon, S.R.; et al. CPX-351 (Cytarabine and Daunorubicin) Liposome for Injection Versus Conventional Cytarabine Plus Daunorubicin in Older Patients with Newly Diagnosed Secondary Acute Myeloid Leukemia. J. Clin. Oncol. 2018, 36, 2684–2692. [Google Scholar] [CrossRef]
- Kantarjian, H.M.; DiNardo, C.D.; Kadia, T.M.; Daver, N.G.; Altman, J.K.; Stein, E.M.; Jabbour, E.; Schiffer, C.A.; Lang, A.; Ravandi, F. Acute Myeloid Leukemia Management and Research in 2025. CA Cancer J. Clin. 2025, 75, 46–67. [Google Scholar] [CrossRef]
- Short, N.J.; Kantarjian, H. When Less Is More: Reevaluating the Role of Intensive Chemotherapy for Older Adults with Acute Myeloid Leukemia in the Modern Era. J. Clin. Oncol. 2021, 39, 3104–3108. [Google Scholar] [CrossRef]
- Kantarjian, H.; Short, N.J.; DiNardo, C.; Stein, E.M.; Daver, N.; Perl, A.E.; Wang, E.S.; Wei, A.; Tallman, M. Harnessing the Benefits of Available Targeted Therapies in Acute Myeloid Leukaemia. Lancet Haematol. 2021, 8, e922–e933. [Google Scholar] [CrossRef] [PubMed]
- DiNardo, C.D.; Jonas, B.A.; Pullarkat, V.; Thirman, M.J.; Garcia, J.S.; Wei, A.H.; Konopleva, M.; Döhner, H.; Letai, A.; Fenaux, P.; et al. Azacitidine and Venetoclax in Previously Untreated Acute Myeloid Leukemia. N. Engl. J. Med. 2020, 383, 617–629. [Google Scholar] [CrossRef] [PubMed]
- Lim, Z.-F.; Ma, P.C. Emerging Insights of Tumor Heterogeneity and Drug Resistance Mechanisms in Lung Cancer Targeted Therapy. J. Hematol. Oncol. 2019, 12, 134. [Google Scholar] [CrossRef] [PubMed]
- O’Reilly, E.; Zeinabad, H.A.; Szegezdi, E. Hematopoietic versus Leukemic Stem Cell Quiescence: Challenges and Therapeutic Opportunities. Blood Rev. 2021, 50, 100850. [Google Scholar] [CrossRef]
- Li, S.; Garrett-Bakelman, F.E.; Chung, S.S.; Sanders, M.A.; Hricik, T.; Rapaport, F.; Patel, J.; Dillon, R.; Vijay, P.; Brown, A.L.; et al. Distinct Evolution and Dynamics of Epigenetic and Genetic Heterogeneity in Acute Myeloid Leukemia. Nat. Med. 2016, 22, 792–799. [Google Scholar] [CrossRef]
- Ma, X.; Liu, Y.; Liu, Y.; Alexandrov, L.B.; Edmonson, M.N.; Gawad, C.; Zhou, X.; Li, Y.; Rusch, M.C.; Easton, J.; et al. Pan-Cancer Genome and Transcriptome Analyses of 1,699 Paediatric Leukaemias and Solid Tumours. Nature 2018, 555, 371–376. [Google Scholar] [CrossRef]
- Takahashi, K.; Tanaka, T. Clonal Evolution and Hierarchy in Myeloid Malignancies. Trends Cancer 2023, 9, 707–715. [Google Scholar] [CrossRef]
- Parkin, B.; Ouillette, P.; Li, Y.; Keller, J.; Lam, C.; Roulston, D.; Li, C.; Shedden, K.; Malek, S.N. Clonal Evolution and Devolution after Chemotherapy in Adult Acute Myelogenous Leukemia. Blood 2013, 121, 369–377. [Google Scholar] [CrossRef]
- Ding, L.; Ley, T.J.; Larson, D.E.; Miller, C.A.; Koboldt, D.C.; Welch, J.S.; Ritchey, J.K.; Young, M.A.; Lamprecht, T.; McLellan, M.D.; et al. Clonal Evolution in Relapsed Acute Myeloid Leukaemia Revealed by Whole-Genome Sequencing. Nature 2012, 481, 506–510. [Google Scholar] [CrossRef]
- Shlush, L.I.; Mitchell, A.; Heisler, L.; Abelson, S.; Ng, S.W.K.; Trotman-Grant, A.; Medeiros, J.J.F.; Rao-Bhatia, A.; Jaciw-Zurakowsky, I.; Marke, R.; et al. Tracing the Origins of Relapse in Acute Myeloid Leukaemia to Stem Cells. Nature 2017, 547, 104–108. [Google Scholar] [CrossRef] [PubMed]
- Wong, T.N.; Ramsingh, G.; Young, A.L.; Miller, C.A.; Touma, W.; Welch, J.S.; Lamprecht, T.L.; Shen, D.; Hundal, J.; Fulton, R.S.; et al. Role of TP53 Mutations in the Origin and Evolution of Therapy-Related Acute Myeloid Leukaemia. Nature 2015, 518, 552–555. [Google Scholar] [CrossRef] [PubMed]
- Döhner, H.; Wei, A.H.; Appelbaum, F.R.; Craddock, C.; DiNardo, C.D.; Dombret, H.; Ebert, B.L.; Fenaux, P.; Godley, L.A.; Hasserjian, R.P.; et al. Diagnosis and Management of AML in Adults: 2022 Recommendations from an International Expert Panel on Behalf of the ELN. Blood 2022, 140, 1345–1377. [Google Scholar] [CrossRef] [PubMed]
- Itzykson, R.; Fournier, E.; Berthon, C.; Röllig, C.; Braun, T.; Marceau-Renaut, A.; Pautas, C.; Nibourel, O.; Lemasle, E.; Micol, J.-B.; et al. Genetic Identification of Patients with AML Older than 60 Years Achieving Long-Term Survival with Intensive Chemotherapy. Blood 2021, 138, 507–519. [Google Scholar] [CrossRef]
- Rücker, F.G.; Schlenk, R.F.; Bullinger, L.; Kayser, S.; Teleanu, V.; Kett, H.; Habdank, M.; Kugler, C.-M.; Holzmann, K.; Gaidzik, V.I.; et al. TP53 Alterations in Acute Myeloid Leukemia with Complex Karyotype Correlate with Specific Copy Number Alterations, Monosomal Karyotype, and Dismal Outcome. Blood 2012, 119, 2114–2121. [Google Scholar] [CrossRef]
- Rajagopalan, A.; Feng, Y.; Gayatri, M.B.; Ranheim, E.A.; Klungness, T.; Matson, D.R.; Lee, M.H.; Jung, M.M.; Zhou, Y.; Gao, X.; et al. A Gain-of-Function P53 Mutant Synergizes with Oncogenic NRAS to Promote Acute Myeloid Leukemia in Mice. J. Clin. Investig. 2003, 133, e173116. [Google Scholar] [CrossRef]
- Rao, A.V.; Valk, P.J.M.; Metzeler, K.H.; Acharya, C.R.; Tuchman, S.A.; Stevenson, M.M.; Rizzieri, D.A.; Delwel, R.; Buske, C.; Bohlander, S.K.; et al. Age-Specific Differences in Oncogenic Pathway Dysregulation in Patients with Acute Myeloid Leukemia. J. Clin. Oncol. 2009, 27, 5580–5586. [Google Scholar] [CrossRef]
- Faber, J.; Krivtsov, A.V.; Stubbs, M.C.; Wright, R.; Davis, T.N.; van den Heuvel-Eibrink, M.; Zwaan, C.M.; Kung, A.L.; Armstrong, S.A. HOXA9 Is Required for Survival in Human MLL-Rearranged Acute Leukemias. Blood 2009, 113, 2375–2385. [Google Scholar] [CrossRef]
- Li, Z.; Chen, P.; Su, R.; Hu, C.; Li, Y.; Elkahloun, A.G.; Zuo, Z.; Gurbuxani, S.; Arnovitz, S.; Weng, H.; et al. PBX3 and MEIS1 Cooperate in Hematopoietic Cells to Drive Acute Myeloid Leukemias Characterized by a Core Transcriptome of the MLL-Rearranged Disease. Cancer Res. 2016, 76, 619–629. [Google Scholar] [CrossRef]
- Stavropoulou, V.; Kaspar, S.; Brault, L.; Sanders, M.A.; Juge, S.; Morettini, S.; Tzankov, A.; Iacovino, M.; Lau, I.-J.; Milne, T.A.; et al. MLL-AF9 Expression in Hematopoietic Stem Cells Drives a Highly Invasive AML Expressing EMT-Related Genes Linked to Poor Outcome. Cancer Cell 2016, 30, 43–58. [Google Scholar] [CrossRef]
- Daver, N.; Schlenk, R.F.; Russell, N.H.; Levis, M.J. Targeting FLT3 Mutations in AML: Review of Current Knowledge and Evidence. Leukemia 2019, 33, 299–312. [Google Scholar] [CrossRef] [PubMed]
- Angenendt, L.; Röllig, C.; Montesinos, P.; Martínez-Cuadrón, D.; Barragan, E.; García, R.; Botella, C.; Martínez, P.; Ravandi, F.; Kadia, T.; et al. Chromosomal Abnormalities and Prognosis in NPM1-Mutated Acute Myeloid Leukemia: A Pooled Analysis of Individual Patient Data From Nine International Cohorts. J. Clin. Oncol. 2019, 37, 2632–2642. [Google Scholar] [CrossRef] [PubMed]
- Krönke, J.; Bullinger, L.; Teleanu, V.; Tschürtz, F.; Gaidzik, V.I.; Kühn, M.W.M.; Rücker, F.G.; Holzmann, K.; Paschka, P.; Kapp-Schwörer, S.; et al. Clonal Evolution in Relapsed NPM1-Mutated Acute Myeloid Leukemia. Blood 2013, 122, 100–108. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.; Yang, H.; Liu, Y.; Yang, Y.; Wang, P.; Kim, S.-H.; Ito, S.; Yang, C.; Wang, P.; Xiao, M.-T.; et al. Oncometabolite 2-Hydroxyglutarate Is a Competitive Inhibitor of α-Ketoglutarate-Dependent Dioxygenases. Cancer Cell 2011, 19, 17–30. [Google Scholar] [CrossRef]
- Chan, S.M.; Thomas, D.; Corces-Zimmerman, M.R.; Xavy, S.; Rastogi, S.; Hong, W.-J.; Zhao, F.; Medeiros, B.C.; Tyvoll, D.A.; Majeti, R. Isocitrate Dehydrogenase 1 and 2 Mutations Induce BCL-2 Dependence in Acute Myeloid Leukemia. Nat. Med. 2015, 21, 178–184. [Google Scholar] [CrossRef]
- Im, A.P.; Sehgal, A.R.; Carroll, M.P.; Smith, B.D.; Tefferi, A.; Johnson, D.E.; Boyiadzis, M. DNMT3A and IDH Mutations in Acute Myeloid Leukemia and Other Myeloid Malignancies: Associations with Prognosis and Potential Treatment Strategies. Leukemia 2014, 28, 1774–1783. [Google Scholar] [CrossRef]
- Shlush, L.I.; Zandi, S.; Mitchell, A.; Chen, W.C.; Brandwein, J.M.; Gupta, V.; Kennedy, J.A.; Schimmer, A.D.; Schuh, A.C.; Yee, K.W.; et al. Identification of Pre-Leukaemic Haematopoietic Stem Cells in Acute Leukaemia. Nature 2014, 506, 328–333. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Zhang, W.; Yan, X.-J.; Lin, X.-Q.; Li, W.; Mi, J.-Q.; Li, J.-M.; Zhu, J.; Chen, Z.; Chen, S.-J. DNMT3A Mutation Leads to Leukemic Extramedullary Infiltration Mediated by TWIST1. J. Hematol. Oncol. 2016, 9, 106. [Google Scholar] [CrossRef]
- Chu, X.; Zhong, L.; Dan, W.; Wang, X.; Zhang, Z.; Liu, Z.; Lu, Y.; Shao, X.; Zhou, Z.; Chen, S.; et al. DNMT3A R882H Mutation Drives Daunorubicin Resistance in Acute Myeloid Leukemia via Regulating NRF2/NQO1 Pathway. Cell Commun. Signal. 2022, 20, 168. [Google Scholar] [CrossRef]
- Guryanova, O.A.; Shank, K.; Spitzer, B.; Luciani, L.; Koche, R.P.; Garrett-Bakelman, F.E.; Ganzel, C.; Durham, B.H.; Mohanty, A.; Hoermann, G.; et al. DNMT3A R882 Mutations Promote Anthracycline Resistance in Acute Myeloid Leukemia through Impaired Nucleosome Remodeling. Nat. Med. 2016, 22, 1488–1495. [Google Scholar] [CrossRef]
- Marcucci, G.; Metzeler, K.H.; Schwind, S.; Becker, H.; Maharry, K.; Mrózek, K.; Radmacher, M.D.; Kohlschmidt, J.; Nicolet, D.; Whitman, S.P.; et al. Age-Related Prognostic Impact of Different Types of DNMT3A Mutations in Adults with Primary Cytogenetically Normal Acute Myeloid Leukemia. J. Clin. Oncol. 2012, 30, 742–750. [Google Scholar] [CrossRef] [PubMed]
- D’Altri, T.; Wilhelmson, A.S.; Schuster, M.B.; Wenzel, A.; Kalvisa, A.; Pundhir, S.; Hansen, A.M.; Porse, B.T. The ASXL1-G643W Variant Accelerates the Development of CEBPA Mutant Acute Myeloid Leukemia. Haematologica 2021, 106, 1000–1007. [Google Scholar] [CrossRef]
- Inoue, D.; Matsumoto, M.; Nagase, R.; Saika, M.; Fujino, T.; Nakayama, K.I.; Kitamura, T. Truncation Mutants of ASXL1 Observed in Myeloid Malignancies Are Expressed at Detectable Protein Levels. Exp. Hematol. 2016, 44, 172–176.e1. [Google Scholar] [CrossRef]
- Ferrando, A.A.; López-Otín, C. Clonal Evolution in Leukemia. Nat. Med. 2017, 23, 1135–1145. [Google Scholar] [CrossRef] [PubMed]
- Thomas, D.; Majeti, R. Biology and Relevance of Human Acute Myeloid Leukemia Stem Cells. Blood 2017, 129, 1577–1585. [Google Scholar] [CrossRef]
- Ho, T.-C.; LaMere, M.; Stevens, B.M.; Ashton, J.M.; Myers, J.R.; O’Dwyer, K.M.; Liesveld, J.L.; Mendler, J.H.; Guzman, M.; Morrissette, J.D.; et al. Evolution of Acute Myelogenous Leukemia Stem Cell Properties after Treatment and Progression. Blood 2016, 128, 1671–1678. [Google Scholar] [CrossRef] [PubMed]
- Ng, S.W.K.; Mitchell, A.; Kennedy, J.A.; Chen, W.C.; McLeod, J.; Ibrahimova, N.; Arruda, A.; Popescu, A.; Gupta, V.; Schimmer, A.D.; et al. A 17-Gene Stemness Score for Rapid Determination of Risk in Acute Leukaemia. Nature 2016, 540, 433–437. [Google Scholar] [CrossRef]
- Saito, Y.; Kitamura, H.; Hijikata, A.; Tomizawa-Murasawa, M.; Tanaka, S.; Takagi, S.; Uchida, N.; Suzuki, N.; Sone, A.; Najima, Y.; et al. Identification of Therapeutic Targets for Quiescent, Chemotherapy-Resistant Human Leukemia Stem Cells. Sci. Transl. Med. 2010, 2, 17ra9. [Google Scholar] [CrossRef]
- Sasca, D.; Szybinski, J.; Schüler, A.; Shah, V.; Heidelberger, J.; Haehnel, P.S.; Dolnik, A.; Kriege, O.; Fehr, E.-M.; Gebhardt, W.H.; et al. NCAM1 (CD56) Promotes Leukemogenesis and Confers Drug Resistance in AML. Blood 2019, 133, 2305–2319. [Google Scholar] [CrossRef]
- Williams, M.S.; Amaral, F.M.; Simeoni, F.; Somervaille, T.C. A Stress-Responsive Enhancer Induces Dynamic Drug Resistance in Acute Myeloid Leukemia. J. Clin. Investig. 2020, 130, 1217–1232. [Google Scholar] [CrossRef]
- Wulf, G.G.; Wang, R.-Y.; Kuehnle, I.; Weidner, D.; Marini, F.; Brenner, M.K.; Andreeff, M.; Goodell, M.A. A Leukemic Stem Cell with Intrinsic Drug Efflux Capacity in Acute Myeloid Leukemia. Blood 2001, 98, 1166–1173. [Google Scholar] [CrossRef] [PubMed]
- Ceraulo, A.; Lapillonne, H.; Cheok, M.H.; Preudhomme, C.; Dombret, H.; Terré, C.; Lambert, J.; Leverger, G.; Bertrand, Y.; Mortreux, F.; et al. Prognostic Impact of ABCA3 Expression in Adult and Pediatric Acute Myeloid Leukemia: An ALFA-ELAM02 Joint Study. Blood Adv. 2022, 6, 2773–2777. [Google Scholar] [CrossRef]
- Steinbach, D.; Gillet, J.-P.; Sauerbrey, A.; Gruhn, B.; Dawczynski, K.; Bertholet, V.; de Longueville, F.; Zintl, F.; Remacle, J.; Efferth, T. ABCA3 as a Possible Cause of Drug Resistance in Childhood Acute Myeloid Leukemia. Clin. Cancer Res. 2006, 12, 4357–4363. [Google Scholar] [CrossRef]
- Shaffer, B.C.; Gillet, J.-P.; Patel, C.; Baer, M.R.; Bates, S.E.; Gottesman, M.M. Drug Resistance: Still a Daunting Challenge to the Successful Treatment of AML. Drug Resist. Updates 2012, 15, 62–69. [Google Scholar] [CrossRef]
- Saadatpour, A.; Guo, G.; Orkin, S.H.; Yuan, G.-C. Characterizing Heterogeneity in Leukemic Cells Using Single-Cell Gene Expression Analysis. Genome Biol. 2014, 15, 525. [Google Scholar] [CrossRef] [PubMed]
- Lagadinou, E.D.; Sach, A.; Callahan, K.; Rossi, R.M.; Neering, S.J.; Minhajuddin, M.; Ashton, J.M.; Pei, S.; Grose, V.; O’Dwyer, K.M.; et al. BCL-2 Inhibition Targets Oxidative Phosphorylation and Selectively Eradicates Quiescent Human Leukemia Stem Cells. Cell Stem Cell 2013, 12, 329–341. [Google Scholar] [CrossRef]
- Jones, C.L.; Stevens, B.M.; D’Alessandro, A.; Reisz, J.A.; Culp-Hill, R.; Nemkov, T.; Pei, S.; Khan, N.; Adane, B.; Ye, H.; et al. Inhibition of Amino Acid Metabolism Selectively Targets Human Leukemia Stem Cells. Cancer Cell 2018, 34, 724–740.e4. [Google Scholar] [CrossRef]
- Pei, S.; Minhajuddin, M.; Adane, B.; Khan, N.; Stevens, B.M.; Mack, S.C.; Lai, S.; Rich, J.N.; Inguva, A.; Shannon, K.M.; et al. AMPK/FIS1-Mediated Mitophagy Is Required for Self-Renewal of Human AML Stem Cells. Cell Stem Cell 2018, 23, 86–100.e6. [Google Scholar] [CrossRef] [PubMed]
- Farge, T.; Saland, E.; De Toni, F.; Aroua, N.; Hosseini, M.; Perry, R.; Bosc, C.; Sugita, M.; Stuani, L.; Fraisse, M.; et al. Chemotherapy-Resistant Human Acute Myeloid Leukemia Cells Are Not Enriched for Leukemic Stem Cells but Require Oxidative Metabolism. Cancer Discov. 2017, 7, 716–735. [Google Scholar] [CrossRef]
- Hassan, N.; Yi, H.; Malik, B.; Gaspard-Boulinc, L.; Samaraweera, S.E.; Casolari, D.A.; Seneviratne, J.; Balachandran, A.; Chew, T.; Duly, A.; et al. Loss of the Stress Sensor GADD45A Promotes Stem Cell Activity and Ferroptosis Resistance in LGR4/HOXA9-Dependent AML. Blood 2024, 144, 84–98. [Google Scholar] [CrossRef]
- Yang, X.; Liu, J.; Liu, W.; Wu, H.; Wei, Y.; Guo, X.; Jia, H.; Can, C.; Wang, D.; Hu, X.; et al. circFAM193B Interaction with PRMT6 Regulates AML Leukemia Stem Cells Chemoresistance through Altering the Oxidative Metabolism and Lipid Peroxidation. Leukemia 2024, 38, 1057–1071. [Google Scholar] [CrossRef] [PubMed]
- Tian, Y.; Huang, Z.; Wang, Z.; Yin, C.; Zhou, L.; Zhang, L.; Huang, K.; Zhou, H.; Jiang, X.; Li, J.; et al. Identification of Novel Molecular Markers for Prognosis Estimation of Acute Myeloid Leukemia: Over-Expression of PDCD7, FIS1 and Ang2 May Indicate Poor Prognosis in Pretreatment Patients with Acute Myeloid Leukemia. PLoS ONE 2014, 9, e84150. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Huang, S.; Chen, J.-L. Understanding of Leukemic Stem Cells and Their Clinical Implications. Mol. Cancer 2017, 16, 2. [Google Scholar] [CrossRef]
- Lechman, E.R.; Gentner, B.; Ng, S.W.; Schoof, E.M.; van Galen, P.; Kennedy, J.A.; Nucera, S.; Ciceri, F.; Kaufmann, K.B.; Takayama, N.; et al. miR-126 Regulates Distinct Self-Renewal Outcomes in Normal and Malignant Hematopoietic Stem Cells. Cancer Cell 2016, 29, 214–228. [Google Scholar] [CrossRef]
- Gu, H.; Chen, C.; Hou, Z.-S.; He, X.-D.; Xie, S.; Ni, J.; Qian, C.; Cheng, X.; Jiang, T.; Yang, C.; et al. PI3Kγ Maintains the Self-Renewal of Acute Myeloid Leukemia Stem Cells by Regulating the Pentose Phosphate Pathway. Blood 2024, 143, 1965–1979. [Google Scholar] [CrossRef] [PubMed]
- Carter, J.L.; Hege, K.; Yang, J.; Kalpage, H.A.; Su, Y.; Edwards, H.; Hüttemann, M.; Taub, J.W.; Ge, Y. Targeting Multiple Signaling Pathways: The New Approach to Acute Myeloid Leukemia Therapy. Signal Transduct. Target. Ther. 2020, 5, 288. [Google Scholar] [CrossRef]
- Freisleben, F.; Behrmann, L.; Thaden, V.; Muschhammer, J.; Bokemeyer, C.; Fiedler, W.; Wellbrock, J. Downregulation of GLI3 Expression Mediates Chemotherapy Resistance in Acute Myeloid Leukemia. Int. J. Mol. Sci. 2020, 21, 5084. [Google Scholar] [CrossRef]
- Lane, S.W.; Wang, Y.J.; Celso, C.L.; Ragu, C.; Bullinger, L.; Sykes, S.M.; Ferraro, F.; Shterental, S.; Lin, C.P.; Gilliland, D.G.; et al. Differential Niche and Wnt Requirements during Acute Myeloid Leukemia Progression. Blood 2011, 118, 2849–2856. [Google Scholar] [CrossRef]
- Méndez-Ferrer, S.; Bonnet, D.; Steensma, D.P.; Hasserjian, R.P.; Ghobrial, I.M.; Gribben, J.G.; Andreeff, M.; Krause, D.S. Bone Marrow Niches in Haematological Malignancies. Nat. Rev. Cancer 2020, 20, 285–298. [Google Scholar] [CrossRef]
- Zhou, H.S.; Carter, B.Z.; Andreeff, M. Bone Marrow Niche-Mediated Survival of Leukemia Stem Cells in Acute Myeloid Leukemia: Yin and Yang. Cancer Biol. Med. 2016, 13, 248–259. [Google Scholar] [CrossRef]
- Konopleva, M.Y.; Jordan, C.T. Leukemia Stem Cells and Microenvironment: Biology and Therapeutic Targeting. J. Clin. Oncol. 2011, 29, 591–599. [Google Scholar] [CrossRef]
- Fei, M.-Y.; Wang, Y.; Chang, B.-H.; Xue, K.; Dong, F.; Huang, D.; Li, X.-Y.; Li, Z.-J.; Hu, C.-L.; Liu, P.; et al. The Non-Cell-Autonomous Function of ID1 Promotes AML Progression via ANGPTL7 from the Microenvironment. Blood 2023, 142, 903–917. [Google Scholar] [CrossRef] [PubMed]
- Welner, R.S.; Amabile, G.; Bararia, D.; Czibere, A.; Yang, H.; Zhang, H.; Pontes, L.L.D.F.; Ye, M.; Levantini, E.; Di Ruscio, A.; et al. Treatment of Chronic Myelogenous Leukemia by Blocking Cytokine Alterations Found in Normal Stem and Progenitor Cells. Cancer Cell 2015, 27, 671–681. [Google Scholar] [CrossRef] [PubMed]
- Kleppe, M.; Kwak, M.; Koppikar, P.; Riester, M.; Keller, M.; Bastian, L.; Hricik, T.; Bhagwat, N.; McKenney, A.S.; Papalexi, E.; et al. JAK–STAT Pathway Activation in Malignant and Nonmalignant Cells Contributes to MPN Pathogenesis and Therapeutic Response. Cancer Discov. 2015, 5, 316–331. [Google Scholar] [CrossRef]
- Flores-Figueroa, E.; Montesinos, J.J.; Flores-Guzmán, P.; Gutiérrez-Espíndola, G.; Arana-Trejo, R.M.; Castillo-Medina, S.; Pérez-Cabrera, A.; Hernández-Estévez, E.; Arriaga, L.; Mayani, H. Functional Analysis of Myelodysplastic Syndromes-Derived Mesenchymal Stem Cells. Leuk. Res. 2008, 32, 1407–1416. [Google Scholar] [CrossRef] [PubMed]
- Medyouf, H.; Mossner, M.; Jann, J.-C.; Nolte, F.; Raffel, S.; Herrmann, C.; Lier, A.; Eisen, C.; Nowak, V.; Zens, B.; et al. Myelodysplastic Cells in Patients Reprogram Mesenchymal Stromal Cells to Establish a Transplantable Stem Cell Niche Disease Unit. Cell Stem Cell 2014, 14, 824–837. [Google Scholar] [CrossRef]
- Jacamo, R.; Chen, Y.; Wang, Z.; Ma, W.; Zhang, M.; Spaeth, E.L.; Wang, Y.; Battula, V.L.; Mak, P.Y.; Schallmoser, K.; et al. Reciprocal Leukemia-Stroma VCAM-1/VLA-4-Dependent Activation of NF-κB Mediates Chemoresistance. Blood 2014, 123, 2691–2702. [Google Scholar] [CrossRef] [PubMed]
- Layani-Bazar, A.; Skornick, I.; Berrebi, A.; Pauker, M.H.; Noy, E.; Silberman, A.; Albeck, M.; Longo, D.L.; Kalechman, Y.; Sredni, B. Redox Modulation of Adjacent Thiols in VLA-4 by AS101 Converts Myeloid Leukemia Cells from a Drug-Resistant to Drug-Sensitive State. Cancer Res. 2014, 74, 3092–3103. [Google Scholar] [CrossRef]
- Matsunaga, T.; Takemoto, N.; Sato, T.; Takimoto, R.; Tanaka, I.; Fujimi, A.; Akiyama, T.; Kuroda, H.; Kawano, Y.; Kobune, M.; et al. Interaction between Leukemic-Cell VLA-4 and Stromal Fibronectin Is a Decisive Factor for Minimal Residual Disease of Acute Myelogenous Leukemia. Nat. Med. 2003, 9, 1158–1165. [Google Scholar] [CrossRef]
- Borella, G.; Da Ros, A.; Borile, G.; Porcù, E.; Tregnago, C.; Benetton, M.; Marchetti, A.; Bisio, V.; Montini, B.; Michielotto, B.; et al. Targeting the Plasticity of Mesenchymal Stromal Cells to Reroute the Course of Acute Myeloid Leukemia. Blood 2021, 138, 557–570. [Google Scholar] [CrossRef] [PubMed]
- Xiao, P.; Sandhow, L.; Heshmati, Y.; Kondo, M.; Bouderlique, T.; Dolinska, M.; Johansson, A.-S.; Sigvardsson, M.; Ekblom, M.; Walfridsson, J.; et al. Distinct Roles of Mesenchymal Stem and Progenitor Cells during the Development of Acute Myeloid Leukemia in Mice. Blood Adv. 2018, 2, 1480–1494. [Google Scholar] [CrossRef] [PubMed]
- Cai, H.; Kondo, M.; Sandhow, L.; Xiao, P.; Johansson, A.-S.; Sasaki, T.; Zawacka-Pankau, J.; Tryggvason, K.; Ungerstedt, J.; Walfridsson, J.; et al. Critical Role of Lama4 for Hematopoiesis Regeneration and Acute Myeloid Leukemia Progression. Blood 2022, 139, 3040–3057. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Niu, C.; Ye, L.; Huang, H.; He, X.; Tong, W.-G.; Ross, J.; Haug, J.; Johnson, T.; Feng, J.Q.; et al. Identification of the Haematopoietic Stem Cell Niche and Control of the Niche Size. Nature 2003, 425, 836–841. [Google Scholar] [CrossRef]
- Kode, A.; Manavalan, J.S.; Mosialou, I.; Bhagat, G.; Rathinam, C.V.; Luo, N.; Khiabanian, H.; Lee, A.; Murty, V.V.; Friedman, R.; et al. Leukaemogenesis Induced by an Activating β-Catenin Mutation in Osteoblasts. Nature 2014, 506, 240–244. [Google Scholar] [CrossRef]
- Liersch, R.; Gerss, J.; Schliemann, C.; Bayer, M.; Schwöppe, C.; Biermann, C.; Appelmann, I.; Kessler, T.; Löwenberg, B.; Büchner, T.; et al. Osteopontin Is a Prognostic Factor for Survival of Acute Myeloid Leukemia Patients. Blood 2012, 119, 5215–5220. [Google Scholar] [CrossRef]
- Galán-Díez, M.; Borot, F.; Ali, A.M.; Zhao, J.; Gil-Iturbe, E.; Shan, X.; Luo, N.; Liu, Y.; Huang, X.-P.; Bisikirska, B.; et al. Subversion of Serotonin Receptor Signaling in Osteoblasts by Kynurenine Drives Acute Myeloid Leukemia. Cancer Discov. 2022, 12, 1106–1127. [Google Scholar] [CrossRef]
- Prendergast, G.C.; Malachowski, W.P.; DuHadaway, J.B.; Muller, A.J. Discovery of IDO1 Inhibitors: From Bench to Bedside. Cancer Res. 2017, 77, 6795–6811. [Google Scholar] [CrossRef] [PubMed]
- Krevvata, M.; Silva, B.C.; Manavalan, J.S.; Galan-Diez, M.; Kode, A.; Matthews, B.G.; Park, D.; Zhang, C.A.; Galili, N.; Nickolas, T.L.; et al. Inhibition of Leukemia Cell Engraftment and Disease Progression in Mice by Osteoblasts. Blood 2014, 124, 2834–2846. [Google Scholar] [CrossRef]
- Xie, X.; Zhang, W.; Xiao, M.; Wei, T.; Qiu, Y.; Qiu, J.; Wang, H.; Qiu, Z.; Zhang, S.; Pan, Y.; et al. TREM2 Acts as a Receptor for IL-34 to Suppress Acute Myeloid Leukemia in Mice. Blood 2023, 141, 3184–3198. [Google Scholar] [CrossRef]
- Candoni, A.; Papayannidis, C.; Martinelli, G.; Simeone, E.; Gottardi, M.; Iacobucci, I.; Gherlinzoni, F.; Visani, G.; Baccarani, M.; Fanin, R. Flai (Fludarabine, Cytarabine, Idarubicin) plus Low-Dose Gemtuzumab Ozogamicin as Induction Therapy in CD33-Positive AML: Final Results and Long Term Outcome of a Phase II Multicenter Clinical Trial. Am. J. Hematol. 2018, 93, 655–663. [Google Scholar] [CrossRef]
- Montillo, M.; Mirto, S.; Petti, M.C.; Latagliata, R.; Magrin, S.; Pinto, A.; Zagonel, V.; Mele, G.; Tedeschi, A.; Ferrara, F. Fludarabine, Cytarabine, and G-CSF (FLAG) for the Treatment of Poor Risk Acute Myeloid Leukemia. Am. J. Hematol. 1998, 58, 105–109. [Google Scholar] [CrossRef]
- Burnett, A.K.; Russell, N.H.; Hills, R.K.; Hunter, A.E.; Kjeldsen, L.; Yin, J.; Gibson, B.E.; Wheatley, K.; Milligan, D. Optimization of Chemotherapy for Younger Patients with Acute Myeloid Leukemia: Results of the Medical Research Council AML15 Trial. J. Clin. Oncol. 2013, 31, 3360–3368. [Google Scholar] [CrossRef]
- Borthakur, G.; Ravandi, F.; Patel, K.; Wang, X.; Kadia, T.; DiNardo, C.; Garcia-Manero, G.; Pemmaraju, N.; Jabbour, E.J.; Takahashi, K.; et al. Retrospective Comparison of Survival and Responses to Fludarabine, Cytarabine, GCSF (FLAG) in Combination with Gemtuzumab Ozogamicin (GO) or Idarubicin (IDA) in Patients with Newly Diagnosed Core Binding Factor (CBF) Acute Myelogenous Leukemia: MD Anderson Experience in 174 Patients. Am. J. Hematol. 2022, 97, 1427–1434. [Google Scholar] [CrossRef] [PubMed]
- Holowiecki, J.; Grosicki, S.; Giebel, S.; Robak, T.; Kyrcz-Krzemien, S.; Kuliczkowski, K.; Skotnicki, A.B.; Hellmann, A.; Sulek, K.; Dmoszynska, A.; et al. Cladribine, But Not Fludarabine, Added to Daunorubicin and Cytarabine During Induction Prolongs Survival of Patients with Acute Myeloid Leukemia: A Multicenter, Randomized Phase III Study. J. Clin. Oncol. 2012, 30, 2441–2448. [Google Scholar] [CrossRef]
- Yates, J.W.; Wallace, H.J., Jr.; Ellison, R.R.; Holland, J.F. Cytosine Arabinoside (NSC-63878) and Daunorubicin (NSC-83142) Therapy in Acute Nonlymphocytic Leukemia. Cancer Chemother. Rep. 1973, 57, 485–488. [Google Scholar] [PubMed]
- Kantarjian, H.M.; Thomas, X.G.; Dmoszynska, A.; Wierzbowska, A.; Mazur, G.; Mayer, J.; Gau, J.-P.; Chou, W.-C.; Buckstein, R.; Cermak, J.; et al. Multicenter, Randomized, Open-Label, Phase III Trial of Decitabine versus Patient Choice, with Physician Advice, of Either Supportive Care or Low-Dose Cytarabine for the Treatment of Older Patients with Newly Diagnosed Acute Myeloid Leukemia. J. Clin. Oncol. 2012, 30, 2670–2677. [Google Scholar] [CrossRef]
- Iwai, T.; Yokota, S.; Nakao, M.; Okamoto, T.; Taniwaki, M.; Onodera, N.; Watanabe, A.; Kikuta, A.; Tanaka, A.; Asami, K.; et al. Internal Tandem Duplication of the FLT3 Gene and Clinical Evaluation in Childhood Acute Myeloid Leukemia. Leukemia 1999, 13, 38–43. [Google Scholar] [CrossRef]
- Macečková, D.; Vaňková, L.; Holubová, M.; Jindra, P.; Klieber, R.; Jandová, E.; Pitule, P. Current Knowledge about FLT3 Gene Mutations, Exploring the Isoforms, and Protein Importance in AML. Mol. Biol. Rep. 2024, 51, 521. [Google Scholar] [CrossRef]
- Stone, R.M.; Fischer, T.; Paquette, R.; Schiller, G.; Schiffer, C.A.; Ehninger, G.; Cortes, J.; Kantarjian, H.M.; DeAngelo, D.J.; Huntsman-Labed, A.; et al. Phase IB Study of the FLT3 Kinase Inhibitor Midostaurin with Chemotherapy in Younger Newly Diagnosed Adult Patients with Acute Myeloid Leukemia. Leukemia 2012, 26, 2061–2068. [Google Scholar] [CrossRef]
- Stone, R.M.; Mandrekar, S.J.; Sanford, B.L.; Laumann, K.; Geyer, S.; Bloomfield, C.D.; Thiede, C.; Prior, T.W.; Döhner, K.; Marcucci, G.; et al. Midostaurin plus Chemotherapy for Acute Myeloid Leukemia with a FLT3 Mutation. N. Engl. J. Med. 2017, 377, 454–464. [Google Scholar] [CrossRef]
- Von Bubnoff, N.; Engh, R.A.; Åberg, E.; Sänger, J.; Peschel, C.; Duyster, J. FMS-Like Tyrosine Kinase 3–Internal Tandem Duplication Tyrosine Kinase Inhibitors Display a Nonoverlapping Profile of Resistance Mutations In Vitro. Cancer Res. 2009, 69, 3032–3041. [Google Scholar] [CrossRef] [PubMed]
- Heidel, F.; Solem, F.K.; Breitenbuecher, F.; Lipka, D.B.; Kasper, S.; Thiede, M.H.; Brandts, C.; Serve, H.; Roesel, J.; Giles, F.; et al. Clinical Resistance to the Kinase Inhibitor PKC412 in Acute Myeloid Leukemia by Mutation of Asn-676 in the FLT3 Tyrosine Kinase Domain. Blood 2006, 107, 293–300. [Google Scholar] [CrossRef] [PubMed]
- Rummelt, C.; Gorantla, S.P.; Meggendorfer, M.; Charlet, A.; Endres, C.; Döhner, K.; Heidel, F.H.; Fischer, T.; Haferlach, T.; Duyster, J.; et al. Activating JAK-Mutations Confer Resistance to FLT3 Kinase Inhibitors in FLT3-ITD Positive AML in Vitro and in Vivo. Leukemia 2021, 35, 2017–2029. [Google Scholar] [CrossRef]
- Schmalbrock, L.K.; Dolnik, A.; Cocciardi, S.; Sträng, E.; Theis, F.; Jahn, N.; Panina, E.; Blätte, T.J.; Herzig, J.; Skambraks, S.; et al. Clonal Evolution of Acute Myeloid Leukemia with FLT3-ITD Mutation under Treatment with Midostaurin. Blood 2021, 137, 3093–3104. [Google Scholar] [CrossRef]
- Döhner, H.; Wei, A.H.; Löwenberg, B. Towards Precision Medicine for AML. Nat. Rev. Clin. Oncol. 2021, 18, 577–590. [Google Scholar] [CrossRef]
- Perl, A.E.; Martinelli, G.; Cortes, J.E.; Neubauer, A.; Berman, E.; Paolini, S.; Montesinos, P.; Baer, M.R.; Larson, R.A.; Ustun, C.; et al. Gilteritinib or Chemotherapy for Relapsed or Refractory FLT3-Mutated AML. N. Engl. J. Med. 2019, 381, 1728–1740. [Google Scholar] [CrossRef] [PubMed]
- Joshi, S.K.; Nechiporuk, T.; Bottomly, D.; Piehowski, P.D.; Reisz, J.A.; Pittsenbarger, J.; Kaempf, A.; Gosline, S.J.; Wang, Y.-T.; Hansen, J.R.; et al. The AML Microenvironment Catalyzes a Stepwise Evolution to Gilteritinib Resistance. Cancer Cell 2021, 39, 999–1014.e8. [Google Scholar] [CrossRef]
- McMahon, C.M.; Ferng, T.; Canaani, J.; Wang, E.S.; Morrissette, J.J.; Eastburn, D.J.; Pellegrino, M.; Durruthy-Durruthy, R.; Watt, C.D.; Asthana, S.; et al. Clonal Selection with RAS Pathway Activation Mediates Secondary Clinical Resistance to Selective FLT3 Inhibition in Acute Myeloid Leukemia. Cancer Discov. 2019, 9, 1050–1063. [Google Scholar] [CrossRef]
- Sabatier, M.; Birsen, R.; Lauture, L.; Mouche, S.; Angelino, P.; Dehairs, J.; Goupille, L.; Boussaid, I.; Heiblig, M.; Boet, E.; et al. C/EBPα Confers Dependence to Fatty Acid Anabolic Pathways and Vulnerability to Lipid Oxidative Stress-Induced Ferroptosis in FLT3-Mutant Leukemia. Cancer Discov. 2023, 13, 1720–1747. [Google Scholar] [CrossRef]
- Smith, C.C.; Wang, Q.; Chin, C.-S.; Salerno, S.; Damon, L.E.; Levis, M.J.; Perl, A.E.; Travers, K.J.; Wang, S.; Hunt, J.P.; et al. Validation of ITD Mutations in FLT3 as a Therapeutic Target in Human Acute Myeloid Leukaemia. Nature 2012, 485, 260–263. [Google Scholar] [CrossRef] [PubMed]
- Smith, C.C.; Paguirigan, A.; Jeschke, G.R.; Lin, K.C.; Massi, E.; Tarver, T.; Chin, C.-S.; Asthana, S.; Olshen, A.; Travers, K.J.; et al. Heterogeneous Resistance to Quizartinib in Acute Myeloid Leukemia Revealed by Single-Cell Analysis. Blood 2017, 130, 48–58. [Google Scholar] [CrossRef] [PubMed]
- Göllner, S.; Oellerich, T.; Agrawal-Singh, S.; Schenk, T.; Klein, H.-U.; Rohde, C.; Pabst, C.; Sauer, T.; Lerdrup, M.; Tavor, S.; et al. Loss of the Histone Methyltransferase EZH2 Induces Resistance to Multiple Drugs in Acute Myeloid Leukemia. Nat. Med. 2017, 23, 69–78. [Google Scholar] [CrossRef] [PubMed]
- Gebru, M.T.; Wang, H.-G. Therapeutic Targeting of FLT3 and Associated Drug Resistance in Acute Myeloid Leukemia. J. Hematol. Oncol. 2020, 13, 155. [Google Scholar] [CrossRef] [PubMed]
- Zarnegar-Lumley, S.; Alonzo, T.A.; Gerbing, R.B.; Othus, M.; Sun, Z.; Ries, R.E.; Wang, J.; Leonti, A.; Kutny, M.A.; Ostronoff, F.; et al. Characteristics and Prognostic Impact of IDH Mutations in AML: A COG, SWOG, and ECOG Analysis. Blood Adv. 2023, 7, 5941–5953. [Google Scholar] [CrossRef]
- Yang, H.; Ye, D.; Guan, K.-L.; Xiong, Y. IDH1 and IDH2 Mutations in Tumorigenesis: Mechanistic Insights and Clinical Perspectives. Clin. Cancer Res. 2012, 18, 5562–5571. [Google Scholar] [CrossRef]
- Stein, E.M.; DiNardo, C.D.; Pollyea, D.A.; Fathi, A.T.; Roboz, G.J.; Altman, J.K.; Stone, R.M.; DeAngelo, D.J.; Levine, R.L.; Flinn, I.W.; et al. Enasidenib in Mutant IDH2 Relapsed or Refractory Acute Myeloid Leukemia. Blood 2017, 130, 722–731. [Google Scholar] [CrossRef]
- DiNardo, C.D.; Stein, E.M.; Pigneux, A.; Altman, J.K.; Collins, R.; Erba, H.P.; Watts, J.M.; Uy, G.L.; Winkler, T.; Wang, H.; et al. Outcomes of Patients with IDH1-Mutant Relapsed or Refractory Acute Myeloid Leukemia Receiving Ivosidenib Who Proceeded to Hematopoietic Stem Cell Transplant. Leukemia 2021, 35, 3278–3281. [Google Scholar] [CrossRef]
- Harding, J.J.; Lowery, M.A.; Shih, A.H.; Schvartzman, J.M.; Hou, S.; Famulare, C.; Patel, M.; Roshal, M.; Do, R.K.; Zehir, A.; et al. Isoform Switching as a Mechanism of Acquired Resistance to Mutant Isocitrate Dehydrogenase Inhibition. Cancer Discov. 2018, 8, 1540–1547. [Google Scholar] [CrossRef]
- Intlekofer, A.M.; Shih, A.H.; Wang, B.; Nazir, A.; Rustenburg, A.S.; Albanese, S.K.; Patel, M.; Famulare, C.; Correa, F.M.; Takemoto, N.; et al. Acquired Resistance to IDH Inhibition through Trans or Cis Dimer-Interface Mutations. Nature 2018, 559, 125–129. [Google Scholar] [CrossRef]
- DiNardo, C.D.; Stein, E.M.; De Botton, S.; Roboz, G.J.; Altman, J.K.; Mims, A.S.; Swords, R.; Collins, R.H.; Mannis, G.N.; Pollyea, D.A.; et al. Durable Remissions with Ivosidenib in IDH1-Mutated Relapsed or Refractory AML. N. Engl. J. Med. 2018, 378, 2386–2398. [Google Scholar] [CrossRef]
- Wang, F.; Morita, K.; DiNardo, C.D.; Furudate, K.; Tanaka, T.; Yan, Y.; Patel, K.P.; MacBeth, K.J.; Wu, B.; Liu, G.; et al. Leukemia Stemness and Co-Occurring Mutations Drive Resistance to IDH Inhibitors in Acute Myeloid Leukemia. Nat. Commun. 2021, 12, 2607. [Google Scholar] [CrossRef] [PubMed]
- Amatangelo, M.D.; Quek, L.; Shih, A.; Stein, E.M.; Roshal, M.; David, M.D.; Marteyn, B.; Farnoud, N.R.; De Botton, S.; Bernard, O.A.; et al. Enasidenib Induces Acute Myeloid Leukemia Cell Differentiation to Promote Clinical Response. Blood 2017, 130, 732–741. [Google Scholar] [CrossRef]
- Stuani, L.; Sabatier, M.; Saland, E.; Cognet, G.; Poupin, N.; Bosc, C.; Castelli, F.A.; Gales, L.; Turtoi, E.; Montersino, C.; et al. Mitochondrial Metabolism Supports Resistance to IDH Mutant Inhibitors in Acute Myeloid Leukemia. J. Exp. Med. 2021, 218, e20200924. [Google Scholar] [CrossRef] [PubMed]
- Wong, S.H.K.; Goode, D.L.; Iwasaki, M.; Wei, M.C.; Kuo, H.-P.; Zhu, L.; Schneidawind, D.; Duque-Afonso, J.; Weng, Z.; Cleary, M.L. The H3K4-Methyl Epigenome Regulates Leukemia Stem Cell Oncogenic Potential. Cancer Cell 2015, 28, 198–209. [Google Scholar] [CrossRef]
- Meyer, C.; Schneider, B.; Jakob, S.; Strehl, S.; Attarbaschi, A.; Schnittger, S.; Schoch, C.; Jansen, M.W.J.C.; van Dongen, J.J.M.; den Boer, M.L.; et al. The MLL Recombinome of Acute Leukemias. Leukemia 2006, 20, 777–784. [Google Scholar] [CrossRef]
- Issa, G.C.; Aldoss, I.; DiPersio, J.; Cuglievan, B.; Stone, R.; Arellano, M.; Thirman, M.J.; Patel, M.R.; Dickens, D.S.; Shenoy, S.; et al. The Menin Inhibitor Revumenib in KMT2A-Rearranged or NPM1-Mutant Leukaemia. Nature 2023, 615, 920–924. [Google Scholar] [CrossRef]
- Perner, F.; Stein, E.M.; Wenge, D.V.; Singh, S.; Kim, J.; Apazidis, A.; Rahnamoun, H.; Anand, D.; Marinaccio, C.; Hatton, C.; et al. MEN1 Mutations Mediate Clinical Resistance to Menin Inhibition. Nature 2023, 615, 913–919. [Google Scholar] [CrossRef] [PubMed]
- Adriaanse, F.R.; Schneider, P.; Arentsen-Peters, S.T.; Fonseca, A.M.N.D.; Stutterheim, J.; Pieters, R.; Zwaan, C.M.; Stam, R.W. Distinct Responses to Menin Inhibition and Synergy with DOT1L Inhibition in KMT2A-Rearranged Acute Lymphoblastic and Myeloid Leukemia. Int. J. Mol. Sci. 2024, 25, 6020. [Google Scholar] [CrossRef]
- Soto-Feliciano, Y.M.; Sánchez-Rivera, F.J.; Perner, F.; Barrows, D.W.; Kastenhuber, E.R.; Ho, Y.-J.; Carroll, T.; Xiong, Y.; Anand, D.; Soshnev, A.A.; et al. A Molecular Switch between Mammalian MLL Complexes Dictates Response to Menin-MLL Inhibition. Cancer Discov. 2023, 13, 146–169. [Google Scholar] [CrossRef]
- Issa, G.C.; Aldoss, I.; Thirman, M.J.; DiPersio, J.; Arellano, M.; Blachly, J.S.; Mannis, G.N.; Perl, A.; Dickens, D.S.; McMahon, C.M.; et al. Menin Inhibition with Revumenib for KMT2A-Rearranged Relapsed or Refractory Acute Leukemia (AUGMENT-101). J. Clin. Oncol. 2025, 43, 75–84. [Google Scholar] [CrossRef]
- Heikamp, E.B.; Henrich, J.A.; Perner, F.; Wong, E.M.; Hatton, C.; Wen, Y.; Barwe, S.P.; Gopalakrishnapillai, A.; Xu, H.; Uckelmann, H.J.; et al. The Menin-MLL1 Interaction Is a Molecular Dependency in NUP98-Rearranged AML. Blood 2022, 139, 894–906. [Google Scholar] [CrossRef]
- Rasouli, M.; Blair, H.; Troester, S.; Szoltysek, K.; Cameron, R.; Ashtiani, M.; Krippner-Heidenreich, A.; Grebien, F.; McGeehan, G.; Zwaan, C.M.; et al. The MLL-Menin Interaction Is a Therapeutic Vulnerability in NUP98-Rearranged AML. Hemasphere 2023, 7, e935. [Google Scholar] [CrossRef] [PubMed]
- Janssens, D.H.; Duran, M.; Otto, D.J.; Wu, W.; Xu, Y.; Kirkey, D.; Mullighan, C.G.; Yi, J.S.; Meshinchi, S.; Sarthy, J.F.; et al. MLL Oncoprotein Levels Influence Leukemia Lineage Identities. Nat. Commun. 2024, 15, 9341. [Google Scholar] [CrossRef] [PubMed]
- Fukushima, N.; Minami, Y.; Kakiuchi, S.; Kuwatsuka, Y.; Hayakawa, F.; Jamieson, C.; Kiyoi, H.; Naoe, T. Small-molecule Hedgehog Inhibitor Attenuates the Leukemia-initiation Potential of Acute Myeloid Leukemia Cells. Cancer Sci. 2016, 107, 1422–1429. [Google Scholar] [CrossRef] [PubMed]
- Litingtung, Y.; Dahn, R.D.; Li, Y.; Fallon, J.F.; Chiang, C. Shh and Gli3 Are Dispensable for Limb Skeleton Formation but Regulate Digit Number and Identity. Nature 2002, 418, 979–983. [Google Scholar] [CrossRef]
- te Welscher, P.; Zuniga, A.; Kuijper, S.; Drenth, T.; Goedemans, H.J.; Meijlink, F.; Zeller, R. Progression of Vertebrate Limb Development through SHH-Mediated Counteraction of GLI3. Science 2002, 298, 827–830. [Google Scholar] [CrossRef]
- Chaudhry, P.; Singh, M.; Triche, T.J.; Guzman, M.; Merchant, A.A. GLI3 Repressor Determines Hedgehog Pathway Activation and Is Required for Response to SMO Antagonist Glasdegib in AML. Blood 2017, 129, 3465–3475. [Google Scholar] [CrossRef]
- Pezzotta, A.; Gentile, I.; Genovese, D.; Totaro, M.G.; Battaglia, C.; Leung, A.Y.-H.; Fumagalli, M.; Parma, M.; Cazzaniga, G.; Fazio, G.; et al. HDAC6 Inhibition Decreases Leukemic Stem Cell Expansion Driven by Hedgehog Hyperactivation by Restoring Primary Ciliogenesis. Pharmacol. Res. 2022, 183, 106378. [Google Scholar] [CrossRef]
- Delbridge, A.R.D.; Grabow, S.; Strasser, A.; Vaux, D.L. Thirty Years of BCL-2: Translating Cell Death Discoveries into Novel Cancer Therapies. Nat. Rev. Cancer 2016, 16, 99–109. [Google Scholar] [CrossRef]
- Khoo, K.H.; Verma, C.S.; Lane, D.P. Drugging the P53 Pathway: Understanding the Route to Clinical Efficacy. Nat. Rev. Drug Discov. 2014, 13, 217–236. [Google Scholar] [CrossRef]
- Youle, R.J.; Strasser, A. The BCL-2 Protein Family: Opposing Activities That Mediate Cell Death. Nat. Rev. Mol. Cell Biol. 2008, 9, 47–59. [Google Scholar] [CrossRef] [PubMed]
- Carter, B.Z.; Tao, W.; Mak, P.Y.; Ostermann, L.B.; Mak, D.; McGeehan, G.; Ordentlich, P.; Andreeff, M. Menin Inhibition Decreases Bcl-2 and Synergizes with Venetoclax in NPM1/FLT3-Mutated AML. Blood 2021, 138, 1637–1641. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Riley-Gillis, B.; Han, L.; Jia, Y.; Lodi, A.; Zhang, H.; Ganesan, S.; Pan, R.; Konoplev, S.N.; Sweeney, S.R.; et al. Activation of RAS/MAPK Pathway Confers MCL-1 Mediated Acquired Resistance to BCL-2 Inhibitor Venetoclax in Acute Myeloid Leukemia. Signal Transduct. Target. Ther. 2022, 7, 51. [Google Scholar] [CrossRef] [PubMed]
- Pan, R.; Ruvolo, V.; Mu, H.; Leverson, J.D.; Nichols, G.; Reed, J.C.; Konopleva, M.; Andreeff, M. Synthetic Lethality of Combined Bcl-2 Inhibition and P53 Activation in AML: Mechanisms and Superior Antileukemic Efficacy. Cancer Cell 2017, 32, 748–760.e6. [Google Scholar] [CrossRef]
- Bhatt, S.; Pioso, M.S.; Olesinski, E.A.; Yilma, B.; Ryan, J.A.; Mashaka, T.; Leutz, B.; Adamia, S.; Zhu, H.; Kuang, Y.; et al. Reduced Mitochondrial Apoptotic Priming Drives Resistance to BH3 Mimetics in Acute Myeloid Leukemia. Cancer Cell 2020, 38, 872–890.e6. [Google Scholar] [CrossRef]
- Nechiporuk, T.; Kurtz, S.E.; Nikolova, O.; Liu, T.; Jones, C.L.; D’Alessandro, A.; Culp-Hill, R.; d’Almeida, A.; Joshi, S.K.; Rosenberg, M.; et al. The TP53 Apoptotic Network Is a Primary Mediator of Resistance to BCL2 Inhibition in AML Cells. Cancer Discov. 2019, 9, 910–925. [Google Scholar] [CrossRef]
- Jones, C.L.; Stevens, B.M.; Pollyea, D.A.; Culp-Hill, R.; Reisz, J.A.; Nemkov, T.; Gehrke, S.; Gamboni, F.; Krug, A.; Winters, A.; et al. Nicotinamide Metabolism Mediates Resistance to Venetoclax in Relapsed Acute Myeloid Leukemia Stem Cells. Cell Stem Cell 2020, 27, 748–764.e4. [Google Scholar] [CrossRef]
- Chen, X.; Glytsou, C.; Zhou, H.; Narang, S.; Reyna, D.E.; Lopez, A.; Sakellaropoulos, T.; Gong, Y.; Kloetgen, A.; Yap, Y.S.; et al. Targeting Mitochondrial Structure Sensitizes Acute Myeloid Leukemia to Venetoclax Treatment. Cancer Discov. 2019, 9, 890–909. [Google Scholar] [CrossRef]
- Zhang, H.; Nakauchi, Y.; Köhnke, T.; Stafford, M.; Bottomly, D.; Thomas, R.; Wilmot, B.; McWeeney, S.K.; Majeti, R.; Tyner, J.W. Integrated Analysis of Patient Samples Identifies Biomarkers for Venetoclax Efficacy and Combination Strategies in Acute Myeloid Leukemia. Nat. Cancer 2020, 1, 826–839. [Google Scholar] [CrossRef]
- Wang, E.; Pineda, J.M.B.; Kim, W.J.; Chen, S.; Bourcier, J.; Stahl, M.; Hogg, S.J.; Bewersdorf, J.P.; Han, C.; Singer, M.E.; et al. Modulation of RNA Splicing Enhances Response to BCL2 Inhibition in Leukemia. Cancer Cell 2023, 41, 164–180.e8. [Google Scholar] [CrossRef]
- Han, L.; Zhang, Q.; Shi, C.; Cavazos, A.; Ruvolo, V.R.; Leverson, J.D.; Dail, M.; Phillips, D.C.; Chen, J.; Jin, S.S.; et al. Targeting MAPK Signaling Pathway with Cobimetinib (GDC-0973) Enhances Anti-Leukemia Efficacy of Venetoclax (ABT-199/GDC-0199) in Acute Myeloid Leukemia Models. Clin. Lymphoma Myeloma Leuk. 2017, 17, S282. [Google Scholar] [CrossRef]
- Bestor, T.H. The DNA Methyltransferases of Mammals. Hum. Mol. Genet. 2000, 9, 2395–2402. [Google Scholar] [CrossRef]
- Zhao, A.; Zhou, H.; Yang, J.; Li, M.; Niu, T. Epigenetic Regulation in Hematopoiesis and Its Implications in the Targeted Therapy of Hematologic Malignancies. Signal Transduct. Target. Ther. 2023, 8, 71. [Google Scholar] [CrossRef]
- Tsai, H.-C.; Li, H.; Van Neste, L.; Cai, Y.; Robert, C.; Rassool, F.V.; Shin, J.J.; Harbom, K.M.; Beaty, R.; Pappou, E.; et al. Transient Low Doses of DNA-Demethylating Agents Exert Durable Antitumor Effects on Hematological and Epithelial Tumor Cells. Cancer Cell 2012, 21, 430–446. [Google Scholar] [CrossRef]
- Messingerova, L.; Imrichova, D.; Kavcova, H.; Turakova, K.; Breier, A.; Sulova, Z. Acute Myeloid Leukemia Cells MOLM-13 and SKM-1 Established for Resistance by Azacytidine Are Crossresistant to P-Glycoprotein Substrates. Toxicol. In Vitro 2015, 29, 1405–1415. [Google Scholar] [CrossRef]
- Valencia, A.; Masala, E.; Rossi, A.; Martino, A.; Sanna, A.; Buchi, F.; Canzian, F.; Cilloni, D.; Gaidano, V.; Voso, M.T.; et al. Expression of Nucleoside-Metabolizing Enzymes in Myelodysplastic Syndromes and Modulation of Response to Azacitidine. Leukemia 2014, 28, 621–628. [Google Scholar] [CrossRef] [PubMed]
- Qin, T.; Jelinek, J.; Si, J.; Shu, J.; Issa, J.P.J. Mechanisms of Resistance to 5-Aza-2′-Deoxycytidine in Human Cancer Cell Lines. Blood 2009, 113, 659–667. [Google Scholar] [CrossRef]
- Gu, X.; Tohme, R.; Tomlinson, B.; Sakre, N.; Hasipek, M.; Durkin, L.; Schuerger, C.; Grabowski, D.; Zidan, A.M.; Radivoyevitch, T.; et al. Decitabine- and 5-Azacytidine Resistance Emerges from Adaptive Responses of the Pyrimidine Metabolism Network. Leukemia 2021, 35, 1023–1036. [Google Scholar] [CrossRef]
- Mahfouz, R.Z.; Jankowska, A.; Ebrahem, Q.; Gu, X.; Visconte, V.; Tabarroki, A.; Terse, P.; Covey, J.; Chan, K.; Ling, Y.; et al. Increased CDA Expression/Activity in Males Contributes to Decreased Cytidine Analog Half-Life and Likely Contributes to Worse Outcomes with 5-Azacytidine or Decitabine Therapy. Clin. Cancer Res. 2013, 19, 938–948. [Google Scholar] [CrossRef]
- Oellerich, T.; Schneider, C.; Thomas, D.; Knecht, K.M.; Buzovetsky, O.; Kaderali, L.; Schliemann, C.; Bohnenberger, H.; Angenendt, L.; Hartmann, W.; et al. Selective Inactivation of Hypomethylating Agents by SAMHD1 Provides a Rationale for Therapeutic Stratification in AML. Nat. Commun. 2019, 10, 3475. [Google Scholar] [CrossRef]
- Penter, L.; Liu, Y.; Wolff, J.O.; Yang, L.; Taing, L.; Jhaveri, A.; Southard, J.; Patel, M.; Cullen, N.M.; Pfaff, K.L.; et al. Mechanisms of Response and Resistance to Combined Decitabine and Ipilimumab for Advanced Myeloid Disease. Blood 2023, 141, 1817–1830. [Google Scholar] [CrossRef] [PubMed]
- Dinndorf, P.A.; Andrews, R.G.; Benjamin, D.; Ridgway, D.; Wolff, L.; Bernstein, I.D. Expression of Normal Myeloid-Associated Antigens by Acute Leukemia Cells. Blood 1986, 67, 1048–1053. [Google Scholar] [CrossRef]
- Pagano, L.; Fianchi, L.; Caira, M.; Rutella, S.; Leone, G. The Role of Gemtuzumab Ozogamicin in the Treatment of Acute Myeloid Leukemia Patients. Oncogene 2007, 26, 3679–3690. [Google Scholar] [CrossRef]
- Matsui, H.; Takeshita, A.; Naito, K.; Shinjo, K.; Shigeno, K.; Maekawa, M.; Yamakawa, Y.; Tanimoto, M.; Kobayashi, M.; Ohnishi, K.; et al. Reduced Effect of Gemtuzumab Ozogamicin (CMA-676) on P-Glycoprotein and/or CD34-Positive Leukemia Cells and Its Restoration by Multidrug Resistance Modifiers. Leukemia 2002, 16, 813–819. [Google Scholar] [CrossRef] [PubMed]
- Walter, R.B.; Raden, B.W.; Hong, T.C.; Flowers, D.A.; Bernstein, I.D.; Linenberger, M.L. Multidrug Resistance Protein Attenuates Gemtuzumab Ozogamicin-Induced Cytotoxicity in Acute Myeloid Leukemia Cells. Blood 2003, 102, 1466–1473. [Google Scholar] [CrossRef]
- Walter, R.B.; Raden, B.W.; Cronk, M.R.; Bernstein, I.D.; Appelbaum, F.R.; Banker, D.E. The Peripheral Benzodiazepine Receptor Ligand PK11195 Overcomes Different Resistance Mechanisms to Sensitize AML Cells to Gemtuzumab Ozogamicin. Blood 2004, 103, 4276–4284. [Google Scholar] [CrossRef] [PubMed]
- Lamba, J.K.; Chauhan, L.; Shin, M.; Loken, M.R.; Pollard, J.A.; Wang, Y.C.; Ries, R.E.; Aplenc, R.; Hirsch, B.A.; Raimondi, S.C.; et al. CD33 Splicing Polymorphism Determines Gemtuzumab Ozogamicin Response in De Novo Acute Myeloid Leukemia: Report from Randomized Phase III Children’s Oncology Group Trial AAML0531. J. Clin. Oncol. 2017, 35, 2674–2682. [Google Scholar] [CrossRef]
- Vago, L.; Gojo, I. Immune Escape and Immunotherapy of Acute Myeloid Leukemia. J. Clin. Investig. 2020, 130, 1552–1564. [Google Scholar] [CrossRef]
- Davids, M.S.; Kim, H.T.; Bachireddy, P.; Costello, C.; Liguori, R.; Savell, A.; Lukez, A.P.; Avigan, D.; Chen, Y.B.; McSweeney, P.; et al. Ipilimumab for Patients with Relapse after Allogeneic Transplantation. N. Engl. J. Med. 2016, 375, 143–153. [Google Scholar] [CrossRef]
- Daver, N.; Alotaibi, A.S.; Bücklein, V.; Subklewe, M. T-Cell-Based Immunotherapy of Acute Myeloid Leukemia: Current Concepts and Future Developments. Leukemia 2021, 35, 1843–1863. [Google Scholar] [CrossRef]
- Rosenblatt, J.; Stone, R.M.; Uhl, L.; Neuberg, D.; Joyce, R.; Levine, J.D.; Arnason, J.; McMasters, M.; Luptakova, K.; Jain, S.; et al. Individualized Vaccination of AML Patients in Remission Is Associated with Induction of Antileukemia Immunity and Prolonged Remissions. Sci. Transl. Med. 2016, 8, 368ra171. [Google Scholar] [CrossRef]
- O’Donnell, J.S.; Teng, M.W.; Smyth, M.J. Cancer Immunoediting and Resistance to T Cell-Based Immunotherapy. Nat. Rev. Clin. Oncol. 2019, 16, 151–167. [Google Scholar] [CrossRef]
- Wang, H.; Zhang, Q.; Teng, Q.; Li, Z.; Liu, H.; Wang, Z.M.; Li, B.; Xia, Y.; Jin, J. A Phase 1b Study Evaluating the Safety and Efficacy of AK117 (Anti-CD47 Monoclonal Antibody) in Combination with Azacitidine in Patients with Treatment-Naïve Acute Myeloid Leukemia. Blood 2023, 142, 4280. [Google Scholar] [CrossRef]
- Mesaros, O.; Onciul, M.; Matei, E.; Joldes, C.; Jimbu, L.; Neaga, A.; Serban, O.; Zdrenghea, M.; Nanut, A.M. Macrophages as Potential Therapeutic Targets in Acute Myeloid Leukemia. Biomedicines 2024, 12, 2306. [Google Scholar] [CrossRef]
- Greiner, J.; Götz, M.; Hofmann, S.; Schrezenmeier, H.; Wiesneth, M.; Bullinger, L.; Döhner, H.; Schneider, V. Specific T-Cell Immune Responses against Colony-Forming Cells Including Leukemic Progenitor Cells of AML Patients Were Increased by Immune Checkpoint Inhibition. Cancer Immunol. Immunother. 2020, 69, 629–640. [Google Scholar] [CrossRef]
- Lichtenegger, F.S.; Krupka, C.; Haubner, S.; Köhnke, T.; Subklewe, M. Recent Developments in Immunotherapy of Acute Myeloid Leukemia. J. Hematol. Oncol. 2017, 10, 142. [Google Scholar] [CrossRef]
- Cunningham, I.; Gee, T.; Reich, L.; Kempin, S.; Naval, A.; Clarkson, B. Acute Promyelocytic Leukemia: Treatment Results during a Decade at Memorial Hospital. Blood 1989, 73, 1116–1122. [Google Scholar] [CrossRef]
- Yilmaz, M.; Kantarjian, H.; Ravandi, F. Acute Promyelocytic Leukemia Current Treatment Algorithms. Blood Cancer J. 2021, 11, 123. [Google Scholar] [CrossRef]
- Vadakekolathu, J.; Minden, M.D.; Hood, T.; Church, S.E.; Reeder, S.; Altmann, H.; Sullivan, A.H.; Viboch, E.J.; Patel, T.; Ibrahimova, N.; et al. Immune Landscapes Predict Chemotherapy Resistance and Immunotherapy Response in Acute Myeloid Leukemia. Sci. Transl. Med. 2020, 12, eaaz0463. [Google Scholar] [CrossRef] [PubMed]
- Jin, J.; Hou, S.; Yao, Y.; Liu, M.; Mao, L.; Yang, M.; Tong, H.; Zeng, T.; Huang, J.; Zhu, Y.; et al. Phosphoproteomic Characterization and Kinase Signature Predict Response to Venetoclax Plus 3+7 Chemotherapy in Acute Myeloid Leukemia. Adv. Sci. 2024, 11, 2305885. [Google Scholar] [CrossRef] [PubMed]
- Moncada, R.; Barkley, D.; Wagner, F.; Chiodin, M.; Devlin, J.C.; Baron, M.; Hajdu, C.H.; Simeone, D.M.; Yanai, I. Integrating Microarray-Based Spatial Transcriptomics and Single-Cell RNA-Seq Reveals Tissue Architecture in Pancreatic Ductal Adenocarcinomas. Nat. Biotechnol. 2020, 38, 333–342. [Google Scholar] [CrossRef]
- Glytsou, C.; Chen, X.; Zacharioudakis, E.; Al-Santli, W.; Zhou, H.; Nadorp, B.; Lee, S.; Lasry, A.; Sun, Z.; Papaioannou, D.; et al. Mitophagy Promotes Resistance to BH3 Mimetics in Acute Myeloid Leukemia. Cancer Discov. 2023, 13, 1656–1677. [Google Scholar] [CrossRef]
- Burd, A.; Levine, R.L.; Ruppert, A.S.; Mims, A.S.; Borate, U.; Stein, E.M.; Patel, P.; Baer, M.R.; Stock, W.; Deininger, M.; et al. Precision Medicine Treatment in Acute Myeloid Leukemia Using Prospective Genomic Profiling: Feasibility and Preliminary Efficacy of the Beat AML Master Trial. Nat. Med. 2020, 26, 1852–1858. [Google Scholar] [CrossRef]
- Yamaura, T.; Nakatani, T.; Uda, K.; Ogura, H.; Shin, W.; Kurokawa, N.; Saito, K.; Fujikawa, N.; Date, T.; Takasaki, M.; et al. A Novel Irreversible FLT3 Inhibitor, FF-10101, Shows Excellent Efficacy against AML Cells with FLT3 Mutations. Blood 2018, 131, 426–438. [Google Scholar] [CrossRef]
- Mellinghoff, I.K.; van den Bent, M.J.; Blumenthal, D.T.; Touat, M.; Peters, K.B.; Clarke, J.; Mendez, J.; Yust-Katz, S.; Welsh, L.; Mason, W.P.; et al. Vorasidenib in IDH1- or IDH2-Mutant Low-Grade Glioma. N. Engl. J. Med. 2023, 389, 589–601. [Google Scholar] [CrossRef]
- Desikan, S.P.; Daver, N.; DiNardo, C.; Kadia, T.; Konopleva, M.; Ravandi, F. Resistance to Targeted Therapies: Delving into FLT3 and IDH. Blood Cancer J. 2022, 12, 91. [Google Scholar] [CrossRef]
- Yeung, Y.A.; Krishnamoorthy, V.; Dettling, D.; Sommer, C.; Poulsen, K.; Ni, I.; Pham, A.; Chen, W.; Liao-Chan, S.; Lindquist, K.; et al. An Optimized Full-Length FLT3/CD3 Bispecific Antibody Demonstrates Potent Anti-Leukemia Activity and Reversible Hematological Toxicity. Mol. Ther. 2020, 28, 889–900. [Google Scholar] [CrossRef] [PubMed]
- Loghavi, S.; Wei, Q.; Ravandi, F.; Quesada, A.E.; Routbort, M.J.; Hu, S.; Toruner, G.A.; Wang, S.A.; Wang, W.; Miranda, R.N.; et al. Optical Genome Mapping Improves the Accuracy of Classification, Risk Stratification, and Personalized Treatment Strategies for Patients with Acute Myeloid Leukemia. Am. J. Hematol. 2024, 99, 1959–1968. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
| Drug | Target | Date of Approval | Monotherapy | Combination | Patient Types |
|---|---|---|---|---|---|
| Midostaurin | FLT3 | 28 April 2017 | Standard cytarabine + daunorubicin induction + cytarabine consolidation | Newly diagnosed AML that is FLT3-mutation-positive | |
| Gilteritinib | FLT3 | 28 November 2018 | √ | Adult patients who have relapsed or refractory AML with an FLT3 mutation | |
| Quizartinib | FLT3 | 20 July 2023 | Standard cytarabine + anthracycline induction + cytarabine consolidation | Adult patients with newly diagnosed AML that is FLT3 ITD-positive | |
| Ivosidenib | IDH1 | 20 July 2018 | √ | Adult patients with relapsed or refractory AML with a susceptible IDH1 mutation | |
| 2 May 2019 | √ | Adult patients with newly diagnosed AML who are ≥75 years old or who have comorbidities that preclude the use of intensive induction chemotherapy | |||
| Olutasidenib | IDH1 | 1 December 2022 | √ | Adult patients with relapsed or refractory AML with a susceptible IDH1 mutation | |
| Enasidenib | IDH2 | 1 August 2017 | √ | Adult patients with relapsed or refractory AML with an IDH2 mutation | |
| Venetoclax | BCL2 | 21 November 2018 | Azacitidine or decitabine or low-dose cytarabine | Newly diagnosed AML in adults who are age 75 years or older, or who have comorbidities that preclude the use of intensive induction chemotherapy | |
| Gemtuzumab Ozogamicin | CD33 | 1 September 2017 | √ | Newly diagnosed CD33-positive AML in adults Relapsed or refractory CD33-positive AML in adults and in pediatric patients 2 years and older | |
| Daunorubicin + cytarabine | Newly diagnosed, de novo AML | ||||
| 16 June 2020 | √ | Newly diagnosed CD33-positive AML in adults and pediatric patients 1 month and older | |||
| Glasdegib | Hedgehog pathway | 21 November 2018 | Low-dose cytarabine | Adult patients who are ≥75 years old or who have comorbidities that preclude the use of intensive induction chemotherapy | |
| Revumenib | Menin | 15 November 2024 | √ | Relapsed or refractory acute leukemia with a KMT2A gene translocation in adult and pediatric patients 1 year and older |
| Mutation | Resistance Mechanisms | Clinical Impact |
|---|---|---|
| TP53 | Disrupts DNA damage repair and apoptosis pathways. Induces genomic instability, accelerating resistant clone expansion. Synergizes with NRAS to promote inflammation via GATA2 downregulation. | Poor prognosis marker. Associated with complex karyotypes. Worse outcomes in elderly patients. |
| RAS | Mutant clones selected under chemotherapy pressure. Synergizes with TP53 to induce inflammation and chemoresistance. | Higher activation frequency in elderly AML patients. Contributes to poor outcomes. |
| MLL | Fusion proteins (e.g., MLL-AF9) activate HOXA9/MEIS1 via DOT1L-mediated H3K79 hypermethylation. Upregulates EMT-related genes (SNAIL, TWIST), enhancing invasiveness. | Prevalent in ~23% of t-AML. Drives relapse through survival pathway activation. |
| FLT3-ITD | Drives clonal expansion with increased allele burden at relapse. Persists post-chemotherapy, dominating relapse. | Independent prognostic factor for chemotherapy failure. Strongly associated with poor survival. |
| IDH1/2 | Neomorphic production of (R)-2-HG inhibits α-KG-dependent enzymes (TET2, Jumonji demethylases), causing epigenetic dysregulation. Promotes leukemic stem cell survival. | Early acquisition and stable retention during progression. Drives relapse and chemoresistance. |
| DNMT3A | Causes global hypomethylation and focal promoter hypermethylation. R882H mutation activates NRF2/NQO1 pathway, reducing daunorubicin sensitivity. Synergizes with NPM1c/FLT3-ITD to upregulate anti-apoptotic genes and disrupt chromatin remodeling. Promotes TWIST1 expression, facilitating extramedullary infiltration. | Higher relapse rates and reduced OS. R882H associated with poor outcomes in younger patients. |
| ASXL1 | Truncation mutations disrupt PRC2-mediated H3K27 methylation, sustaining LSCs in an undifferentiated state. Synergizes with CEBPA mutations to suppress ribosome biogenesis, DNA damage response, and immune activation. | Associated with poor prognosis and chemoresistance. Accelerates AML progression in synergy with other mutations. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cao, S.; Wang, Q.; Zhu, G. From Chemotherapy to Targeted Therapy: Unraveling Resistance in Acute Myeloid Leukemia Through Genetic and Non-Genetic Insights. Int. J. Mol. Sci. 2025, 26, 4005. https://doi.org/10.3390/ijms26094005
Cao S, Wang Q, Zhu G. From Chemotherapy to Targeted Therapy: Unraveling Resistance in Acute Myeloid Leukemia Through Genetic and Non-Genetic Insights. International Journal of Molecular Sciences. 2025; 26(9):4005. https://doi.org/10.3390/ijms26094005
Chicago/Turabian StyleCao, Shuting, Qiuxia Wang, and Ganqian Zhu. 2025. "From Chemotherapy to Targeted Therapy: Unraveling Resistance in Acute Myeloid Leukemia Through Genetic and Non-Genetic Insights" International Journal of Molecular Sciences 26, no. 9: 4005. https://doi.org/10.3390/ijms26094005
APA StyleCao, S., Wang, Q., & Zhu, G. (2025). From Chemotherapy to Targeted Therapy: Unraveling Resistance in Acute Myeloid Leukemia Through Genetic and Non-Genetic Insights. International Journal of Molecular Sciences, 26(9), 4005. https://doi.org/10.3390/ijms26094005