
Triplexes Color the Chromaverse by Modulating Nucleosome Phasing and Anchoring Chromatin Condensates


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Genomes Encode Information by Both Shape and Sequence
3. T-Flipons and TPX Forming Sequences
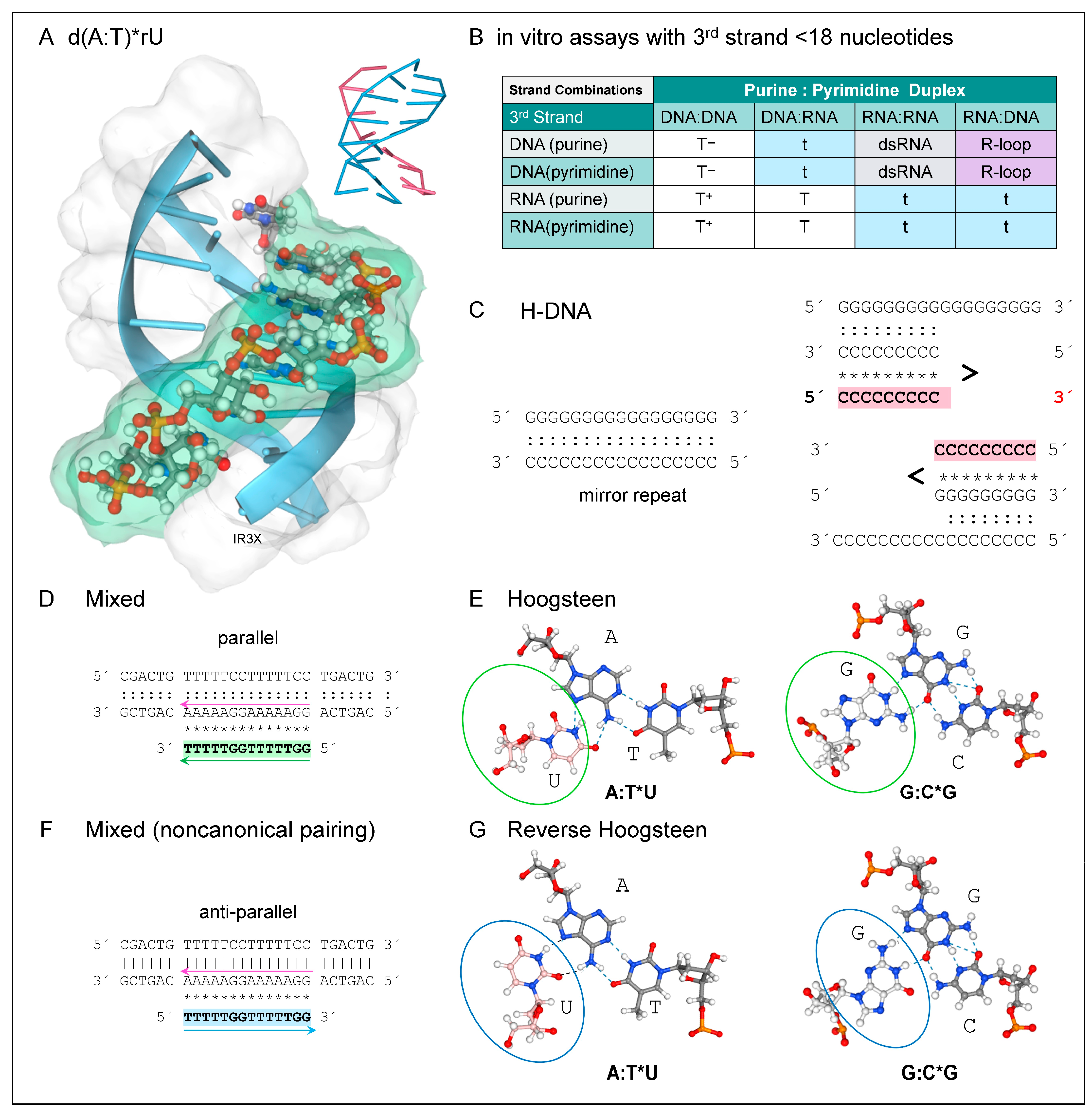
4. DNA and Hybrid TPX
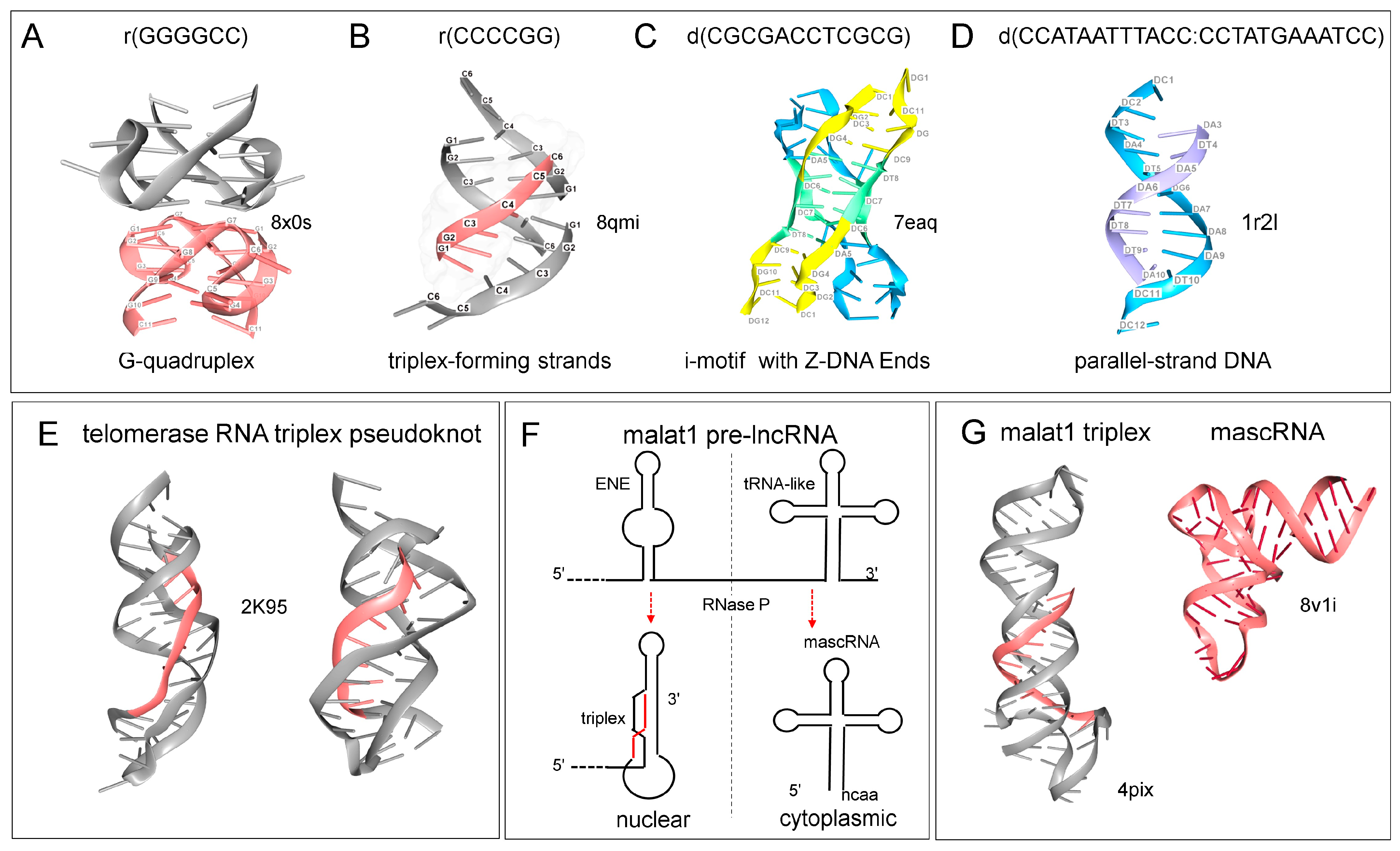
5. Flipons That Fold in Many Different Ways
6. RNA Only TPXs and Other RNA Folds
7. Resolution of TPX
8. Genome-Wide Prediction of TPXs by Computational Approaches
9. Physical Mapping of TPXs In Vivo
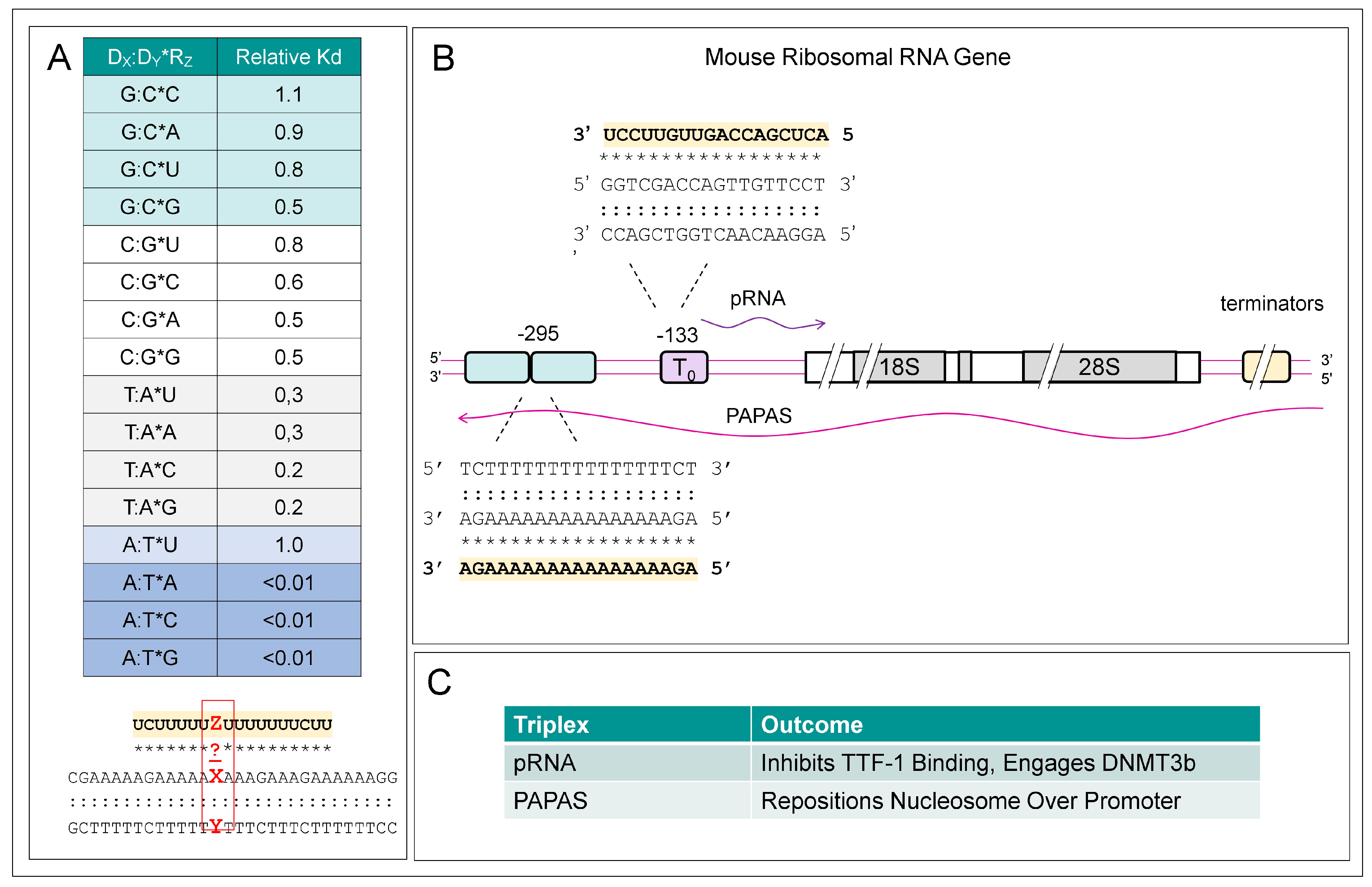
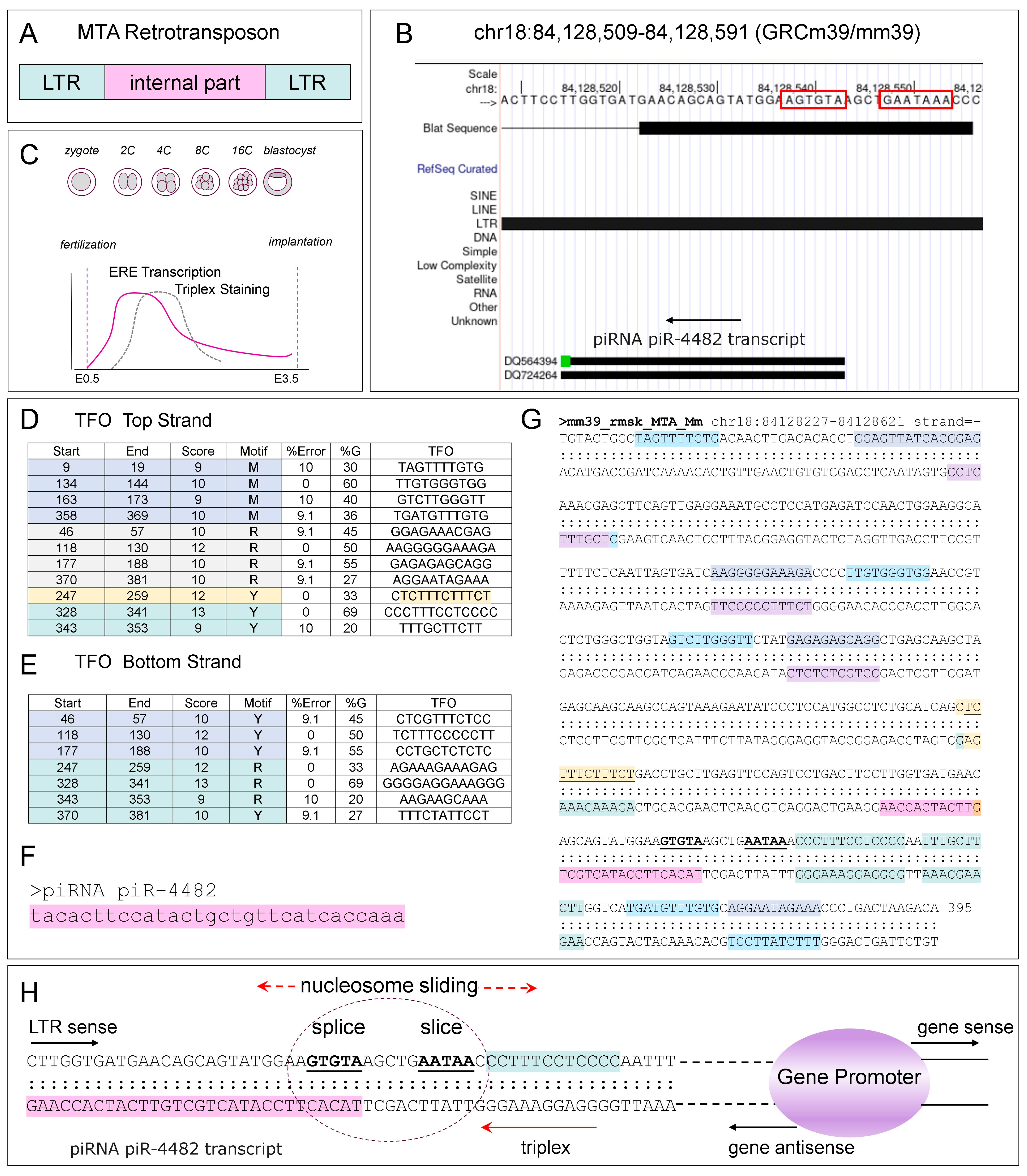
10. TPXs Forming Sequences and NFRs
11. miRNAs and TPXs
12. lncRNAs and TPXs
13. TPXs and EREs
14. lncRNAs, sRNAs, and Chromatin Condensates
15. Helicases and Enhancer–Promoter RNA Interactions
16. Summary
17. Future
Funding
Conflicts of Interest
References
- Judd, B.H. Genes and chromomeres: A puzzle in three dimensions. Genetics 1998, 150, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Filion, G.J.; van Bemmel, J.G.; Braunschweig, U.; Talhout, W.; Kind, J.; Ward, L.D.; Brugman, W.; de Castro, I.J.; Kerkhoven, R.M.; Bussemaker, H.J.; et al. Systematic protein location mapping reveals five principal chromatin types in Drosophila cells. Cell 2010, 143, 212–224. [Google Scholar] [CrossRef] [PubMed]
- Crick, F. Central dogma of molecular biology. Nature 1970, 227, 561–563. [Google Scholar] [CrossRef] [PubMed]
- van Bakel, H.; Nislow, C.; Blencowe, B.J.; Hughes, T.R. Most “dark matter” transcripts are associated with known genes. PLoS Biol. 2010, 8, e1000371. [Google Scholar] [CrossRef]
- Eisen, M.B.; van Bakel, H.; Nislow, C.; Blencowe, B.J.; Hughes, T.R. Response to “The Reality of Pervasive Transcription”. PLoS Biol. 2011, 9, e1001102. [Google Scholar] [CrossRef]
- Eisen, M.B.; Clark, M.B.; Amaral, P.P.; Schlesinger, F.J.; Dinger, M.E.; Taft, R.J.; Rinn, J.L.; Ponting, C.P.; Stadler, P.F.; Morris, K.V.; et al. The Reality of Pervasive Transcription. PLoS Biol. 2011, 9, e1000625. [Google Scholar] [CrossRef]
- Djebali, S.; Davis, C.A.; Merkel, A.; Dobin, A.; Lassmann, T.; Mortazavi, A.; Tanzer, A.; Lagarde, J.; Lin, W.; Schlesinger, F.; et al. Landscape of transcription in human cells. Nature 2012, 489, 101–108. [Google Scholar] [CrossRef]
- Consortium, E.P.; Moore, J.E.; Purcaro, M.J.; Pratt, H.E.; Epstein, C.B.; Shoresh, N.; Adrian, J.; Kawli, T.; Davis, C.A.; Dobin, A.; et al. Expanded encyclopaedias of DNA elements in the human and mouse genomes. Nature 2020, 583, 699–710. [Google Scholar] [CrossRef]
- Consortium, E.P.; Snyder, M.P.; Gingeras, T.R.; Moore, J.E.; Weng, Z.; Gerstein, M.B.; Ren, B.; Hardison, R.C.; Stamatoyannopoulos, J.A.; Graveley, B.R.; et al. Perspectives on ENCODE. Nature 2020, 583, 693–698. [Google Scholar] [CrossRef]
- Li, Y.; Syed, J.; Sugiyama, H. RNA-DNA Triplex Formation by Long Noncoding RNAs. Cell Chem. Biol. 2016, 23, 1325–1333. [Google Scholar] [CrossRef]
- Mattick, J.S.; Amaral, P.P.; Carninci, P.; Carpenter, S.; Chang, H.Y.; Chen, L.L.; Chen, R.; Dean, C.; Dinger, M.E.; Fitzgerald, K.A.; et al. Long non-coding RNAs: Definitions, functions, challenges and recommendations. Nat. Rev. Mol. Cell Biol. 2023, 24, 430–447. [Google Scholar] [CrossRef] [PubMed]
- Velazquez-Flores, M.; Ruiz Esparza-Garrido, R. Fragments derived from non-coding RNAs: How complex is genome regulation? Genome 2024, 67, 292–306. [Google Scholar] [CrossRef] [PubMed]
- Meister, G. Argonaute proteins: Functional insights and emerging roles. Nat. Rev. Genet. 2013, 14, 447–459. [Google Scholar] [CrossRef] [PubMed]
- Ozata, D.M.; Gainetdinov, I.; Zoch, A.; O’Carroll, D.; Zamore, P.D. PIWI-interacting RNAs: Small RNAs with big functions. Nat. Rev. Genet. 2019, 20, 89–108. [Google Scholar] [CrossRef]
- Zhang, H.; Sim, G.; Kehling, A.C.; Adhav, V.A.; Savidge, A.; Pastore, B.; Tang, W.; Nakanishi, K. Target cleavage and gene silencing by Argonautes with cityRNAs. Cell Rep. 2024, 43, 114806. [Google Scholar] [CrossRef]
- Zhang, Y.; Shi, J.; Chen, Q. tsRNAs: New players in mammalian retrotransposon control. Cell Res. 2017, 27, 1307–1308. [Google Scholar] [CrossRef]
- Di Fazio, A.; Gullerova, M. An old friend with a new face: tRNA-derived small RNAs with big regulatory potential in cancer biology. Br. J. Cancer 2023, 128, 1625–1635. [Google Scholar] [CrossRef]
- Sarkar, N.; Kumar, A. Paradigm shift: microRNAs interact with target gene promoters to cause transcriptional gene activation or silencing. Exp. Cell Res. 2025, 444, 114372. [Google Scholar] [CrossRef]
- Li, L.C.; Okino, S.T.; Zhao, H.; Pookot, D.; Place, R.F.; Urakami, S.; Enokida, H.; Dahiya, R. Small dsRNAs induce transcriptional activation in human cells. Proc. Natl. Acad. Sci. USA 2006, 103, 17337–17342. [Google Scholar] [CrossRef]
- Janowski, B.A.; Younger, S.T.; Hardy, D.B.; Ram, R.; Huffman, K.E.; Corey, D.R. Activating gene expression in mammalian cells with promoter-targeted duplex RNAs. Nat. Chem. Biol. 2007, 3, 166–173. [Google Scholar] [CrossRef]
- Brenner, S. Refuge of spandrels. Curr. Biol. 1998, 8, R669. [Google Scholar] [CrossRef] [PubMed]
- Britten, R.J.; Davidson, E.H. Gene Regulation for Higher Cells: A Theory. Science 1969, 165, 349–357. [Google Scholar] [CrossRef] [PubMed]
- Huang, R.; Zhou, P.K. DNA damage repair: Historical perspectives, mechanistic pathways and clinical translation for targeted cancer therapy. Signal Transduct. Target. Ther. 2021, 6, 254. [Google Scholar] [CrossRef] [PubMed]
- Felsenfeld, G.; Davies, D.R.; Rich, A. Formation of a Three-Stranded Polynucleotide Molecule. J. Am. Chem. Soc. 1957, 79, 2023–2024. [Google Scholar] [CrossRef]
- Leisegang, M.S.; Warwick, T.; Stotzel, J.; Brandes, R.P. RNA-DNA triplexes: Molecular mechanisms and functional relevance. Trends Biochem. Sci. 2024, 49, 532–544. [Google Scholar] [CrossRef]
- Hisey, J.A.; Masnovo, C.; Mirkin, S.M. Triplex H-DNA structure: The long and winding road from the discovery to its role in human disease. NAR Mol. Med. 2024, 1, ugae024. [Google Scholar] [CrossRef]
- Arnott, S.; Chandrasekaran, R.; Marttila, C.M. Structures for polyinosinic acid and polyguanylic acid. Biochem. J. 1974, 141, 537–543. [Google Scholar] [CrossRef]
- Chen, M.C.; Tippana, R.; Demeshkina, N.A.; Murat, P.; Balasubramanian, S.; Myong, S.; Ferre-D’Amare, A.R. Structural basis of G-quadruplex unfolding by the DEAH/RHA helicase DHX36. Nature 2018, 558, 465–469. [Google Scholar] [CrossRef]
- Pohl, F.M.; Jovin, T.M. Salt-induced co-operative conformational change of a synthetic DNA: Equilibrium and kinetic studies with poly (dG-dC). J. Mol. Biol. 1972, 67, 375–396. [Google Scholar] [CrossRef]
- Wang, A.H.; Hakoshima, T.; van der Marel, G.; van Boom, J.H.; Rich, A. AT base pairs are less stable than GC base pairs in Z-DNA: The crystal structure of d(m5CGTAm5CG). Cell 1984, 37, 321–331. [Google Scholar] [CrossRef]
- Herbert, A. Flipons and the Logic of Soft-Wired Genomes, 1st ed.; CRC Press: Boca Raton, FL, USA, 2024. [Google Scholar]
- Gehring, K.; Leroy, J.L.; Gueron, M. A tetrameric DNA structure with protonated cytosine.cytosine base pairs. Nature 1993, 363, 561–565. [Google Scholar] [CrossRef] [PubMed]
- Guneri, D.; Alexandrou, E.; El Omari, K.; Dvorakova, Z.; Chikhale, R.V.; Pike, D.T.S.; Waudby, C.A.; Morris, C.J.; Haider, S.; Parkinson, G.N.; et al. Structural insights into i-motif DNA structures in sequences from the insulin-linked polymorphic region. Nat. Commun. 2024, 15, 7119. [Google Scholar] [CrossRef] [PubMed]
- Herbert, A. A Genetic Instruction Code Based on DNA Conformation. Trends Genet. 2019, 35, 887–890. [Google Scholar] [CrossRef] [PubMed]
- Herbert, A. The Simple Biology of Flipons and Condensates Enhances the Evolution of Complexity. Molecules 2021, 26, 4881. [Google Scholar] [CrossRef]
- Lightfoot, H.L.; Hagen, T.; Tatum, N.J.; Hall, J. The diverse structural landscape of quadruplexes. FEBS Lett. 2019, 593, 2083–2102. [Google Scholar] [CrossRef]
- Spiegel, J.; Cuesta, S.M.; Adhikari, S.; Hansel-Hertsch, R.; Tannahill, D.; Balasubramanian, S. G-quadruplexes are transcription factor binding hubs in human chromatin. Genome Biol. 2021, 22, 117. [Google Scholar] [CrossRef]
- Herbert, A. A Compendium of G-Flipon Biological Functions That Have Experimental Validation. Int. J. Mol. Sci. 2024, 25, 10299. [Google Scholar] [CrossRef]
- Zhang, T.; Yin, C.; Fedorov, A.; Qiao, L.; Bao, H.; Beknazarov, N.; Wang, S.; Gautam, A.; Williams, R.M.; Crawford, J.C.; et al. ADAR1 masks the cancer immunotherapeutic promise of ZBP1-driven necroptosis. Nature 2022, 606, 594–602. [Google Scholar] [CrossRef]
- Jiao, H.; Wachsmuth, L.; Wolf, S.; Lohmann, J.; Nagata, M.; Kaya, G.G.; Oikonomou, N.; Kondylis, V.; Rogg, M.; Diebold, M.; et al. ADAR1 averts fatal type I interferon induction by ZBP1. Nature 2022, 607, 776–783. [Google Scholar] [CrossRef]
- Hubbard, N.W.; Ames, J.M.; Maurano, M.; Chu, L.H.; Somfleth, K.Y.; Gokhale, N.S.; Werner, M.; Snyder, J.M.; Lichauco, K.; Savan, R.; et al. ADAR1 mutation causes ZBP1-dependent immunopathology. Nature 2022, 607, 769–775. [Google Scholar] [CrossRef]
- de Reuver, R.; Verdonck, S.; Dierick, E.; Nemegeer, J.; Hessmann, E.; Ahmad, S.; Jans, M.; Blancke, G.; Van Nieuwerburgh, F.; Botzki, A.; et al. ADAR1 prevents autoinflammation by suppressing spontaneous ZBP1 activation. Nature 2022, 607, 784–789. [Google Scholar] [CrossRef] [PubMed]
- Wittig, B.; Dorbic, T.; Rich, A. The level of Z-DNA in metabolically active, permeabilized mammalian cell nuclei is regulated by torsional strain. J. Cell Biol. 1989, 108, 755–764. [Google Scholar] [CrossRef] [PubMed]
- Wittig, B.; Wolfl, S.; Dorbic, T.; Vahrson, W.; Rich, A. Transcription of human c-myc in permeabilized nuclei is associated with formation of Z-DNA in three discrete regions of the gene. EMBO J. 1992, 11, 4653–4663. [Google Scholar] [CrossRef]
- Wolfl, S.; Martinez, C.; Rich, A.; Majzoub, J.A. Transcription of the human corticotropin-releasing hormone gene in NPLC cells is correlated with Z-DNA formation. Proc. Natl. Acad. Sci. USA 1996, 93, 3664–3668. [Google Scholar] [CrossRef]
- Kwon, J.A.; Rich, A. Biological function of the vaccinia virus Z-DNA-binding protein E3L: Gene transactivation and antiapoptotic activity in HeLa cells. Proc. Natl. Acad. Sci. USA 2005, 102, 12759–12764. [Google Scholar] [CrossRef]
- Zhang, J.; Ohta, T.; Maruyama, A.; Hosoya, T.; Nishikawa, K.; Maher, J.M.; Shibahara, S.; Itoh, K.; Yamamoto, M. BRG1 interacts with Nrf2 to selectively mediate HO-1 induction in response to oxidative stress. Mol. Cell. Biol. 2006, 26, 7942–7952. [Google Scholar] [CrossRef]
- Georgakopoulos-Soares, I.; Victorino, J.; Parada, G.E.; Agarwal, V.; Zhao, J.; Wong, H.Y.; Umar, M.I.; Elor, O.; Muhwezi, A.; An, J.Y.; et al. High-throughput characterization of the role of non-B DNA motifs on promoter function. Cell Genom. 2022, 2, 100111. [Google Scholar] [CrossRef]
- Herbert, A. Flipons and small RNAs accentuate the asymmetries of pervasive transcription by the reset and sequence-specific microcoding of promoter conformation. J. Biol. Chem. 2023, 299, 105140. [Google Scholar] [CrossRef]
- Herbert, A. The ancient Z-DNA and Z-RNA specific Zalpha fold has evolved modern roles in immunity and transcription through the natural selection of flipons. R. Soc. Open Sci. 2024, 11, 240080. [Google Scholar] [CrossRef]
- Herbert, A. ALU non-B-DNA conformations, flipons, binary codes and evolution. R. Soc. Open Sci. 2020, 7, 200222. [Google Scholar] [CrossRef]
- Maldonado, R.; Langst, G. The chromatin—Triple helix connection. Biol. Chem. 2023, 404, 1037–1049. [Google Scholar] [CrossRef] [PubMed]
- Roberts, R.W.; Crothers, D.M. Stability and properties of double and triple helices: Dramatic effects of RNA or DNA backbone composition. Science 1992, 258, 1463–1466. [Google Scholar] [CrossRef] [PubMed]
- Han, H.; Dervan, P.B. Sequence-specific recognition of double helical RNA and RNA.DNA by triple helix formation. Proc. Natl. Acad. Sci. USA 1993, 90, 3806–3810. [Google Scholar] [CrossRef]
- Escudeé, C.; Francçois, J.-C.; Sun, J.-S.; Ott, G.; Sprinzl, M.; Garestier, T.s.; Heélene, J.-C. Stability of triple helices containing RNA and DNA strands: Experimental and molecular modeling studies. Nucleic Acids Res. 1993, 21, 5547–5553. [Google Scholar] [CrossRef]
- Sandstrom, K.; Warmlander, S.; Graslund, A.; Leijon, M. A-tract DNA disfavours triplex formation. J. Mol. Biol. 2002, 315, 737–748. [Google Scholar] [CrossRef]
- Cox, R.; Mirkin, S.M. Characteristic enrichment of DNA repeats in different genomes. Proc. Natl. Acad. Sci. USA 1997, 94, 5237–5242. [Google Scholar] [CrossRef]
- Beal, P.A.; Dervan, P.B. Second Structural Motif for Recognition of DNA by Oligonucleotide-Directed Triple-Helix Formation. Science 1991, 251, 1360–1363. [Google Scholar] [CrossRef]
- Escudé, C.; Sun, J.-S. DNA Major Groove Binders: Triple Helix-Forming Oligonucleotides, Triple Helix-Specific DNA Ligands and Cleaving Agents. In DNA Binders and Related Subjects; Topics in Current Chemistry; Springer: Berlin/Heidelberg, Germany, 2005; pp. 109–148. [Google Scholar]
- Mills, M.; Arimondo, P.B.; Lacroix, L.; Garestier, T.; Klump, H.; Mergny, J.L. Chemical modification of the third strand: Differential effects on purine and pyrimidine triple helix formation. Biochemistry 2002, 41, 357–366. [Google Scholar] [CrossRef]
- Sun, J.S.; Mergny, J.L.; Lavery, R.; Montenay-Garestier, T.; Helene, C. Triple helix structures: Sequence dependence, flexibility and mismatch effects. J. Biomol. Struct. Dyn. 1991, 9, 411–424. [Google Scholar] [CrossRef]
- Kunkler, C.N.; Hulewicz, J.P.; Hickman, S.C.; Wang, M.C.; McCown, P.J.; Brown, J.A. Stability of an RNA*DNA-DNA triple helix depends on base triplet composition and length of the RNA third strand. Nucleic Acids Res. 2019, 47, 7213–7222. [Google Scholar] [CrossRef]
- Abu Almakarem, A.S.; Petrov, A.I.; Stombaugh, J.; Zirbel, C.L.; Leontis, N.B. Comprehensive survey and geometric classification of base triples in RNA structures. Nucleic Acids Res. 2012, 40, 1407–1423. [Google Scholar] [CrossRef] [PubMed]
- Fakharzadeh, A.; Zhang, J.; Roland, C.; Sagui, C. Novel eGZ-motif formed by regularly extruded guanine bases in a left-handed Z-DNA helix as a major motif behind CGG trinucleotide repeats. Nucleic Acids Res. 2022, 50, 4860–4876. [Google Scholar] [CrossRef] [PubMed]
- Pan, F.; Xu, P.; Roland, C.; Sagui, C.; Weninger, K. Structural and Dynamical Properties of Nucleic Acid Hairpins Implicated in Trinucleotide Repeat Expansion Diseases. Biomolecules 2024, 14, 1278. [Google Scholar] [CrossRef] [PubMed]
- Cooney, M.; Czernuszewicz, G.; Postel, E.H.; Flint, S.J.; Hogan, M.E. Site-Specific Oligonucleotide Binding Represses Transcription of the Human c-myc Gene In Vitro. Science 1988, 241, 456–459. [Google Scholar] [CrossRef]
- Wang, G.; Vasquez, K.M. Naturally occurring H-DNA-forming sequences are mutagenic in mammalian cells. Proc. Natl. Acad. Sci. USA 2004, 101, 13448–13453. [Google Scholar] [CrossRef]
- Esain-Garcia, I.; Kirchner, A.; Melidis, L.; Tavares, R.C.A.; Dhir, S.; Simeone, A.; Yu, Z.; Madden, S.K.; Hermann, R.; Tannahill, D.; et al. G-quadruplex DNA structure is a positive regulator of MYC transcription. Proc. Natl. Acad. Sci. USA 2024, 121, e2320240121. [Google Scholar] [CrossRef]
- Brown, J.A. Unraveling the structure and biological functions of RNA triple helices. Wiley Interdiscip. Rev. RNA 2020, 11, e1598. [Google Scholar] [CrossRef]
- Kim, N.K.; Zhang, Q.; Zhou, J.; Theimer, C.A.; Peterson, R.D.; Feigon, J. Solution structure and dynamics of the wild-type pseudoknot of human telomerase RNA. J. Mol. Biol. 2008, 384, 1249–1261. [Google Scholar] [CrossRef]
- Ghanim, G.E.; Fountain, A.J.; van Roon, A.M.; Rangan, R.; Das, R.; Collins, K.; Nguyen, T.H.D. Structure of human telomerase holoenzyme with bound telomeric DNA. Nature 2021, 593, 449–453. [Google Scholar] [CrossRef]
- Skeparnias, I.; Bou-Nader, C.; Anastasakis, D.G.; Fan, L.; Wang, Y.X.; Hafner, M.; Zhang, J. Structural basis of MALAT1 RNA maturation and mascRNA biogenesis. Nat. Struct. Mol. Biol. 2024, 31, 1655–1668. [Google Scholar] [CrossRef]
- Gast, M.; Nageswaran, V.; Kuss, A.W.; Tzvetkova, A.; Wang, X.; Mochmann, L.H.; Rad, P.R.; Weiss, S.; Simm, S.; Zeller, T.; et al. tRNA-like Transcripts from the NEAT1-MALAT1 Genomic Region Critically Influence Human Innate Immunity and Macrophage Functions. Cells 2022, 11, 3970. [Google Scholar] [CrossRef] [PubMed]
- Brown, J.A.; Kinzig, C.G.; DeGregorio, S.J.; Steitz, J.A. Methyltransferase-like protein 16 binds the 3′-terminal triple helix of MALAT1 long noncoding RNA. Proc. Natl. Acad. Sci. USA 2016, 113, 14013–14018. [Google Scholar] [CrossRef] [PubMed]
- Satterwhite, E.R.; Mansfield, K.D. RNA methyltransferase METTL16: Targets and function. Wiley Interdiscip. Rev. RNA 2022, 13, e1681. [Google Scholar] [CrossRef]
- Schievelbein, M.J.; Resende, C.; Glennon, M.M.; Kerosky, M.; Brown, J.A. Global RNA modifications to the MALAT1 triple helix differentially affect thermostability and weaken binding to METTL16. J. Biol. Chem. 2024, 300, 105548. [Google Scholar] [CrossRef]
- Nomura, M. Assembly of bacterial ribosomes. Science 1973, 179, 864–873. [Google Scholar] [CrossRef]
- Tapescu, I.; Cherry, S. DDX RNA helicases: Key players in cellular homeostasis and innate antiviral immunity. J. Virol. 2024, 98, e0004024. [Google Scholar] [CrossRef]
- Bohnsack, K.E.; Yi, S.; Venus, S.; Jankowsky, E.; Bohnsack, M.T. Cellular functions of eukaryotic RNA helicases and their links to human diseases. Nat. Rev. Mol. Cell Biol. 2023, 24, 749–769. [Google Scholar] [CrossRef]
- Toyama, Y.; Shimada, I. NMR characterization of RNA binding property of the DEAD-box RNA helicase DDX3X and its implications for helicase activity. Nat. Commun. 2024, 15, 3303. [Google Scholar] [CrossRef]
- Jolma, A.; Zhang, J.; Mondragon, E.; Morgunova, E.; Kivioja, T.; Laverty, K.U.; Yin, Y.; Zhu, F.; Bourenkov, G.; Morris, Q.; et al. Binding specificities of human RNA-binding proteins toward structured and linear RNA sequences. Genome Res. 2020, 30, 962–973. [Google Scholar] [CrossRef]
- Klein, D.J.; Schmeing, T.M.; Moore, P.B.; Steitz, T.A. The kink-turn: A new RNA secondary structure motif. EMBO J. 2001, 20, 4214–4221. [Google Scholar] [CrossRef]
- Schroeder, K.T.; McPhee, S.A.; Ouellet, J.; Lilley, D.M. A structural database for k-turn motifs in RNA. RNA 2010, 16, 1463–1468. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Liu, S.; Zheng, W.; Liu, A.; Yu, P.; Wu, D.; Zhou, J.; Zhang, P.; Liu, C.; Lin, Q.; et al. RIP-PEN-seq identifies a class of kink-turn RNAs as splicing regulators. Nat. Biotechnol. 2024, 42, 119–131. [Google Scholar] [CrossRef] [PubMed]
- Ge, P.; Islam, S.; Zhong, C.; Zhang, S. De novo discovery of structural motifs in RNA 3D structures through clustering. Nucleic Acids Res. 2018, 46, 4783–4793. [Google Scholar] [CrossRef]
- D’Ascenzo, L.; Vicens, Q.; Auffinger, P. Identification of receptors for UNCG and GNRA Z-turns and their occurrence in rRNA. Nucleic Acids Res. 2018, 46, 7989–7997. [Google Scholar] [CrossRef]
- Troisi, R.; Sica, F. Structural overview of DNA and RNA G-quadruplexes in their interaction with proteins. Curr. Opin. Struct. Biol. 2024, 87, 102846. [Google Scholar] [CrossRef]
- Studer, M.K.; Ivanovic, L.; Weber, M.E.; Marti, S.; Jonas, S. Structural basis for DEAH-helicase activation by G-patch proteins. Proc. Natl. Acad. Sci. USA 2020, 117, 7159–7170. [Google Scholar] [CrossRef]
- Warwick, T.; Brandes, R.P.; Leisegang, M.S. Computational Methods to Study DNA:DNA:RNA Triplex Formation by lncRNAs. Noncoding RNA 2023, 9, 10. [Google Scholar] [CrossRef]
- Buske, F.A.; Bauer, D.C.; Mattick, J.S.; Bailey, T.L. Triplexator: Detecting nucleic acid triple helices in genomic and transcriptomic data. Genome Res. 2012, 22, 1372–1381. [Google Scholar] [CrossRef]
- Kuo, C.C.; Hanzelmann, S.; Senturk Cetin, N.; Frank, S.; Zajzon, B.; Derks, J.P.; Akhade, V.S.; Ahuja, G.; Kanduri, C.; Grummt, I.; et al. Detection of RNA-DNA binding sites in long noncoding RNAs. Nucleic Acids Res. 2019, 47, e32. [Google Scholar] [CrossRef]
- Amatria-Barral, I.; Gonzalez-Dominguez, J.; Tourino, J. PATO: Genome-wide prediction of lncRNA-DNA triple helices. Bioinformatics 2023, 39, btad134. [Google Scholar] [CrossRef]
- Cicconetti, C.; Lauria, A.; Proserpio, V.; Masera, M.; Tamburrini, A.; Maldotti, M.; Oliviero, S.; Molineris, I. 3plex enables deep computational investigation of triplex forming lncRNAs. Comput. Struct. Biotechnol. J. 2023, 21, 3091–3102. [Google Scholar] [CrossRef] [PubMed]
- He, S.; Zhang, H.; Liu, H.; Zhu, H. LongTarget: A tool to predict lncRNA DNA-binding motifs and binding sites via Hoogsteen base-pairing analysis. Bioinformatics 2015, 31, 178–186. [Google Scholar] [CrossRef] [PubMed]
- Biffi, G.; Tannahill, D.; McCafferty, J.; Balasubramanian, S. Quantitative visualization of DNA G-quadruplex structures in human cells. Nat. Chem. 2013, 5, 182–186. [Google Scholar] [CrossRef]
- Javadekar, S.M.; Nilavar, N.M.; Paranjape, A.; Das, K.; Raghavan, S.C. Characterization of G-quadruplex antibody reveals differential specificity for G4 DNA forms. DNA Res. 2020, 27, dsaa024. [Google Scholar] [CrossRef]
- Stollar, B.D.; Raso, V. Antibodies recognise specific structures of triple-helical polynucleotides built on poly(A) or poly(dA). Nature 1974, 250, 231–234. [Google Scholar] [CrossRef]
- Gorab, E.; Amabis, J.M.; Stocker, A.J.; Drummond, L.; Stollar, B.D. Potential sites of triple-helical nucleic acid formation in chromosomes of Rhynchosciara (Diptera: Sciaridae) and Drosophila melanogaster. Chromosome Res. 2009, 17, 821–832. [Google Scholar] [CrossRef]
- Lee, J.S.; Burkholder, G.D.; Latimer, L.J.; Haug, B.L.; Braun, R.P. A monoclonal antibody to triplex DNA binds to eucaryotic chromosomes. Nucleic Acids Res. 1987, 15, 1047–1061. [Google Scholar] [CrossRef]
- Agazie, Y.M.; Lee, J.S.; Burkholder, G.D. Characterization of a new monoclonal antibody to triplex DNA and immunofluorescent staining of mammalian chromosomes. J. Biol. Chem. 1994, 269, 7019–7023. [Google Scholar] [CrossRef]
- Senturk Cetin, N.; Kuo, C.C.; Ribarska, T.; Li, R.; Costa, I.G.; Grummt, I. Isolation and genome-wide characterization of cellular DNA:RNA triplex structures. Nucleic Acids Res. 2019, 47, 2306–2321. [Google Scholar] [CrossRef]
- Bonetti, A.; Agostini, F.; Suzuki, A.M.; Hashimoto, K.; Pascarella, G.; Gimenez, J.; Roos, L.; Nash, A.J.; Ghilotti, M.; Cameron, C.J.F.; et al. RADICL-seq identifies general and cell type-specific principles of genome-wide RNA-chromatin interactions. Nat. Commun. 2020, 11, 1018. [Google Scholar] [CrossRef]
- Xu, H.; Ye, J.; Zhang, K.X.; Hu, Q.; Cui, T.; Tong, C.; Wang, M.; Geng, H.; Shui, K.M.; Sun, Y.; et al. Chemoproteomic profiling unveils binding and functional diversity of endogenous proteins that interact with endogenous triplex DNA. Nat. Chem. 2024, 16, 1811–1821. [Google Scholar] [CrossRef] [PubMed]
- Rosu, F.; Nguyen, C.H.; De Pauw, E.; Gabelica, V. Ligand binding mode to duplex and triplex DNA assessed by combining electrospray tandem mass spectrometry and molecular modeling. J. Am. Soc. Mass. Spectrom. 2007, 18, 1052–1062. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, T.C.; Cao, X.; Yu, P.; Xiao, S.; Lu, J.; Biase, F.H.; Sridhar, B.; Huang, N.; Zhang, K.; Zhong, S. Mapping RNA-RNA interactome and RNA structure in vivo by MARIO. Nat. Commun. 2016, 7, 12023. [Google Scholar] [CrossRef] [PubMed]
- Sridhar, B.; Rivas-Astroza, M.; Nguyen, T.C.; Chen, W.; Yan, Z.; Cao, X.; Hebert, L.; Zhong, S. Systematic Mapping of RNA-Chromatin Interactions In Vivo. Curr. Biol. 2017, 27, 602–609. [Google Scholar] [CrossRef]
- Liang, L.; Cao, C.; Ji, L.; Cai, Z.; Wang, D.; Ye, R.; Chen, J.; Yu, X.; Zhou, J.; Bai, Z.; et al. Complementary Alu sequences mediate enhancer-promoter selectivity. Nature 2023, 619, 868–875. [Google Scholar] [CrossRef]
- Espinas, M.L.; Jimenez-Garcia, E.; Martinez-Balbas, A.; Azorin, F. Formation of triple-stranded DNA at d(GA.TC)n sequences prevents nucleosome assembly and is hindered by nucleosomes. J. Biol. Chem. 1996, 271, 31807–31812. [Google Scholar] [CrossRef]
- Brown, P.M.; Fox, K.R. DNA triple-helix formation on nucleosome core particles. Effect of length of the oligopurine tract. Eur. J. Biochem. 1999, 261, 301–310. [Google Scholar] [CrossRef]
- Segal, E.; Widom, J. Poly(dA:dT) tracts: Major determinants of nucleosome organization. Curr. Opin. Struct. Biol. 2009, 19, 65–71. [Google Scholar] [CrossRef]
- Kubik, S.; Bruzzone, M.J.; Jacquet, P.; Falcone, J.L.; Rougemont, J.; Shore, D. Nucleosome Stability Distinguishes Two Different Promoter Types at All Protein-Coding Genes in Yeast. Mol. Cell 2015, 60, 422–434. [Google Scholar] [CrossRef]
- Struhl, K.; Segal, E. Determinants of nucleosome positioning. Nat. Struct. Mol. Biol. 2013, 20, 267–273. [Google Scholar] [CrossRef]
- Chereji, R.V.; Bryson, T.D.; Henikoff, S. Quantitative MNase-seq accurately maps nucleosome occupancy levels. Genome Biol. 2019, 20, 198. [Google Scholar] [CrossRef] [PubMed]
- Prajapati, H.K.; Ocampo, J.; Clark, D.J. Interplay among ATP-Dependent Chromatin Remodelers Determines Chromatin Organisation in Yeast. Biology 2020, 9, 190. [Google Scholar] [CrossRef] [PubMed]
- de Boer, C.G.; Hughes, T.R. Poly-dA:dT tracts form an in vivo nucleosomal turnstile. PLoS ONE 2014, 9, e110479. [Google Scholar] [CrossRef] [PubMed]
- Barnes, T.; Korber, P. The Active Mechanism of Nucleosome Depletion by Poly(dA:dT) Tracts In Vivo. Int. J. Mol. Sci. 2021, 22, 8233. [Google Scholar] [CrossRef]
- Amigo, R.; Raiqueo, F.; Tarifeno, E.; Farkas, C.; Gutierrez, J.L. Poly(dA:dT) Tracts Differentially Modulate Nucleosome Remodeling Activity of RSC and ISW1a Complexes, Exerting Tract Orientation-Dependent and -Independent Effects. Int. J. Mol. Sci. 2023, 24, 15245. [Google Scholar] [CrossRef]
- Hampel, K.J.; Crosson, P.; Lee, J.S. Polyamines favor DNA triplex formation at neutral pH. Biochemistry 1991, 30, 4455–4459. [Google Scholar] [CrossRef]
- Maldonado, R.; Schwartz, U.; Silberhorn, E.; Langst, G. Nucleosomes Stabilize ssRNA-dsDNA Triple Helices in Human Cells. Mol. Cell 2019, 73, 1243–1254.e6. [Google Scholar] [CrossRef]
- Jalali, S.; Singh, A.; Maiti, S.; Scaria, V. Genome-wide computational analysis of potential long noncoding RNA mediated DNA:DNA:RNA triplexes in the human genome. J. Transl. Med. 2017, 15, 186. [Google Scholar] [CrossRef]
- Paugh, S.W.; Coss, D.R.; Bao, J.; Laudermilk, L.T.; Grace, C.R.; Ferreira, A.M.; Waddell, M.B.; Ridout, G.; Naeve, D.; Leuze, M.; et al. MicroRNAs Form Triplexes with Double Stranded DNA at Sequence-Specific Binding Sites; a Eukaryotic Mechanism via which microRNAs Could Directly Alter Gene Expression. PLoS Comput. Biol. 2016, 12, e1004744. [Google Scholar] [CrossRef]
- Schwammle, V.; Sidoli, S.; Ruminowicz, C.; Wu, X.; Lee, C.F.; Helin, K.; Jensen, O.N. Systems Level Analysis of Histone H3 Post-translational Modifications (PTMs) Reveals Features of PTM Crosstalk in Chromatin Regulation. Mol. Cell. Proteom. 2016, 15, 2715–2729. [Google Scholar] [CrossRef]
- Kohestani, H.; Wereszczynski, J. The effects of RNA.DNA-DNA triple helices on nucleosome structures and dynamics. Biophys. J. 2023, 122, 1229–1239. [Google Scholar] [CrossRef] [PubMed]
- Ludwig, N.; Leidinger, P.; Becker, K.; Backes, C.; Fehlmann, T.; Pallasch, C.; Rheinheimer, S.; Meder, B.; Stahler, C.; Meese, E.; et al. Distribution of miRNA expression across human tissues. Nucleic Acids Res. 2016, 44, 3865–3877. [Google Scholar] [CrossRef] [PubMed]
- Toscano-Garibay, J.D.; Aquino-Jarquin, G. Transcriptional regulation mechanism mediated by miRNA-DNA*DNA triplex structure stabilized by Argonaute. Biochim. Biophys. Acta 2014, 1839, 1079–1083. [Google Scholar] [CrossRef]
- Piriyapongsa, J.; Marino-Ramirez, L.; Jordan, I.K. Origin and evolution of human microRNAs from transposable elements. Genetics 2007, 176, 1323–1337. [Google Scholar] [CrossRef]
- Lehnert, S.; Van Loo, P.; Thilakarathne, P.J.; Marynen, P.; Verbeke, G.; Schuit, F.C. Evidence for co-evolution between human microRNAs and Alu-repeats. PLoS ONE 2009, 4, e4456. [Google Scholar] [CrossRef]
- Girard, A.; Sachidanandam, R.; Hannon, G.J.; Carmell, M.A. A germline-specific class of small RNAs binds mammalian Piwi proteins. Nature 2006, 442, 199–202. [Google Scholar] [CrossRef]
- Schorn, A.J.; Gutbrod, M.J.; LeBlanc, C.; Martienssen, R. LTR-Retrotransposon Control by tRNA-Derived Small RNAs. Cell 2017, 170, 61–71.e11. [Google Scholar] [CrossRef]
- Stamidis, N.; Zylicz, J.J. RNA-mediated heterochromatin formation at repetitive elements in mammals. EMBO J. 2023, 42, e111717. [Google Scholar] [CrossRef]
- Mayer, C.; Schmitz, K.M.; Li, J.; Grummt, I.; Santoro, R. Intergenic transcripts regulate the epigenetic state of rRNA genes. Mol. Cell 2006, 22, 351–361. [Google Scholar] [CrossRef]
- Mayer, C.; Neubert, M.; Grummt, I. The structure of NoRC-associated RNA is crucial for targeting the chromatin remodelling complex NoRC to the nucleolus. EMBO Rep. 2008, 9, 774–780. [Google Scholar] [CrossRef]
- Schmitz, K.M.; Mayer, C.; Postepska, A.; Grummt, I. Interaction of noncoding RNA with the rDNA promoter mediates recruitment of DNMT3b and silencing of rRNA genes. Genes Dev. 2010, 24, 2264–2269. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.; Senturk, N.; Song, C.; Grummt, I. lncRNA PAPAS tethered to the rDNA enhancer recruits hypophosphorylated CHD4/NuRD to repress rRNA synthesis at elevated temperatures. Genes Dev. 2018, 32, 836–848. [Google Scholar] [CrossRef] [PubMed]
- Blank-Giwojna, A.; Postepska-Igielska, A.; Grummt, I. lncRNA KHPS1 Activates a Poised Enhancer by Triplex-Dependent Recruitment of Epigenomic Regulators. Cell Rep. 2019, 26, 2904–2915.e2904. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.; Giles, K.E.; Felsenfeld, G. DNA.RNA triple helix formation can function as a cis-acting regulatory mechanism at the human beta-globin locus. Proc. Natl. Acad. Sci. USA 2019, 116, 6130–6139. [Google Scholar] [CrossRef]
- Li, W.; Notani, D.; Rosenfeld, M.G. Enhancers as non-coding RNA transcription units: Recent insights and future perspectives. Nat. Rev. Genet. 2016, 17, 207–223. [Google Scholar] [CrossRef]
- Hon, C.C.; Ramilowski, J.A.; Harshbarger, J.; Bertin, N.; Rackham, O.J.; Gough, J.; Denisenko, E.; Schmeier, S.; Poulsen, T.M.; Severin, J.; et al. An atlas of human long non-coding RNAs with accurate 5′ ends. Nature 2017, 543, 199–204. [Google Scholar] [CrossRef]
- Mondal, T.; Subhash, S.; Vaid, R.; Enroth, S.; Uday, S.; Reinius, B.; Mitra, S.; Mohammed, A.; James, A.R.; Hoberg, E.; et al. MEG3 long noncoding RNA regulates the TGF-beta pathway genes through formation of RNA-DNA triplex structures. Nat. Commun. 2015, 6, 7743. [Google Scholar] [CrossRef]
- Li, Y.; Tan, Z.; Zhang, Y.; Zhang, Z.; Hu, Q.; Liang, K.; Jun, Y.; Ye, Y.; Li, Y.C.; Li, C.; et al. A noncoding RNA modulator potentiates phenylalanine metabolism in mice. Science 2021, 373, 662–673. [Google Scholar] [CrossRef]
- Chen, L.L.; Kim, V.N. Small and long non-coding RNAs: Past, present, and future. Cell 2024, 187, 6451–6485. [Google Scholar] [CrossRef]
- Tanaka, Y.; Yamashita, R.; Suzuki, Y.; Nakai, K. Effects of Alu elements on global nucleosome positioning in the human genome. BMC Genom. 2010, 11, 309. [Google Scholar] [CrossRef]
- Conti, A.; Carnevali, D.; Bollati, V.; Fustinoni, S.; Pellegrini, M.; Dieci, G. Identification of RNA polymerase III-transcribed Alu loci by computational screening of RNA-Seq data. Nucleic Acids Res. 2015, 43, 817–835. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.O.; Gingeras, T.R.; Weng, Z. Genome-wide analysis of polymerase III-transcribed Alu elements suggests cell-type-specific enhancer function. Genome Res. 2019, 29, 1402–1414. [Google Scholar] [CrossRef] [PubMed]
- Policarpi, C.; Crepaldi, L.; Brookes, E.; Nitarska, J.; French, S.M.; Coatti, A.; Riccio, A. Enhancer SINEs Link Pol III to Pol II Transcription in Neurons. Cell Rep. 2017, 21, 2879–2894. [Google Scholar] [CrossRef] [PubMed]
- Hu, Q.; Tanasa, B.; Trabucchi, M.; Li, W.; Zhang, J.; Ohgi, K.A.; Rose, D.W.; Glass, C.K.; Rosenfeld, M.G. DICER- and AGO3-dependent generation of retinoic acid-induced DR2 Alu RNAs regulates human stem cell proliferation. Nat. Struct. Mol. Biol. 2012, 19, 1168–1175. [Google Scholar] [CrossRef]
- Hoffman, Y.; Dahary, D.; Bublik, D.R.; Oren, M.; Pilpel, Y. The majority of endogenous microRNA targets within Alu elements avoid the microRNA machinery. Bioinformatics 2013, 29, 894–902. [Google Scholar] [CrossRef]
- Smalheiser, N.R.; Torvik, V.I. Alu elements within human mRNAs are probable microRNA targets. Trends Genet. 2006, 22, 532–536. [Google Scholar] [CrossRef]
- Soibam, B. Super-lncRNAs: Identification of lncRNAs that target super-enhancers via RNA:DNA:DNA triplex formation. RNA 2017, 23, 1729–1742. [Google Scholar] [CrossRef]
- Allo, M.; Agirre, E.; Bessonov, S.; Bertucci, P.; Gomez Acuna, L.; Buggiano, V.; Bellora, N.; Singh, B.; Petrillo, E.; Blaustein, M.; et al. Argonaute-1 binds transcriptional enhancers and controls constitutive and alternative splicing in human cells. Proc. Natl. Acad. Sci. USA 2014, 111, 15622–15629. [Google Scholar] [CrossRef]
- Shuaib, M.; Parsi, K.M.; Thimma, M.; Adroub, S.A.; Kawaji, H.; Seridi, L.; Ghosheh, Y.; Fort, A.; Fallatah, B.; Ravasi, T.; et al. Nuclear AGO1 Regulates Gene Expression by Affecting Chromatin Architecture in Human Cells. Cell Syst. 2019, 9, 446–458.e446. [Google Scholar] [CrossRef]
- Zhang, X.; Jiang, Q.; Li, J.; Zhang, S.; Cao, Y.; Xia, X.; Cai, D.; Tan, J.; Chen, J.; Han, J.J. KCNQ1OT1 promotes genome-wide transposon repression by guiding RNA-DNA triplexes and HP1 binding. Nat. Cell Biol. 2022, 24, 1617–1629. [Google Scholar] [CrossRef]
- Fadloun, A.; Le Gras, S.; Jost, B.; Ziegler-Birling, C.; Takahashi, H.; Gorab, E.; Carninci, P.; Torres-Padilla, M.E. Chromatin signatures and retrotransposon profiling in mouse embryos reveal regulation of LINE-1 by RNA. Nat. Struct. Mol. Biol. 2013, 20, 332–338. [Google Scholar] [CrossRef] [PubMed]
- Tohonen, V.; Katayama, S.; Vesterlund, L.; Jouhilahti, E.M.; Sheikhi, M.; Madissoon, E.; Filippini-Cattaneo, G.; Jaconi, M.; Johnsson, A.; Burglin, T.R.; et al. Novel PRD-like homeodomain transcription factors and retrotransposon elements in early human development. Nat. Commun. 2015, 6, 8207. [Google Scholar] [CrossRef] [PubMed]
- Jachowicz, J.W.; Bing, X.; Pontabry, J.; Boskovic, A.; Rando, O.J.; Torres-Padilla, M.E. LINE-1 activation after fertilization regulates global chromatin accessibility in the early mouse embryo. Nat. Genet. 2017, 49, 1502–1510. [Google Scholar] [CrossRef] [PubMed]
- Peaston, A.E.; Evsikov, A.V.; Graber, J.H.; de Vries, W.N.; Holbrook, A.E.; Solter, D.; Knowles, B.B. Retrotransposons regulate host genes in mouse oocytes and preimplantation embryos. Dev. Cell 2004, 7, 597–606. [Google Scholar] [CrossRef]
- Napoli, S. Targeting Promoter-Associated RNAs by siRNAs. Methods Mol. Biol. 2017, 1543, 209–219. [Google Scholar] [CrossRef]
- Martianov, I.; Ramadass, A.; Serra Barros, A.; Chow, N.; Akoulitchev, A. Repression of the human dihydrofolate reductase gene by a non-coding interfering transcript. Nature 2007, 445, 666–670. [Google Scholar] [CrossRef]
- Rom, A.; Melamed, L.; Gil, N.; Goldrich, M.J.; Kadir, R.; Golan, M.; Biton, I.; Perry, R.B.; Ulitsky, I. Regulation of CHD2 expression by the Chaserr long noncoding RNA gene is essential for viability. Nat. Commun. 2019, 10, 5092. [Google Scholar] [CrossRef]
- Latos, P.A.; Pauler, F.M.; Koerner, M.V.; Senergin, H.B.; Hudson, Q.J.; Stocsits, R.R.; Allhoff, W.; Stricker, S.H.; Klement, R.M.; Warczok, K.E.; et al. Airn transcriptional overlap, but not its lncRNA products, induces imprinted Igf2r silencing. Science 2012, 338, 1469–1472. [Google Scholar] [CrossRef]
- Song, J.; Gooding, A.R.; Hemphill, W.O.; Love, B.D.; Robertson, A.; Yao, L.; Zon, L.I.; North, T.E.; Kasinath, V.; Cech, T.R. Structural basis for inactivation of PRC2 by G-quadruplex RNA. Science 2023, 381, 1331–1337. [Google Scholar] [CrossRef]
- Chu, C.; Qu, K.; Zhong, F.L.; Artandi, S.E.; Chang, H.Y. Genomic maps of long noncoding RNA occupancy reveal principles of RNA-chromatin interactions. Mol. Cell 2011, 44, 667–678. [Google Scholar] [CrossRef]
- Grote, P.; Wittler, L.; Hendrix, D.; Koch, F.; Wahrisch, S.; Beisaw, A.; Macura, K.; Blass, G.; Kellis, M.; Werber, M.; et al. The tissue-specific lncRNA Fendrr is an essential regulator of heart and body wall development in the mouse. Dev. Cell 2013, 24, 206–214. [Google Scholar] [CrossRef] [PubMed]
- O’Leary, V.B.; Ovsepian, S.V.; Carrascosa, L.G.; Buske, F.A.; Radulovic, V.; Niyazi, M.; Moertl, S.; Trau, M.; Atkinson, M.J.; Anastasov, N. PARTICLE, a Triplex-Forming Long ncRNA, Regulates Locus-Specific Methylation in Response to Low-Dose Irradiation. Cell Rep. 2015, 11, 474–485. [Google Scholar] [CrossRef]
- Kim, E.Z.; Wespiser, A.R.; Caffrey, D.R. The domain structure and distribution of Alu elements in long noncoding RNAs and mRNAs. RNA 2016, 22, 254–264. [Google Scholar] [CrossRef]
- Shaath, H.; Vishnubalaji, R.; Elango, R.; Kardousha, A.; Islam, Z.; Qureshi, R.; Alam, T.; Kolatkar, P.R.; Alajez, N.M. Long non-coding RNA and RNA-binding protein interactions in cancer: Experimental and machine learning approaches. Semin. Cancer Biol. 2022, 86, 325–345. [Google Scholar] [CrossRef]
- Thakur, J.; Henikoff, S. Architectural RNA in chromatin organization. Biochem. Soc. Trans. 2020, 48, 1967–1978. [Google Scholar] [CrossRef]
- Bartolomei, M.S.; Zemel, S.; Tilghman, S.M. Parental imprinting of the mouse H19 gene. Nature 1991, 351, 153–155. [Google Scholar] [CrossRef]
- Cai, X.; Cullen, B.R. The imprinted H19 noncoding RNA is a primary microRNA precursor. RNA 2007, 13, 313–316. [Google Scholar] [CrossRef]
- Dey, B.K.; Pfeifer, K.; Dutta, A. The H19 long noncoding RNA gives rise to microRNAs miR-675-3p and miR-675-5p to promote skeletal muscle differentiation and regeneration. Genes Dev. 2014, 28, 491–501. [Google Scholar] [CrossRef]
- Ouvrard, J.; Muniz, L.; Nicolas, E.; Trouche, D. Small Interfering RNAs Targeting a Chromatin-Associated RNA Induce Its Transcriptional Silencing in Human Cells. Mol. Cell. Biol. 2022, 42, e0027122. [Google Scholar] [CrossRef]
- Xiao, M.; Li, J.; Li, W.; Wang, Y.; Wu, F.; Xi, Y.; Zhang, L.; Ding, C.; Luo, H.; Li, Y.; et al. MicroRNAs activate gene transcription epigenetically as an enhancer trigger. RNA Biol. 2017, 14, 1326–1334. [Google Scholar] [CrossRef]
- Liang, Y.; Zou, Q.; Yu, W. Steering Against Wind: A New Network of NamiRNAs and Enhancers. Genom. Proteom. Bioinform. 2017, 15, 331–337. [Google Scholar] [CrossRef] [PubMed]
- Kozomara, A.; Griffiths-Jones, S. miRBase: Annotating high confidence microRNAs using deep sequencing data. Nucleic Acids Res. 2014, 42, D68–D73. [Google Scholar] [CrossRef] [PubMed]
- Ohno, S.I.; Oikawa, K.; Tsurui, T.; Harada, Y.; Ono, K.; Tateishi, M.; Mirza, A.; Takanashi, M.; Kanekura, K.; Nagase, K.; et al. Nuclear microRNAs release paused Pol II via the DDX21-CDK9 complex. Cell Rep. 2022, 39, 110673. [Google Scholar] [CrossRef]
- Flynn, R.A.; Do, B.T.; Rubin, A.J.; Calo, E.; Lee, B.; Kuchelmeister, H.; Rale, M.; Chu, C.; Kool, E.T.; Wysocka, J.; et al. 7SK-BAF axis controls pervasive transcription at enhancers. Nat. Struct. Mol. Biol. 2016, 23, 231–238. [Google Scholar] [CrossRef]
- Hainer, S.J.; Gu, W.; Carone, B.R.; Landry, B.D.; Rando, O.J.; Mello, C.C.; Fazzio, T.G. Suppression of pervasive noncoding transcription in embryonic stem cells by esBAF. Genes Dev. 2015, 29, 362–378. [Google Scholar] [CrossRef]
- Ho, L.; Ronan, J.L.; Wu, J.; Staahl, B.T.; Chen, L.; Kuo, A.; Lessard, J.; Nesvizhskii, A.I.; Ranish, J.; Crabtree, G.R. An embryonic stem cell chromatin remodeling complex, esBAF, is essential for embryonic stem cell self-renewal and pluripotency. Proc. Natl. Acad. Sci. USA 2009, 106, 5181–5186. [Google Scholar] [CrossRef]
- Gagnon, K.T.; Li, L.; Chu, Y.; Janowski, B.A.; Corey, D.R. RNAi factors are present and active in human cell nuclei. Cell Rep. 2014, 6, 211–221. [Google Scholar] [CrossRef]
- Egloff, S.; Studniarek, C.; Kiss, T. 7SK small nuclear RNA, a multifunctional transcriptional regulatory RNA with gene-specific features. Transcription 2018, 9, 95–101. [Google Scholar] [CrossRef]
- Xing, Y.H.; Yao, R.W.; Zhang, Y.; Guo, C.J.; Jiang, S.; Xu, G.; Dong, R.; Yang, L.; Chen, L.L. SLERT Regulates DDX21 Rings Associated with Pol I Transcription. Cell 2017, 169, 664–678.e616. [Google Scholar] [CrossRef]
- Haberman, N.; Digby, H.; Faraway, R.; Cheung, R.; Chakrabarti, A.M.; Jobbins, A.M.; Parr, C.; Yasuzawa, K.; Kasukawa, T.; Yip, C.W.; et al. Widespread 3′UTR capped RNAs derive from G-rich regions in proximity to AGO2 binding sites. BMC Biol. 2024, 22, 254. [Google Scholar] [CrossRef]
- Aktas, T.; Avsar Ilik, I.; Maticzka, D.; Bhardwaj, V.; Pessoa Rodrigues, C.; Mittler, G.; Manke, T.; Backofen, R.; Akhtar, A. DHX9 suppresses RNA processing defects originating from the Alu invasion of the human genome. Nature 2017, 544, 115–119. [Google Scholar] [CrossRef]
- Chakraborty, P.; Huang, J.T.J.; Hiom, K. DHX9 helicase promotes R-loop formation in cells with impaired RNA splicing. Nat. Commun. 2018, 9, 4346. [Google Scholar] [CrossRef] [PubMed]
- Herbert, A. To “Z” or not to “Z”: Z-RNA, self-recognition, and the MDA5 helicase. PLoS Genet. 2021, 17, e1009513. [Google Scholar] [CrossRef] [PubMed]
- Herbert, A.; Pavlov, F.; Konovalov, D.; Poptsova, M. Conserved microRNAs and Flipons Shape Gene Expression during Development by Altering Promoter Conformations. Int. J. Mol. Sci. 2023, 24, 4884. [Google Scholar] [CrossRef] [PubMed]
- Bridge, K.S.; Shah, K.M.; Li, Y.; Foxler, D.E.; Wong, S.C.K.; Miller, D.C.; Davidson, K.M.; Foster, J.G.; Rose, R.; Hodgkinson, M.R.; et al. Argonaute Utilization for miRNA Silencing Is Determined by Phosphorylation-Dependent Recruitment of LIM-Domain-Containing Proteins. Cell Rep. 2017, 20, 173–187. [Google Scholar] [CrossRef]
- Hock, J.; Weinmann, L.; Ender, C.; Rudel, S.; Kremmer, E.; Raabe, M.; Urlaub, H.; Meister, G. Proteomic and functional analysis of Argonaute-containing mRNA-protein complexes in human cells. EMBO Rep. 2007, 8, 1052–1060. [Google Scholar] [CrossRef]
- Griffin, K.N.; Walters, B.W.; Li, H.; Wang, H.; Biancon, G.; Tebaldi, T.; Kaya, C.B.; Kanyo, J.; Lam, T.T.; Cox, A.L.; et al. Widespread association of the Argonaute protein AGO2 with meiotic chromatin suggests a distinct nuclear function in mammalian male reproduction. Genome Res. 2022, 32, 1655–1668. [Google Scholar] [CrossRef]
- Egli, M.; Manoharan, M. Chemistry, structure and function of approved oligonucleotide therapeutics. Nucleic Acids Res. 2023, 51, 2529–2573. [Google Scholar] [CrossRef]
- Duca, M.; Vekhoff, P.; Oussedik, K.; Halby, L.; Arimondo, P.B. The triple helix: 50 years later, the outcome. Nucleic Acids Res. 2008, 36, 5123–5138. [Google Scholar] [CrossRef]
- Zhu, Q.; Schlick, T. A Fiedler Vector Scoring Approach for Novel RNA Motif Selection. J. Phys. Chem. B 2021, 125, 1144–1155. [Google Scholar] [CrossRef]
- Stein, C.A.; Hansen, J.B.; Lai, J.; Wu, S.; Voskresenskiy, A.; Hog, A.; Worm, J.; Hedtjarn, M.; Souleimanian, N.; Miller, P.; et al. Efficient gene silencing by delivery of locked nucleic acid antisense oligonucleotides, unassisted by transfection reagents. Nucleic Acids Res. 2010, 38, e3. [Google Scholar] [CrossRef]
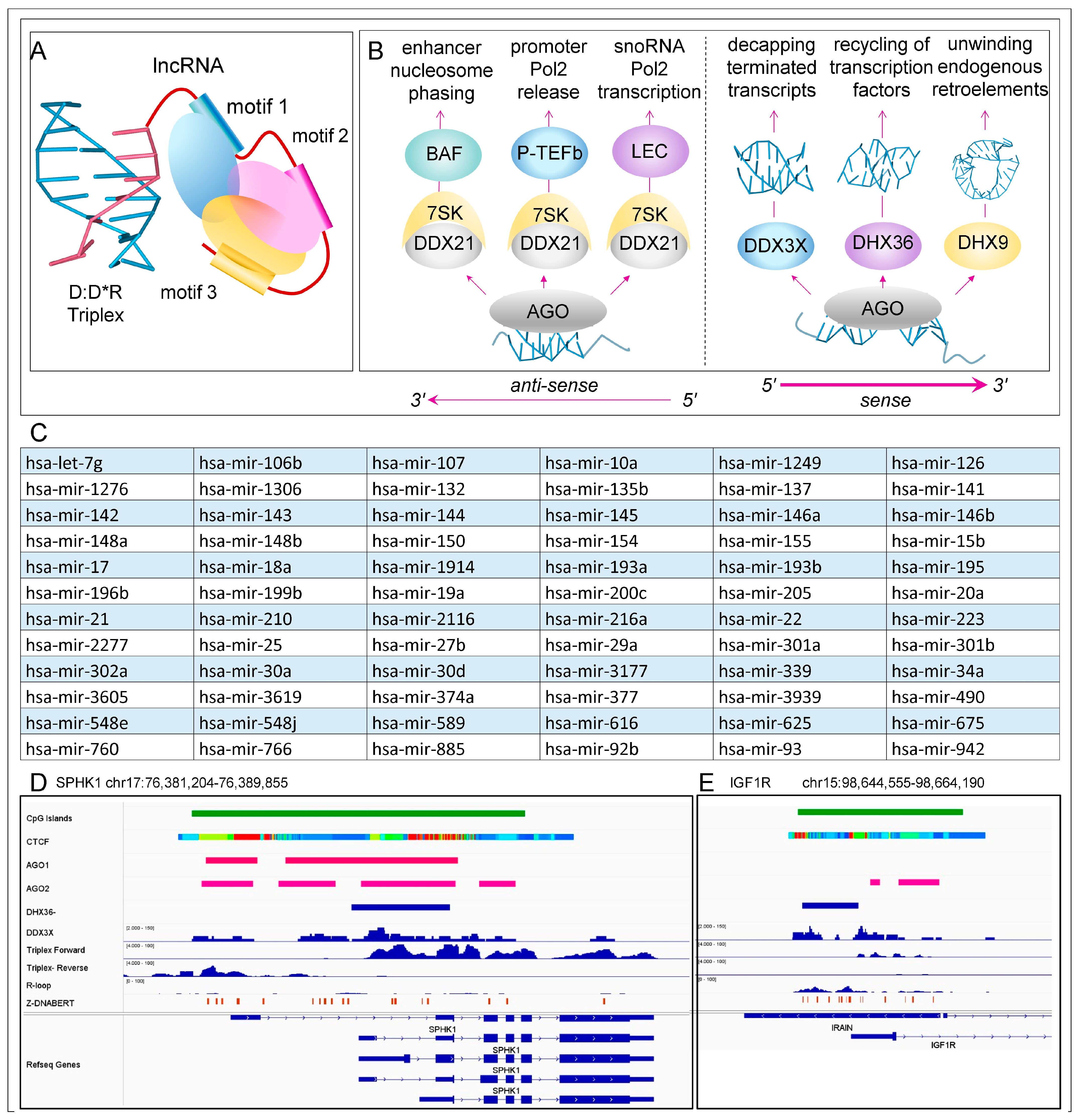

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Herbert, A. Triplexes Color the Chromaverse by Modulating Nucleosome Phasing and Anchoring Chromatin Condensates. Int. J. Mol. Sci. 2025, 26, 4032. https://doi.org/10.3390/ijms26094032
Herbert A. Triplexes Color the Chromaverse by Modulating Nucleosome Phasing and Anchoring Chromatin Condensates. International Journal of Molecular Sciences. 2025; 26(9):4032. https://doi.org/10.3390/ijms26094032
Chicago/Turabian StyleHerbert, Alan. 2025. "Triplexes Color the Chromaverse by Modulating Nucleosome Phasing and Anchoring Chromatin Condensates" International Journal of Molecular Sciences 26, no. 9: 4032. https://doi.org/10.3390/ijms26094032
APA StyleHerbert, A. (2025). Triplexes Color the Chromaverse by Modulating Nucleosome Phasing and Anchoring Chromatin Condensates. International Journal of Molecular Sciences, 26(9), 4032. https://doi.org/10.3390/ijms26094032