
Alterations in Immune Cell Profiles in the Liver in Diabetes Mellitus: A Systematic Review


Abstract
1. Introduction
2. Materials and Methods
2.1. Search Protocol and Study Eligibility
2.2. Data Extraction
2.3. Risk of Bias Assessment
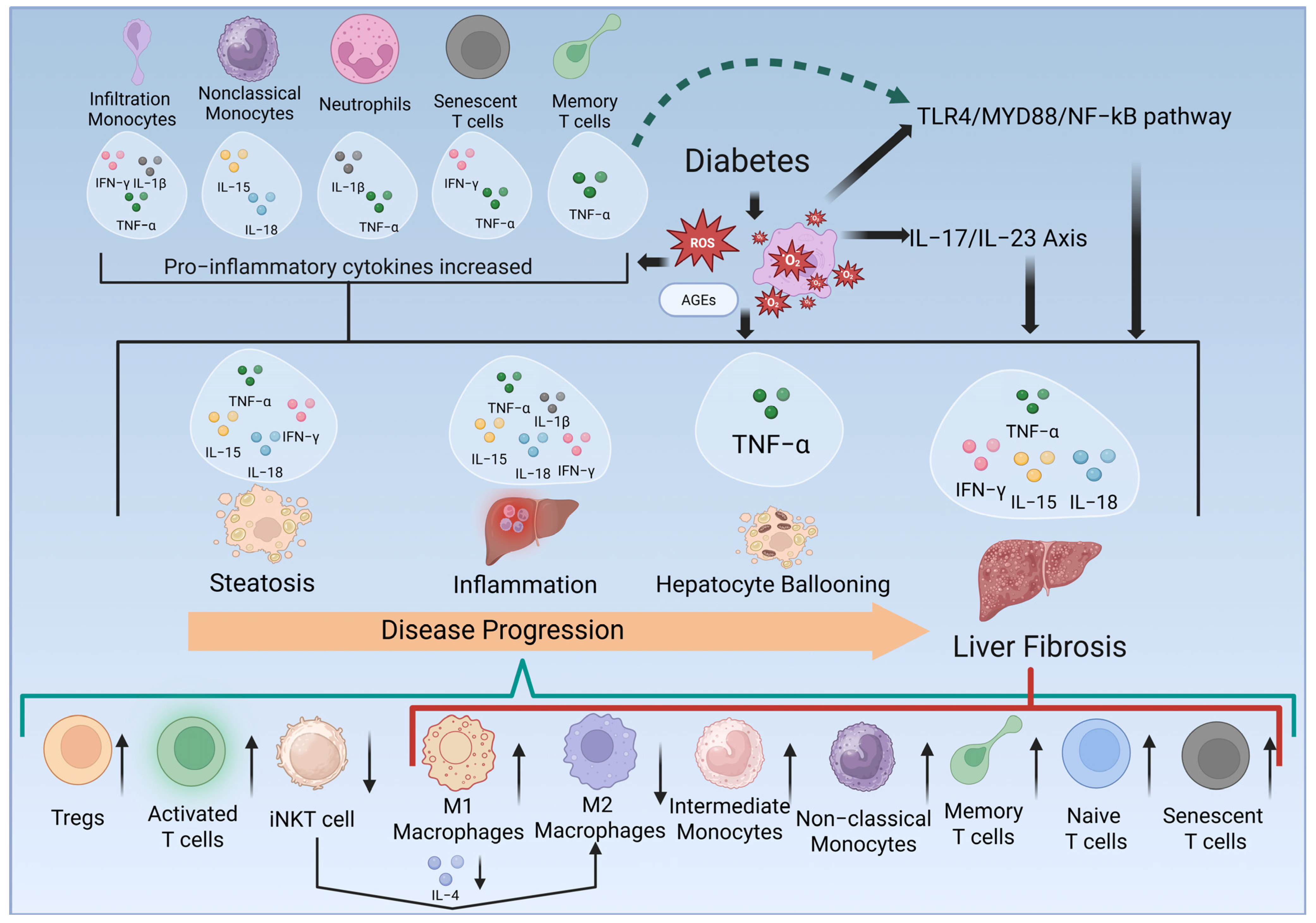
3. Results
3.1. Search Results
3.2. Monocytes/Macrophages
3.3. Neutrophils
3.4. iNKT Cells
3.5. T Cells
4. Discussion
4.1. Innate Immune Cells
4.2. Adaptive Immune Cells
4.3. Related Cytokines and Signalling Pathways
4.4. Limitations
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| AP-1 | activator protein-1 |
| AGEs | advanced glycation end products |
| AMPK | AMP-activated protein kinase |
| ECM | extracellular matrix |
| Foxp3 | forkhead box P3 |
| HSCs | hepatic stellate cells |
| IFN-ɣ | interferon-ɣ |
| IL | interleukin |
| iNKT | invariant natural killer T |
| mTOR | mammalian target of rapamycin |
| MMPs | matrix metalloproteinases |
| MAFLD | metabolic dysfunction-associated fatty liver disease |
| MASH | metabolic dysfunction-associated steatohepatitis |
| MASLD | metabolic dysfunction-associated steatotic liver disease |
| MyD88 | myeloid differentiation primary response 88 |
| NOS | Newcastle-Ottawa Scale |
| NAFLD | non-alcoholic fatty liver disease |
| NASH | non-alcoholic steatohepatitis |
| NF-κB | nuclear factor-kappa B |
| PRISMA | Preferred Reporting Items for Systematic Reviews and Meta-Analyses |
| PD-1 | programmed cell death protein-1 |
| ROS | reactive oxygen species |
| Tregs | regulatory T cells |
| SYRCLE | Systematic Review Centre for Laboratory Animal Experimentation |
| TIMPs | tissue inhibitors of metalloproteinases |
| TLR4 | toll-like receptor 4 |
| TGF-β | transforming growth factor-β |
| TNF-α | tumor necrosis factor-α |
| T1DM | type 1 diabetes mellitus |
| T2DM | type 2 diabetes mellitus |
References
- Vernon, G.; Baranova, A.; Younossi, Z. Systematic review: The epidemiology and natural history of non-alcoholic fatty liver disease and non-alcoholic steatohepatitis in adults. Aliment. Pharmacol. Ther. 2011, 34, 274–285. [Google Scholar] [CrossRef] [PubMed]
- Rinella, M.E.; Lazarus, J.V.; Ratziu, V.; Francque, S.M.; Sanyal, A.J.; Kanwal, F.; Romero, D.; Abdelmalek, M.F.; Anstee, Q.M.; Arab, J.P. A multisociety Delphi consensus statement on new fatty liver disease nomenclature. Hepatology 2023, 78, 1966–1986. [Google Scholar] [CrossRef] [PubMed]
- Cho, E.E.L.; Ang, C.Z.; Quek, J.; Fu, C.E.; Lim, L.K.E.; Heng, Z.E.Q.; Tan, D.J.H.; Lim, W.H.; Yong, J.N.; Zeng, R. Global prevalence of non-alcoholic fatty liver disease in type 2 diabetes mellitus: An updated systematic review and meta-analysis. Gut 2023, 72, 2138–2148. [Google Scholar]
- Stepanova, M.; Clement, S.; Wong, R.; Saab, S.; Ahmed, A.; Younossi, Z.M. Patients with diabetes and chronic liver disease are at increased risk for overall mortality: A population study from the United States. Clin. Diabetes 2017, 35, 79–83. [Google Scholar] [CrossRef]
- Kaneto, H.; Katakami, N.; Matsuhisa, M.; Matsuoka, T.-a. Role of reactive oxygen species in the progression of type 2 diabetes and atherosclerosis. Mediat. Inflamm. 2010, 2010, 453892. [Google Scholar] [CrossRef]
- Hu, H.; Jiang, H.; Ren, H.; Hu, X.; Wang, X.; Han, C. AGEs and chronic subclinical inflammation in diabetes: Disorders of immune system. Diabetes/Metab. Res. Rev. 2015, 31, 127–137. [Google Scholar] [CrossRef]
- Henderson, N.C.; Rieder, F.; Wynn, T.A. Fibrosis: From mechanisms to medicines. Nature 2020, 587, 555–566. [Google Scholar] [CrossRef]
- Chiang, D.J.; Pritchard, M.T.; Nagy, L.E. Obesity, diabetes mellitus, and liver fibrosis. Am. J. Physiol.-Gastrointest. Liver Physiol. 2011, 300, G697–G702. [Google Scholar] [CrossRef]
- Xu, R.; Zhang, Z.; Wang, F.-S. Liver fibrosis: Mechanisms of immune-mediated liver injury. Cell. Mol. Immunol. 2012, 9, 296–301. [Google Scholar] [CrossRef]
- Murray, P.J.; Wynn, T.A. Protective and pathogenic functions of macrophage subsets. Nat. Rev. Immunol. 2011, 11, 723–737. [Google Scholar] [CrossRef]
- Horckmans, M.; Ring, L.; Duchene, J.; Santovito, D.; Schloss, M.J.; Drechsler, M.; Weber, C.; Soehnlein, O.; Steffens, S. Neutrophils orchestrate post-myocardial infarction healing by polarizing macrophages towards a reparative phenotype. Eur. Heart J. 2017, 38, 187–197. [Google Scholar] [CrossRef] [PubMed]
- Kubes, P. The enigmatic neutrophil: What we do not know. Cell Tissue Res. 2018, 371, 399–406. [Google Scholar] [CrossRef] [PubMed]
- Cumpelik, A.; Ankli, B.; Zecher, D.; Schifferli, J.A. Neutrophil microvesicles resolve gout by inhibiting C5a-mediated priming of the inflammasome. Ann. Rheum. Dis. 2016, 75, 1236–1245. [Google Scholar] [CrossRef] [PubMed]
- Hammerich, L.; Tacke, F. Hepatic inflammatory responses in liver fibrosis. Nat. Rev. Gastroenterol. Hepatol. 2023, 20, 633–646. [Google Scholar] [CrossRef]
- Dowey, R.; Iqbal, A.; Heller, S.R.; Sabroe, I.; Prince, L.R. A bittersweet response to infection in diabetes; targeting neutrophils to modify inflammation and improve host immunity. Front. Immunol. 2021, 12, 678771. [Google Scholar] [CrossRef]
- Gu, X.; Chu, Q.; Ma, X.; Wang, J.; Chen, C.; Guan, J.; Ren, Y.; Wu, S.; Zhu, H. New insights into iNKT cells and their roles in liver diseases. Front. Immunol. 2022, 13, 1035950. [Google Scholar] [CrossRef]
- Breuer, D.A.; Pacheco, M.C.; Washington, M.K.; Montgomery, S.A.; Hasty, A.H.; Kennedy, A.J. CD8+ T cells regulate liver injury in obesity-related nonalcoholic fatty liver disease. Am. J. Physiol.-Gastrointest. Liver Physiol. 2020, 318, G211–G224. [Google Scholar] [CrossRef]
- Gong, T.; Liu, L.; Jiang, W.; Zhou, R. DAMP-sensing receptors in sterile inflammation and inflammatory diseases. Nat. Rev. Immunol. 2020, 20, 95–112. [Google Scholar] [CrossRef]
- Liberati, A.; Altman, D.G.; Tetzlaff, J.; Mulrow, C.; Gøtzsche, P.C.; Ioannidis, J.P.; Clarke, M.; Devereaux, P.; Moher, D.; Kleijnen, J. PRISMA Statement per il reporting di revisioni sistematiche e meta-analisi degli studi che valutano gli interventi sanitari: Spiegazione ed elaborazione. Evidence 2015, 7, e1000115. [Google Scholar]
- Hooijmans, C.R.; Rovers, M.M.; De Vries, R.B.; Leenaars, M.; Ritskes-Hoitinga, M.; Langendam, M.W. SYRCLE’s risk of bias tool for animal studies. BMC Med. Res. Methodol. 2014, 14, 43. [Google Scholar] [CrossRef]
- Wells, G.A.; Shea, B.; O’Connell, D.; Peterson, J.; Welch, V.; Losos, M.; Tugwell, P. The Newcastle-Ottawa Scale (NOS) for assessing the quality of nonrandomised studies in meta-analyses. In Proceedings of the Symposiun on Systematic Reviews: Beyond the Basics, Ottawa, ON, Canada, 13 January 2014. [Google Scholar]
- Sim, B.C.; Kang, Y.E.; You, S.K.; Lee, S.E.; Nga, H.T.; Lee, H.Y.; Nguyen, T.L.; Moon, J.S.; Tian, J.; Jang, H.J. Hepatic T-cell senescence and exhaustion are implicated in the progression of fatty liver disease in patients with type 2 diabetes and mouse model with nonalcoholic steatohepatitis. Cell Death Dis. 2023, 14, 618. [Google Scholar] [CrossRef] [PubMed]
- Yi, H.-S.; Kim, S.Y.; Kim, J.T.; Lee, Y.-S.; Moon, J.S.; Kim, M.; Kang, Y.E.; Joung, K.H.; Lee, J.H.; Kim, H.J. T-cell senescence contributes to abnormal glucose homeostasis in humans and mice. Cell Death Dis. 2019, 10, 249. [Google Scholar] [CrossRef] [PubMed]
- Korn, A.; Nadeem, C.; Bos, E.N.; Niessen, H.W.; Simsek, S.; Krijnen, P.A. Hepatic Fat and Macrophages Are Increased in Livers of Diabetic Patients without Non-Alcoholic Fatty Liver Disease. Pathobiology 2023, 90, 409–416. [Google Scholar] [CrossRef]
- Lee, Y.-S.; Eun, H.S.; Kim, S.Y.; Jeong, J.-M.; Seo, W.; Byun, J.-S.; Jeong, W.-I.; Yi, H.-S. Hepatic immunophenotyping for streptozotocin-induced hyperglycemia in mice. Sci. Rep. 2016, 6, 30656. [Google Scholar] [CrossRef]
- Meng, Z.; Liu, X.; Li, T.; Fang, T.; Cheng, Y.; Han, L.; Sun, B.; Chen, L. The SGLT2 inhibitor empagliflozin negatively regulates IL-17/IL-23 axis-mediated inflammatory responses in T2DM with NAFLD via the AMPK/mTOR/autophagy pathway. Int. Immunopharmacol. 2021, 94, 107492. [Google Scholar] [CrossRef]
- Sheikh, R.; Shakerian, S.; Tabatabaei, S.R.F.; Habibi, A. Moderate and high-intensity interval training protect against diabetes-induced modulation of hepatic CD86 and CD206 expression associated with the amelioration of insulin resistance and inflammation in rats. Immunobiology 2023, 228, 152745. [Google Scholar] [CrossRef]
- Han, L.-P.; Li, C.-J.; Sun, B.; Xie, Y.; Guan, Y.; Ma, Z.-J.; Chen, L.-M. Protective effects of celastrol on diabetic liver injury via TLR4/MyD88/NF-κB signaling pathway in type 2 diabetic rats. J. Diabetes Res. 2016, 2016, 2641248. [Google Scholar] [CrossRef]
- Takashima, S.; Ikejima, K.; Arai, K.; Yokokawa, J.; Kon, K.; Yamashina, S.; Watanabe, S. Glycine prevents metabolic steatohepatitis in diabetic KK-Ay mice through modulation of hepatic innate immunity. Am. J. Physiol.-Gastrointest. Liver Physiol. 2016, 311, G1105–G1113. [Google Scholar] [CrossRef]
- Liu, Y.; Cao, Z.-T.; Xu, C.-F.; Lu, Z.-D.; Luo, Y.-L.; Wang, J. Optimization of lipid-assisted nanoparticle for disturbing neutrophils-related inflammation. Biomaterials 2018, 172, 92–104. [Google Scholar] [CrossRef]
- Sbierski-Kind, J.; Schmidt-Bleek, K.; Streitz, M.; Kath, J.; Spranger, J.; Volk, H.-D. An Advanced Murine Model for Nonalcoholic Steatohepatitis in Association with Type 2 Diabetes. J. Vis. Exp. (JoVE) 2019. [Google Scholar] [CrossRef]
- Serrano, D.; Crookshank, J.; Morgan, B.; Mueller, R.; Paré, M.-F.; Marandi, L.; Poussier, P.; Scott, F. Dysregulated liver lipid metabolism and innate immunity associated with hepatic steatosis in neonatal BBdp rats and NOD mice. Sci. Rep. 2019, 9, 14594. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.-C.; Pai, M.-H.; Chen, Y.-T.; Hou, Y.-C. Dietary exposure to chlorpyrifos affects systemic and hepatic immune-cell phenotypes in diabetic mice. Toxicology 2021, 452, 152698. [Google Scholar] [CrossRef]
- Xu, J.; Morinaga, H.; Oh, D.; Li, P.; Chen, A.; Talukdar, S.; Lazarowski, E.; Olefsky, J.M.; Kim, J.J. GPR105 ablation prevents inflammation and improves insulin sensitivity in mice with diet-induced obesity. J. Immunol. 2012, 189, 1992–1999. [Google Scholar] [CrossRef]
- Heymann, F.; Hammerich, L.; Storch, D.; Bartneck, M.; Huss, S.; Rüsseler, V.; Gassler, N.; Lira, S.A.; Luedde, T.; Trautwein, C. Hepatic macrophage migration and differentiation critical for liver fibrosis is mediated by the chemokine receptor C-C motif chemokine receptor 8 in mice. Hepatology 2012, 55, 898–909. [Google Scholar] [CrossRef]
- Serbina, N.V.; Pamer, E.G. Monocyte emigration from bone marrow during bacterial infection requires signals mediated by chemokine receptor CCR2. Nat. Immunol. 2006, 7, 311–317. [Google Scholar] [CrossRef]
- Imamura, M.; Ogawa, T.; Sasaguri, Y.; Chayama, K.; Ueno, H. Suppression of macrophage infiltration inhibits activation of hepatic stellate cells and liver fibrogenesis in rats. Gastroenterology 2005, 128, 138–146. [Google Scholar] [CrossRef]
- Karlmark, K.R.; Weiskirchen, R.; Zimmermann, H.W.; Gassler, N.; Ginhoux, F.; Weber, C.; Merad, M.; Luedde, T.; Trautwein, C.; Tacke, F. Hepatic recruitment of the inflammatory Gr1+ monocyte subset upon liver injury promotes hepatic fibrosis. Hepatology 2009, 50, 261–274. [Google Scholar] [CrossRef]
- Meng, F.; Wang, K.; Aoyama, T.; Grivennikov, S.I.; Paik, Y.; Scholten, D.; Cong, M.; Iwaisako, K.; Liu, X.; Zhang, M. Interleukin-17 signaling in inflammatory, Kupffer cells, and hepatic stellate cells exacerbates liver fibrosis in mice. Gastroenterology 2012, 143, 765–776.e3. [Google Scholar] [CrossRef]
- Chiu, Y.S.; Wei, C.C.; Lin, Y.J.; Hsu, Y.H.; Chang, M.S. IL-20 and IL-20R1 antibodies protect against liver fibrosis. Hepatology 2014, 60, 1003–1014. [Google Scholar] [CrossRef]
- Nagareddy, P.R.; Murphy, A.J.; Stirzaker, R.A.; Hu, Y.; Yu, S.; Miller, R.G.; Ramkhelawon, B.; Distel, E.; Westerterp, M.; Huang, L.-S. Hyperglycemia promotes myelopoiesis and impairs the resolution of atherosclerosis. Cell Metab. 2013, 17, 695–708. [Google Scholar] [CrossRef]
- Zhang, J.; Liu, Y.; Chen, H.; Yuan, Q.; Wang, J.; Niu, M.; Hou, L.; Gu, J.; Zhang, J. MyD88 in hepatic stellate cells enhances liver fibrosis via promoting macrophage M1 polarization. Cell Death Dis. 2022, 13, 411. [Google Scholar] [CrossRef] [PubMed]
- Satoh, N.; Shimatsu, A.; Himeno, A.; Sasaki, Y.; Yamakage, H.; Yamada, K.; Suganami, T.; Ogawa, Y. Unbalanced M1/M2 phenotype of peripheral blood monocytes in obese diabetic patients: Effect of pioglitazone. Diabetes Care 2010, 33, e7. [Google Scholar] [CrossRef] [PubMed]
- Becker, K.J. Strain-related differences in the immune response: Relevance to human stroke. Transl. Stroke Res. 2016, 7, 303–312. [Google Scholar] [CrossRef] [PubMed]
- Liaskou, E.; Zimmermann, H.W.; Li, K.K.; Oo, Y.H.; Suresh, S.; Stamataki, Z.; Qureshi, O.; Lalor, P.F.; Shaw, J.; Syn, W.k. Monocyte subsets in human liver disease show distinct phenotypic and functional characteristics. Hepatology 2013, 57, 385–398. [Google Scholar] [CrossRef]
- Xu, R.; Huang, H.; Zhang, Z.; Wang, F.-S. The role of neutrophils in the development of liver diseases. Cell. Mol. Immunol. 2014, 11, 224–231. [Google Scholar] [CrossRef]
- Zimmermann, H.W.; Tacke, F. Modification of chemokine pathways and immune cell infiltration as a novel therapeutic approach in liver inflammation and fibrosis. Inflamm. Allergy-Drug Targets (Former. Curr. Drug Targets-Inflamm. Allergy) (Discontin.) 2011, 10, 509–536. [Google Scholar] [CrossRef]
- Yang, L.; Jhaveri, R.; Huang, J.; Qi, Y.; Diehl, A.M. Endoplasmic reticulum stress, hepatocyte CD1d and NKT cell abnormalities in murine fatty livers. Lab. Investig. 2007, 87, 927–937. [Google Scholar] [CrossRef]
- Zhu, H.; Zhang, Q.; Chen, G. CXCR6 deficiency ameliorates ischemia-reperfusion injury by reducing the recruitment and cytokine production of hepatic NKT cells in a mouse model of non-alcoholic fatty liver disease. Int. Immunopharmacol. 2019, 72, 224–234. [Google Scholar] [CrossRef]
- Tajiri, K.; Shimizu, Y. Role of NKT cells in the pathogenesis of NAFLD. Int. J. Hepatol. 2012, 2012, 850836. [Google Scholar] [CrossRef]
- Wang, H.; Feng, D.; Park, O.; Yin, S.; Gao, B. Invariant NKT cell activation induces neutrophil accumulation and hepatitis: Opposite regulation by IL-4 and IFN-γ. Hepatology 2013, 58, 1474–1485. [Google Scholar] [CrossRef]
- Park, O.; Jeong, W.I.; Wang, L.; Wang, H.; Lian, Z.X.; Gershwin, M.E.; Gao, B. Diverse roles of invariant natural killer T cells in liver injury and fibrosis induced by carbon tetrachloride. Hepatology 2009, 49, 1683–1694. [Google Scholar] [CrossRef] [PubMed]
- Akbar, A.N.; Henson, S.M. Are senescence and exhaustion intertwined or unrelated processes that compromise immunity? Nat. Rev. Immunol. 2011, 11, 289–295. [Google Scholar] [CrossRef] [PubMed]
- Raphael, I.; Nalawade, S.; Eagar, T.N.; Forsthuber, T.G. T cell subsets and their signature cytokines in autoimmune and inflammatory diseases. Cytokine 2015, 74, 5–17. [Google Scholar] [CrossRef] [PubMed]
- Madakamutil, L.T.; Maricic, I.; Sercarz, E.; Kumar, V. Regulatory T cells control autoimmunity in vivo by inducing apoptotic depletion of activated pathogenic lymphocytes. J. Immunol. 2003, 170, 2985–2992. [Google Scholar] [CrossRef]
- Jiang, G.; Yang, H.-R.; Wang, L.; Wildey, G.M.; Fung, J.; Qian, S.; Lu, L. Hepatic stellate cells preferentially expand allogeneic CD4+ CD25+ FoxP3+ regulatory T cells in an IL-2-dependent manner. Transplantation 2008, 86, 1492–1502. [Google Scholar] [CrossRef]
- Zhang, X.; Feng, M.; Liu, X.; Bai, L.; Kong, M.; Chen, Y.; Zheng, S.; Liu, S.; Wan, Y.-J.Y.; Duan, Z. Persistence of cirrhosis is maintained by intrahepatic regulatory T cells that inhibit fibrosis resolution by regulating the balance of tissue inhibitors of metalloproteinases and matrix metalloproteinases. Transl. Res. 2016, 169, 67–79.e2. [Google Scholar] [CrossRef]
- Wu, K.-J.; Qian, Q.-F.; Zhou, J.-R.; Sun, D.-L.; Duan, Y.-F.; Zhu, X.; Sartorius, K.; Lu, Y.-J. Regulatory T cells (Tregs) in liver fibrosis. Cell Death Discov. 2023, 9, 53. [Google Scholar] [CrossRef]
- Wangoo, A.; Laban, C.; Cook, H.T.; Glenville, B.; Shaw, R. Interleukin-10-and corticosteroid-induced reduction in type I procollagen in a human ex vivo scar culture. Int. J. Exp. Pathol. 1997, 78, 33–41. [Google Scholar] [CrossRef]
- Breous, E.; Somanathan, S.; Vandenberghe, L.H.; Wilson, J.M. Hepatic regulatory T cells and Kupffer cells are crucial mediators of systemic T cell tolerance to antigens targeting murine liver. Hepatology 2009, 50, 612–621. [Google Scholar] [CrossRef]
- Li, J.; Qiu, S.-J.; She, W.-M.; Wang, F.-P.; Gao, H.; Li, L.; Tu, C.-T.; Wang, J.-Y.; Shen, X.-Z.; Jiang, W. Significance of the balance between regulatory T (Treg) and T helper 17 (Th17) cells during hepatitis B virus related liver fibrosis. PLoS ONE 2012, 7, e39307. [Google Scholar] [CrossRef]
- Qiao, Y.-C.; Shen, J.; He, L.; Hong, X.-Z.; Tian, F.; Pan, Y.-H.; Liang, L.; Zhang, X.-X.; Zhao, H.-L. Changes of regulatory T cells and of proinflammatory and immunosuppressive cytokines in patients with type 2 diabetes mellitus: A systematic review and meta-analysis. J. Diabetes Res. 2016, 2016, 3694957. [Google Scholar] [CrossRef] [PubMed]
- Kakino, S.; Ohki, T.; Nakayama, H.; Yuan, X.; Otabe, S.; Hashinaga, T.; Wada, N.; Kurita, Y.; Tanaka, K.; Hara, K. Pivotal role of TNF-α in the development and progression of nonalcoholic fatty liver disease in a murine model. Horm. Metab. Res. 2018, 50, 80–87. [Google Scholar] [CrossRef] [PubMed]
- Zelová, H.; Hošek, J. TNF-α signalling and inflammation: Interactions between old acquaintances. Inflamm. Res. 2013, 62, 641–651. [Google Scholar] [CrossRef] [PubMed]
- Seki, E.; Schwabe, R.F. Hepatic inflammation and fibrosis: Functional links and key pathways. Hepatology 2015, 61, 1066–1079. [Google Scholar] [CrossRef]
- Cloutier, M.; Variya, B.; Akbari, S.A.; Rexhepi, F.; Ilangumaran, S.; Ramanathan, S. Profibrogenic role of IL-15 through IL-15 receptor alpha-mediated trans-presentation in the carbon tetrachloride-induced liver fibrosis model. Front. Immunol. 2024, 15, 1404891. [Google Scholar] [CrossRef]
- Cepero-Donates, Y.; Lacraz, G.; Ghobadi, F.; Rakotoarivelo, V.; Orkhis, S.; Mayhue, M.; Chen, Y.-G.; Rola-Pleszczynski, M.; Menendez, A.; Ilangumaran, S. Interleukin-15-mediated inflammation promotes non-alcoholic fatty liver disease. Cytokine 2016, 82, 102–111. [Google Scholar] [CrossRef]
- Knorr, J.; Kaufmann, B.; Inzaugarat, M.E.; Holtmann, T.M.; Geisler, L.; Hundertmark, J.; Kohlhepp, M.S.; Boosheri, L.M.; Chilin-Fuentes, D.R.; Birmingham, A. Interleukin-18 signaling promotes activation of hepatic stellate cells in mouse liver fibrosis. Hepatology 2023, 77, 1968–1982. [Google Scholar] [CrossRef]
- Scott, T.E.; Lewis, C.V.; Zhu, M.; Wang, C.; Samuel, C.S.; Drummond, G.R.; Kemp-Harper, B.K. IL-4 and IL-13 induce equivalent expression of traditional M2 markers and modulation of reactive oxygen species in human macrophages. Sci. Rep. 2023, 13, 19589. [Google Scholar] [CrossRef]
- Wang, N.; Wang, H.; Yao, H.; Wei, Q.; Mao, X.-M.; Jiang, T.; Xiang, J.; Dila, N. Expression and activity of the TLR4/NF-κB signaling pathway in mouse intestine following administration of a short-term high-fat diet. Exp. Ther. Med. 2013, 6, 635–640. [Google Scholar] [CrossRef]
- Wang, Y.; Tu, Q.; Yan, W.; Xiao, D.; Zeng, Z.; Ouyang, Y.; Huang, L.; Cai, J.; Zeng, X.; Chen, Y.-J. CXC195 suppresses proliferation and inflammatory response in LPS-induced human hepatocellular carcinoma cells via regulating TLR4-MyD88-TAK1-mediated NF-κB and MAPK pathway. Biochem. Biophys. Res. Commun. 2015, 456, 373–379. [Google Scholar] [CrossRef]
- Iwakura, Y.; Ishigame, H. The IL-23/IL-17 axis in inflammation. J. Clin. Investig. 2006, 116, 1218–1222. [Google Scholar] [CrossRef] [PubMed]
- Bluestone, J.A.; Herold, K.; Eisenbarth, G. Genetics, pathogenesis and clinical interventions in type 1 diabetes. Nature 2010, 464, 1293–1300. [Google Scholar] [CrossRef] [PubMed]
- Herder, C.; Baumert, J.; Thorand, B.; Koenig, W.; De Jager, W.; Meisinger, C.; Illig, T.; Martin, S.; Kolb, H. Chemokines as risk factors for type 2 diabetes: Results from the MONICA/KORA Augsburg study, 1984–2002. Diabetologia 2006, 49, 921–929. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
| Cell Types | Marker Detected | Direction of Change | Fold-Change | p Value | Altered Cytokines | Activated Pathways | Mice/Rats Models */Human Studies | Reported Liver Pathology |
|---|---|---|---|---|---|---|---|---|
| Monocytes/Macrophages | CD11b+Ly6Chigh | Increased | 2.8 [25] | <0.01 | IL-17/IL-23 Axis [26] and TLR4/MyD88/NF-κB pathway [28] | Normal chow-fed, STZ-induced diabetic mice vs. normal chow-fed mice | Inflammation | |
| CD11b+F4/80int | Increased | 4.6 [25] | <0.01 | ↑IFN-γ [25] | ||||
| ↑TNF-α [25] | ||||||||
| ↑IL-1β [25] | ||||||||
| CD11b+F4/80high | No significant change | N/A [25] | >0.05 | |||||
| F4/80+CD11c+CD206- | Increased | 6.5 [26] | <0.01 | High-fat-diet-fed, STZ-induced T2DM mice vs. normal-chow-diet-fed mice | Steatosis and inflammation | |||
| F4/80+CD11c-CD206+ | No significant change | N/A [26] | >0.05 | |||||
| CD86+ | Increased | 4.5 [26] | <0.01 | |||||
| 2.2 [27] | <0.05 | High-fat-diet-fed, STZ-induced diabetic Wistar rats vs. normal-chow-diet-fed Wistar rats | Inflammation and necrosis and fibrosis | |||||
| CD206+ | Decreased | 3.9 [27] | <0.05 | |||||
| 2 [32] | <0.01 | Diabetes-prone Bio Breeding rats (T1DM) vs. Bio Breeding rats | Steatosis and inflammation | |||||
| CD14++ CD16+int | Increased | NASH: 2.8 [22] | <0.001 | Humans with T2DM and with or without NASH or liver cirrhosis | Steatosis and inflammation and fibrosis | |||
| Cirrhosis:2.3 [22] | <0.001 | |||||||
| CD14+CD16++ | Increased | NAHS: 1.3 [22] | <0.05 | ↑IL-18 and ↑IL-15 [22] | ||||
| Cirrhosis:2.5 [22] | <0.01 | |||||||
| CD68+ | Increased | 2 [24] | <0.001 | Humans with or without T1DM/T2DM | Steatosis observed in T2DM | |||
| N/A [28] | N/A | High-fat-diet-fed, STZ-induced T2DM rats vs. normal-chow-fed rats | Steatosis and inflammation and hepatocyte ballooning and fibrosis | |||||
| F4/80+ | Increased | 1.3 [34] | <0.005 | High-fat-diet-fed T2DM mice vs. normal-chow-diet-fed mice | Inflammation | |||
| Neutrophils | CD11b+Ly6G+ | Increased | 1.5 [25] | <0.05 | ↑TNF-α [25] | Normal chow-fed, STZ-induced diabetic mice vs. normal chow-fed mice | ||
| ↑IL-1β [25] | ||||||||
| 1.5 [30] | <0.05 | |||||||
| 6 [33] | <0.001 | High-fat-diet-fed T2DM mice vs. normal-chow-diet-fed mice | Steatosis and inflammation and hepatocyte ballooning | |||||
| iNKT cells | CD3+NK1.1+ | Decreased | 1.8 [29] | <0.01 | ↓IL-4 [29] | Normal chow-fed, KK-Ay diabetic mice vs. glycine-containing diet-fed KK-Ay mice | Steatosis and inflammation | |
| T cells | CD45+CD4+ | No significant change | N/A [25] | >0.05 | IL-17/IL-23 Axis [26] | Normal chow-fed, STZ-induced diabetic mice vs. normal chow-fed mice | Inflammation | |
| CD45+CD8+ | No significant change | N/A [25] | >0.05 | |||||
| CD4+CD69+ | Increased | 2.5 [33] | <0.05 | Steatosis and inflammation and hepatocyte ballooning | ||||
| CD8+CD69+ | Increased | 2 [33] | <0.05 | |||||
| CD4+CD28-CD57+ | Increased | 2.9 [22] | <0.05 | ↑TNF-α [22] | Individuals with T2DM and with or without NASH or liver cirrhosis | Steatosis and inflammation and fibrosis | ||
| CD8+CD28-CD57+ | Increased | 1.3 [22] | <0.05 | ↑IFN-γ [22] | ||||
| ↑TNF-α [22] | ||||||||
| 1.4 [23] | <0.05 | Individuals with or without T2DM | Inflammation | |||||
| CD4+PD-1+ | Increased | NASH: 1.3 [22] | <0.01 | Individuals with T2DM and with or without NASH or liver cirrhosis | Steatosis and inflammation and fibrosis | |||
| Cirrhosis:1.7 [22] | <0.01 | |||||||
| CD8+PD-1+ | Increased | NASH: 1.4 [22] | <0.05 | |||||
| Cirrhosis:2 [22] | <0.01 | |||||||
| CD4+CD44+CD62L- | Increased | 1.4 [31] | <0.01 | ↑TNF-α [31] | High-fat-diet-fed diabetic mice within specific-pathogen-free (T2DM) environment vs. exposed to open environment | Steatosis and inflammation and hepatocyte ballooning and fibrosis | ||
| CD8+CD44+CD62L- | Increased | 1.5 [31] | <0.001 | ↑TNF-α [31] | ||||
| CD4+CD44-CD62L+ | Decreased | 8.5 [31] | <0.001 | |||||
| CD8+CD44-CD62L+ | Decreased | 3.4 [31] | <0.01 | |||||
| CD25+Foxp3+ | Increased | 4.5 [25] | <0.01 | Normal chow-fed, STZ-induced diabetic mice vs. normal chow-fed mice | Inflammation |
| Liver Pathology | Steatosis | Inflammation | Hepatocyte Ballooning | Necrosis | Fibrosis | |
|---|---|---|---|---|---|---|
| Immune Factors | ||||||
| Immune cells increased | M1 macrophages [26] | Infiltration monocytes [25] | Neutrophils [34] | M1 macrophages [27] | M1 macrophages [27] | |
| M1 macrophages [26,27] | ||||||
| Intermediate monocytes [22] | Intermediate monocytes [22] | Intermediate monocytes [22] | ||||
| Non-classical monocytes [22] | Non-classical monocytes [22] | Non-classical monocytes [22] | ||||
| Neutrophils [34] | Neutrophils [25,33,34] | |||||
| Activated CD4+/CD8+ T cells [33] | Activated CD4+/CD8+ T cells [33] | Activated CD4+/CD8+ T cells [33] | ||||
| Senescent CD4+/CD8+ T cells [22] | Senescent CD4+/CD8+ T cells [22,23] | Senescent CD4+/CD8+ T cells [22] | ||||
| PD-1+CD4+/CD8+ T cells [22] | PD-1+CD4+/CD8+ T cells [22] | Memory CD4+/CD8+ T cells [31] | PD-1+CD4+/CD8+ T cells [22] | |||
| Memory CD4+/CD8+ T cells [31] | Memory CD4+/CD8+ T cells [31] | Memory CD4+/CD8+ T cells [31] | ||||
| T regulatory cells [25] | ||||||
| Immune cells decreased | M2 macrophages [32] | M2 macrophages [27,32] | Naïve CD4+/CD8+ T cells [31] | M2 macrophages [27] | M2 macrophages [27] Naïve CD4+/CD8+ T cells [31] | |
| iNKT cells [29] | iNKT cells [29] | |||||
| Naïve CD4+/CD8+ T cells [31] | Naïve CD4+/CD8+ T cells [31] | |||||
| Cytokines upregulated | IFN-γ [22] TNF-α [22,31] IL-18 [22] IL-15 [22] | IFN-γ [22,25] TNF-α [22,25,31] IL-18 [22] IL-15 [22] IL-1β [25] | TNF-α [31] | IFN-γ [22] TNF-α [22,31] IL-18 [22] IL-15 [22] | ||
| Cytokines downregulated | IL-4 [29] | IL-4 [29] | ||||
| Signaling pathways activated | IL-17/IL-23 axis [26] | IL-17/IL-23 axis [26] | TLR4/MyD88 pathways [28] | TLR4/MyD88 pathways [28] | ||
| TLR4/MyD88 pathways [28] | TLR4/MyD88 pathways [28] | |||||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Du, W.; Siwan, E.; Twigg, S.M.; Min, D. Alterations in Immune Cell Profiles in the Liver in Diabetes Mellitus: A Systematic Review. Int. J. Mol. Sci. 2025, 26, 4027. https://doi.org/10.3390/ijms26094027
Du W, Siwan E, Twigg SM, Min D. Alterations in Immune Cell Profiles in the Liver in Diabetes Mellitus: A Systematic Review. International Journal of Molecular Sciences. 2025; 26(9):4027. https://doi.org/10.3390/ijms26094027
Chicago/Turabian StyleDu, Wanying, Elisha Siwan, Stephen M. Twigg, and Danqing Min. 2025. "Alterations in Immune Cell Profiles in the Liver in Diabetes Mellitus: A Systematic Review" International Journal of Molecular Sciences 26, no. 9: 4027. https://doi.org/10.3390/ijms26094027
APA StyleDu, W., Siwan, E., Twigg, S. M., & Min, D. (2025). Alterations in Immune Cell Profiles in the Liver in Diabetes Mellitus: A Systematic Review. International Journal of Molecular Sciences, 26(9), 4027. https://doi.org/10.3390/ijms26094027