
Targets and Gene Therapy of ALS (Part 1)

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Familial Forms of ALS
2.1. Mutations in the SOD1 Gene
2.2. Mutations in the TARDBP Gene
2.3. Mutations in the FUS Gene
2.4. Mutations in the C9orf72 Gene
3. Gene Therapy Methods
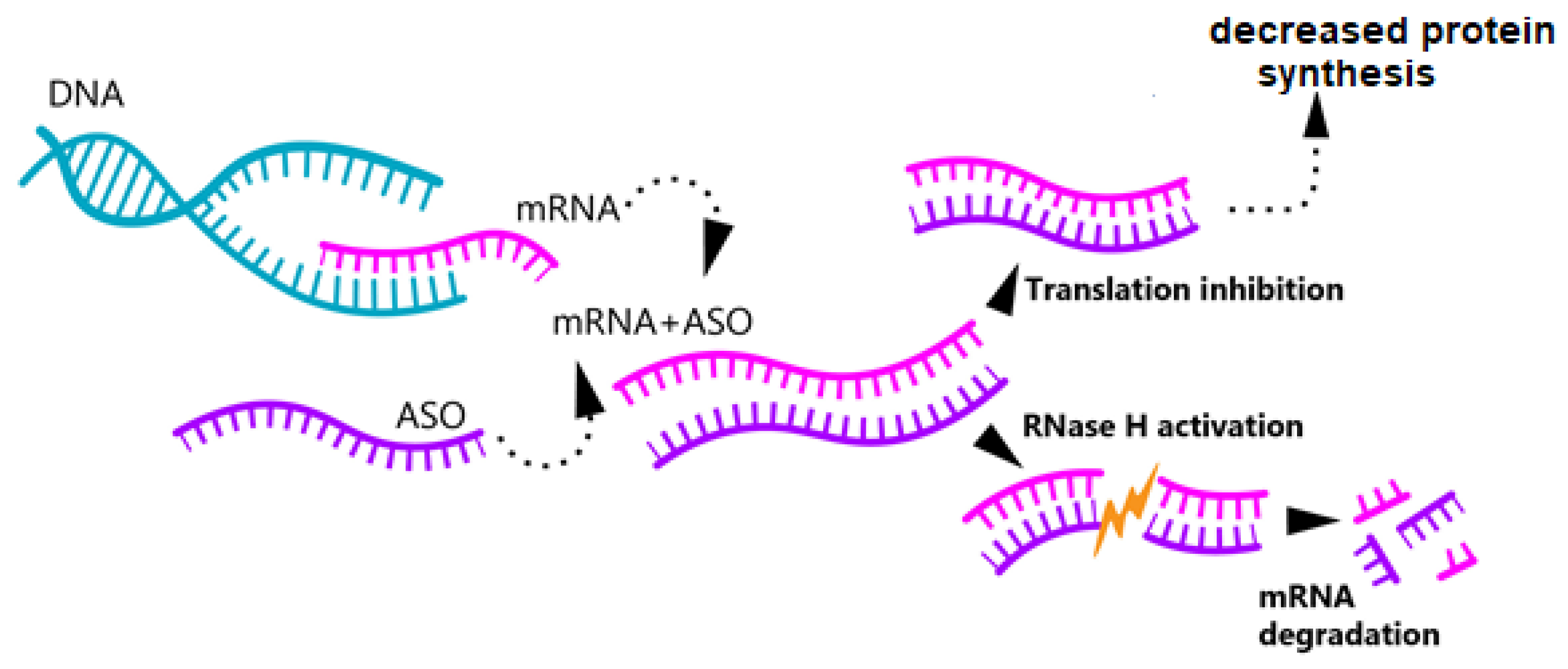
3.1. Antisense Oligonucleotide Therapy
3.1.1. SOD1
Clinical Studies
3.1.2. C9orf72
Clinical Studies
3.1.3. FUS
Clinical Studies
3.1.4. TARDBP and ATXN2
3.2. Therapy with Small Interfering RNAs and miRNAs
Small Interfering RNAs and miRNAs in ALS Treatment
Clinical Studies
3.3. CRISPR/Cas9 Method
- Target selection, i.e., the selection of a specific gene or sequence that needs to be edited.
- Design of guide RNA (sgRNA), which is an RNA complementary to the selected DNA sequence necessary for directing the Cas protein towards the target for subsequent correction [108].
- The third stage is to deliver the CRISPR/Cas9 system to the cell. Viral and non-viral vectors can be used for this purpose.
- Formation of the Cas9–sgRNA complex can be divided into two major stages:
- (1)
- Binding stage:
- (2)
- Target recognition stage:
- 5.
- Cleavage of DNA chains:
- 6.
- The resulting break triggers a reaction aimed at its repair. To correct double-stranded breaks, cells can involve one of two major mechanisms: (1) non-homologous end joining (NHEJ), which can lead to small insertions or deletions at the break site and, as a result, to gene knockout; (2) homology directed repair (HDR), allowing for the accurate repair of the break using a template, which provides an opportunity to make specific changes to the genome or even add new genes.
- 7.
- The last stage is an analysis to assess the accuracy of the changes made. For this, the genome or its specific site is sequenced.
Use of the CRISPR/Cas9 Method for ALS Treatment
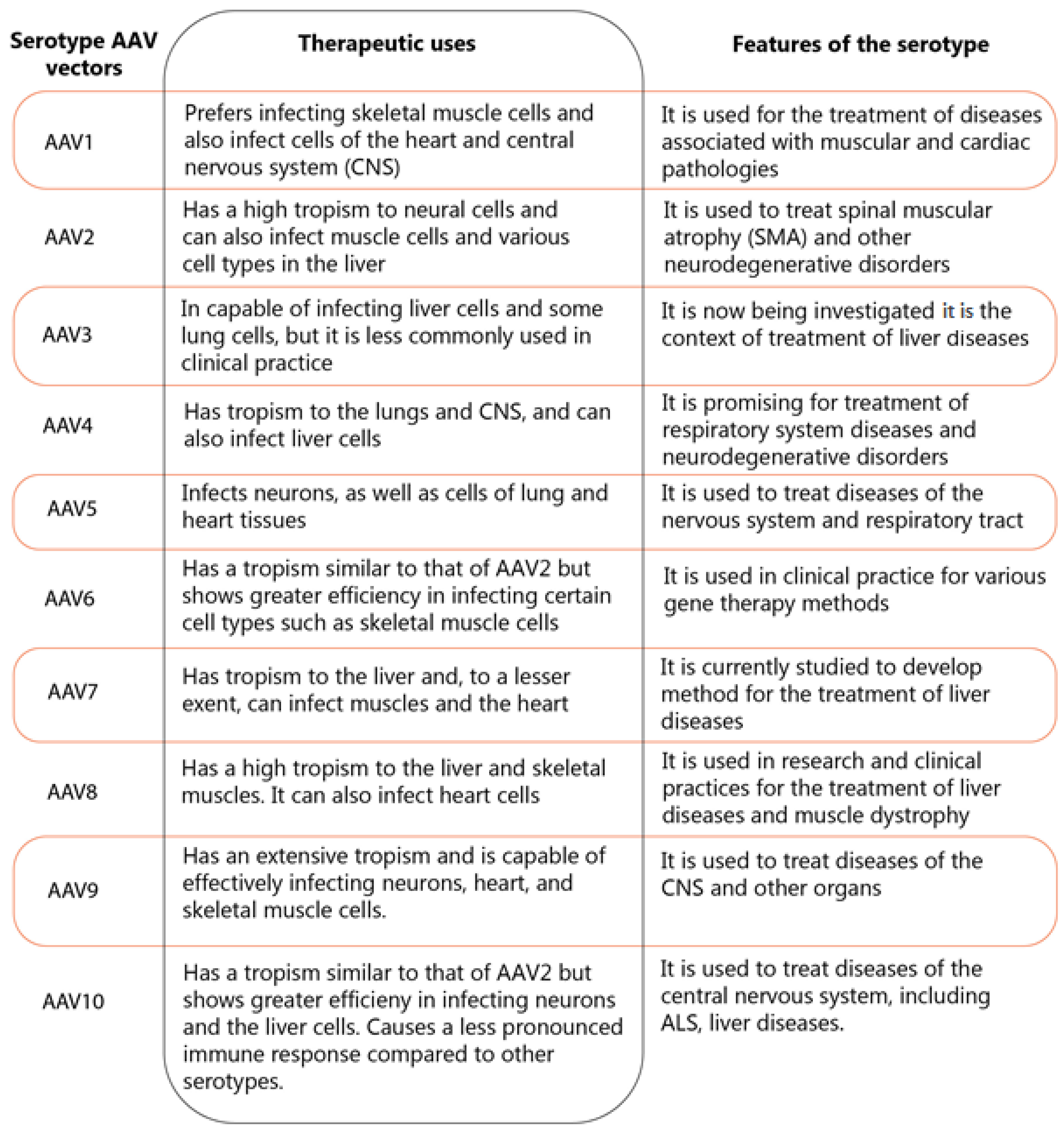
3.4. Vector-Based Gene Therapy
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Ruf, W.P.; Boros, M.; Freischmidt, A.; Brenner, D.; Grozdanov, V.; de Meirelles, J.; Meyer, T.; Grehl, T.; Petri, S.; Grosskreutz, J.; et al. Spectrum and frequency of genetic variants in sporadic amyotrophic lateral sclerosis. Brain Commun. 2023, 5, fcad152. [Google Scholar] [CrossRef] [PubMed]
- Lu, L.; Zheng, L.; Viera, L.; Suswam, E.; Li, Y.; Li, X.; Estévez, A.G.; King, P.H. Mutant Cu/Zn-Superoxide Dismutase Associated with Amyotrophic Lateral Sclerosis Destabilizes Vascular Endothelial Growth Factor mRNA and Downregulates Its Expression. J. Neurosci. 2007, 27, 7929–7938. [Google Scholar] [CrossRef] [PubMed]
- Lu, L.; Wang, S.; Zheng, L.; Li, X.; Suswam, E.A.; Zhang, X.; Wheeler, C.G.; Nabors, L.B.; Filippova, N.; King, P.H. Amyotrophic Lateral Sclerosis-linked Mutant SOD1 Sequesters Hu Antigen R (HuR) and TIA-1-related Protein (TIAR). J. Biol. Chem. 2009, 284, 33989–33998. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Qian, K.; Du, Z.; Cao, J.; Petersen, A.; Liu, H.; Blackbourn, L.W.; Huang, C.-L.; Errigo, A.; Yin, Y.; et al. Modeling ALS with iPSCs Reveals that Mutant SOD1 Misregulates Neurofilament Balance in Motor Neurons. Cell Stem Cell 2014, 14, 796–809. [Google Scholar] [CrossRef]
- Buratti, E.; Baralle, F.E. Characterization and Functional Implications of the RNA Binding Properties of Nuclear Factor TDP-43, a Novel Splicing Regulator ofCFTR Exon 9 *. J. Biol. Chem. 2001, 276, 36337–36343. [Google Scholar] [CrossRef]
- Tan, A.Y.; Riley, T.R.; Coady, T.; Bussemaker, H.J.; Manley, J.L. TLS/FUS (translocated in liposarcoma/fused in sarcoma) regulates target gene transcription via single-stranded DNA response elements. Proc. Natl. Acad. Sci. USA 2012, 109, 6030–6035. [Google Scholar] [CrossRef]
- Forsberg, K.; Andersen, P.M.; Marklund, S.L.; Brännström, T. Glial nuclear aggregates of superoxide dismutase-1 are regularly present in patients with amyotrophic lateral sclerosis. Acta Neuropathol. 2011, 121, 623–634. [Google Scholar] [CrossRef]
- Szewczyk, B.; Günther, R.; Japtok, J.; Frech, M.J.; Naumann, M.; Lee, H.O.; Hermann, A. FUS ALS neurons activate major stress pathways and reduce translation as an early protective mechanism against neurodegeneration. Cell Rep. 2023, 42, 112025. [Google Scholar] [CrossRef]
- Bose, J.K.; Huang, C.-C.; Shen, C.-K.J. Regulation of Autophagy by Neuropathological Protein TDP-43 *. J. Biol. Chem. 2011, 286, 44441–44448. [Google Scholar] [CrossRef]
- Farg, M.A.; Sundaramoorthy, V.; Sultana, J.M.; Yang, S.; Atkinson, R.A.K.; Levina, V.; Halloran, M.A.; Gleeson, P.A.; Blair, I.P.; Soo, K.Y.; et al. C9ORF72, implicated in amytrophic lateral sclerosis and frontotemporal dementia, regulates endosomal trafficking. Hum. Mol. Genet. 2014, 23, 3579–3595. [Google Scholar] [CrossRef]
- Cp, W.; Ef, S.; Aj, G.; Kj, D.V. C9orf72 plays a central role in Rab GTPase-dependent regulation of autophagy. Small GTPases 2018, 9, 399–408. [Google Scholar] [CrossRef]
- Konopka, A.; Whelan, D.R.; Jamali, M.S.; Perri, E.; Shahheydari, H.; Toth, R.P.; Parakh, S.; Robinson, T.; Cheong, A.; Mehta, P.; et al. Impaired NHEJ repair in amyotrophic lateral sclerosis is associated with TDP-43 mutations. Mol. Neurodegener. 2020, 15, 51. [Google Scholar] [CrossRef] [PubMed]
- Mitra, J.; Guerrero, E.N.; Hegde, P.M.; Liachko, N.F.; Wang, H.; Vasquez, V.; Gao, J.; Pandey, A.; Taylor, J.P.; Kraemer, B.C.; et al. Motor neuron disease-associated loss of nuclear TDP-43 is linked to DNA double-strand break repair defects. Proc. Natl. Acad. Sci. USA 2019, 116, 4696–4705. [Google Scholar] [CrossRef]
- Sama, R.R.K.; Ward, C.L.; Bosco, D.A. Functions of FUS/TLS From DNA Repair to Stress Response: Implications for ALS. ASN Neuro 2014, 6, 1759091414544472. [Google Scholar] [CrossRef] [PubMed]
- Sharma, A.; Lyashchenko, A.K.; Lu, L.; Nasrabady, S.E.; Elmaleh, M.; Mendelsohn, M.; Nemes, A.; Tapia, J.C.; Mentis, G.Z.; Shneider, N.A. ALS-associated mutant FUS induces selective motor neuron degeneration through toxic gain of function. Nat. Commun. 2016, 7, 10465. [Google Scholar] [CrossRef]
- Miller, T.; Pestronk, A.; David, W.; Rothstein, J.; Simpson, E.; Appel, S.H.; Andres, P.L.; Mahoney, K.; Allred, P.; Alexander, K.; et al. A Phase I, Randomised, First-in-Human Study of an Antisense Oligonucleotide Directed Against SOD1 Delivered Intrathecally in SOD1-Familial ALS Patients. Lancet Neurol. 2013, 12, 435–442. [Google Scholar] [CrossRef]
- Miller, T.; Cudkowicz, M.; Shaw, P.J.; Andersen, P.M.; Atassi, N.; Bucelli, R.C.; Genge, A.; Glass, J.; Ladha, S.; Ludolph, A.L.; et al. Phase 1-2 Trial of Antisense Oligonucleotide Tofersen for SOD1 ALS. N. Engl. J. Med. 2020, 383, 109–119. [Google Scholar] [CrossRef]
- Miller, T.M.; Cudkowicz, M.E.; Genge, A.; Shaw, P.J.; Sobue, G.; Bucelli, R.C.; Chiò, A.; Damme, P.V.; Ludolph, A.C.; Glass, J.D.; et al. Trial of Antisense Oligonucleotide Tofersen for SOD1 ALS. N. Engl. J. Med. 2022, 387, 1099–1110. [Google Scholar] [CrossRef]
- Korobeynikov, V.A.; Lyashchenko, A.K.; Blanco-Redondo, B.; Jafar-Nejad, P.; Shneider, N.A. Antisense oligonucleotide silencing of FUS expression as a therapeutic approach in amyotrophic lateral sclerosis. Nat. Med. 2022, 28, 104–116. [Google Scholar] [CrossRef]
- Tran, H.; Moazami, M.P.; Yang, H.; McKenna-Yasek, D.; Douthwright, C.L.; Pinto, C.; Metterville, J.; Shin, M.; Sanil, N.; Dooley, C.; et al. Suppression of mutant C9orf72 expression by a potent mixed backbone antisense oligonucleotide. Nat. Med. 2022, 28, 117–124. [Google Scholar] [CrossRef]
- Wang, L.; Yi, F.; Fu, L.; Yang, J.; Wang, S.; Wang, Z.; Suzuki, K.; Sun, L.; Xu, X.; Yu, Y.; et al. CRISPR/Cas9-mediated targeted gene correction in amyotrophic lateral sclerosis patient iPSCs. Protein Cell 2017, 8, 365–378. [Google Scholar] [CrossRef] [PubMed]
- Lim, C.K.; Gapinske, M.; Brooks, A.K.; Woods, W.S.; Powell, J.E.; Winter, J.; Perez-Pinera, P.; Gaj, T. Treatment of a Mouse Model of ALS by In Vivo Base Editing. Mol. Ther. 2020, 28, 1177–1189. [Google Scholar] [CrossRef] [PubMed]
- Aishwarya, R.; Abdullah, C.S.; Remex, N.S.; Nitu, S.; Hartman, B.; King, J.; Bhuiyan, M.A.N.; Rom, O.; Miriyala, S.; Panchatcharam, M.; et al. Pathological Sequelae Associated with Skeletal Muscle Atrophy and Histopathology in G93A*SOD1 Mice. Muscles 2023, 2, 51–74. [Google Scholar] [CrossRef] [PubMed]
- Saccon, R.A.; Bunton-Stasyshyn, R.K.A.; Fisher, E.M.C.; Fratta, P. Is SOD1 loss of function involved in amyotrophic lateral sclerosis? Brain 2013, 136, 2342–2358. [Google Scholar] [CrossRef]
- Tsekrekou, M.; Giannakou, M.; Papanikolopoulou, K.; Skretas, G. Protein aggregation and therapeutic strategies in SOD1- and TDP-43- linked ALS. Front. Mol. Biosci. 2024, 11, 1383453. [Google Scholar] [CrossRef]
- Duranti, E.; Villa, C. Molecular Investigations of Protein Aggregation in the Pathogenesis of Amyotrophic Lateral Sclerosis. Int. J. Mol. Sci. 2022, 24, 704. [Google Scholar] [CrossRef]
- Rakhit, R.; Chakrabartty, A. Structure, folding, and misfolding of Cu,Zn superoxide dismutase in amyotrophic lateral sclerosis. Biochim. Biophys. Acta BBA-Mol. Basis Dis. 2006, 1762, 1025–1037. [Google Scholar] [CrossRef]
- Trist, B.G.; Hilton, J.B.; Hare, D.J.; Crouch, P.J.; Double, K.L. Superoxide Dismutase 1 in Health and Disease: How a Frontline Antioxidant Becomes Neurotoxic. Angew. Chem. Int. Ed Engl. 2021, 60, 9215–9246. [Google Scholar] [CrossRef]
- Pasinelli, P.; Belford, M.E.; Lennon, N.; Bacskai, B.J.; Hyman, B.T.; Trotti, D.; Brown, R.H. Amyotrophic Lateral Sclerosis-Associated SOD1 Mutant Proteins Bind and Aggregate with Bcl-2 in Spinal Cord Mitochondria. Neuron 2004, 43, 19–30. [Google Scholar] [CrossRef]
- Pedrini, S.; Sau, D.; Guareschi, S.; Bogush, M.; Brown, R.H.; Naniche, N.; Kia, A.; Trotti, D.; Pasinelli, P. ALS-linked mutant SOD1 damages mitochondria by promoting conformational changes in Bcl-2. Hum. Mol. Genet. 2010, 19, 2974–2986. [Google Scholar] [CrossRef]
- Tan, W.; Pasinelli, P.; Trotti, D. Role of mitochondria in mutant SOD1 linked amyotrophic lateral sclerosis. Biochim. Biophys. Acta 2014, 1842, 1295–1301. [Google Scholar] [CrossRef] [PubMed]
- Andersen, P.M. Amyotrophic lateral sclerosis associated with mutations in the Cu,Zn superoxide dismutase gene. Curr. Neurol. Neurosci. Rep. 2006, 6, 37–46. [Google Scholar] [CrossRef] [PubMed]
- Tsang, C.K.; Liu, Y.; Thomas, J.; Zhang, Y.; Zheng, X.F.S. Superoxide dismutase 1 acts as a nuclear transcription factor to regulate oxidative stress resistance. Nat. Commun. 2014, 5, 3446. [Google Scholar] [CrossRef] [PubMed]
- Menzies, F.M.; Grierson, A.J.; Cookson, M.R.; Heath, P.R.; Tomkins, J.; Figlewicz, D.A.; Ince, P.G.; Shaw, P.J. Selective loss of neurofilament expression in Cu/Zn superoxide dismutase (SOD1) linked amyotrophic lateral sclerosis. J. Neurochem. 2002, 82, 1118–1128. [Google Scholar] [CrossRef]
- Takeuchi, R.; Tada, M.; Shiga, A.; Toyoshima, Y.; Konno, T.; Sato, T.; Nozaki, H.; Kato, T.; Horie, M.; Shimizu, H.; et al. Heterogeneity of cerebral TDP-43 pathology in sporadic amyotrophic lateral sclerosis: Evidence for clinico-pathologic subtypes. Acta Neuropathol. Commun. 2016, 4, 61. [Google Scholar] [CrossRef]
- Giordana, M.T.; Piccinini, M.; Grifoni, S.; Marco, G.D.; Vercellino, M.; Magistrello, M.; Pellerino, A.; Buccinna, B.; Lupino, E.; Rinaudo, M.T. TDP-43 Redistribution is an Early Event in Sporadic Amyotrophic Lateral Sclerosis. Brain Pathol. 2009, 20, 351–360. [Google Scholar] [CrossRef]
- Conforti, F.L.; Sproviero, W.; Simone, I.L.; Mazzei, R.; Valentino, P.; Ungaro, C.; Magariello, A.; Patitucci, A.; La Bella, V.; Sprovieri, T.; et al. TARDBP gene mutations in south Italian patients with amyotrophic lateral sclerosis. J. Neurol. Neurosurg. Psychiatry 2011, 82, 587–588. [Google Scholar] [CrossRef]
- Ou, S.H.; Wu, F.; Harrich, D.; García-Martínez, L.F.; Gaynor, R.B. Cloning and characterization of a novel cellular protein, TDP-43, that binds to human immunodeficiency virus type 1 TAR DNA sequence motifs. J. Virol. 1995, 69, 3584–3596. [Google Scholar] [CrossRef]
- Arai, T.; Hasegawa, M.; Akiyama, H.; Ikeda, K.; Nonaka, T.; Mori, H.; Mann, D.; Tsuchiya, K.; Yoshida, M.; Hashizume, Y.; et al. TDP-43 is a component of ubiquitin-positive tau-negative inclusions in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Biochem. Biophys. Res. Commun. 2006, 351, 602–611. [Google Scholar] [CrossRef]
- Neumann, M.; Sampathu, D.M.; Kwong, L.K.; Truax, A.C.; Micsenyi, M.C.; Chou, T.T.; Bruce, J.; Schuck, T.; Grossman, M.; Clark, C.M.; et al. Ubiquitinated TDP-43 in Frontotemporal Lobar Degeneration and Amyotrophic Lateral Sclerosis. Science 2006, 314, 130–133. [Google Scholar] [CrossRef]
- Schipper, L.J.; Raaphorst, J.; Aronica, E.; Baas, F.; de Haan, R.; de Visser, M.; Troost, D. Prevalence of brain and spinal cord inclusions, including dipeptide repeat proteins, in patients with the C9ORF72 hexanucleotide repeat expansion: A systematic neuropathological review. Neuropathol. Appl. Neurobiol. 2016, 42, 547–560. [Google Scholar] [CrossRef] [PubMed]
- Ayala, Y.M.; Zago, P.; D’Ambrogio, A.; Xu, Y.-F.; Petrucelli, L.; Buratti, E.; Baralle, F.E. Structural determinants of the cellular localization and shuttling of TDP-43. J. Cell Sci. 2008, 121, 3778–3785. [Google Scholar] [CrossRef] [PubMed]
- Winton, M.J.; Igaz, L.M.; Wong, M.M.; Kwong, L.K.; Trojanowski, J.Q.; Lee, V.M.-Y. Disturbance of Nuclear and Cytoplasmic TAR DNA-binding Protein (TDP-43) Induces Disease-like Redistribution, Sequestration, and Aggregate Formation *. J. Biol. Chem. 2008, 283, 13302–13309. [Google Scholar] [CrossRef]
- Wang, W.; Wang, L.; Lu, J.; Siedlak, S.L.; Fujioka, H.; Liang, J.; Jiang, S.; Ma, X.; Jiang, Z.; da Rocha, E.L.; et al. The inhibition of TDP-43 mitochondrial localization blocks its neuronal toxicity. Nat. Med. 2016, 22, 869–878. [Google Scholar] [CrossRef]
- Walker, A.K.; Spiller, K.J.; Ge, G.; Zheng, A.; Xu, Y.; Zhou, M.; Tripathy, K.; Kwong, L.K.; Trojanowski, J.Q.; Lee, V.M.-Y. Functional recovery in new mouse models of ALS/FTLD after clearance of pathological cytoplasmic TDP-43. Acta Neuropathol. 2015, 130, 643–660. [Google Scholar] [CrossRef]
- Buratti, E.; Baralle, F.E. Multiple roles of TDP-43 in gene expression, splicing regulation, and human disease. Front. Biosci.-Landmark 2008, 13, 867–878. [Google Scholar] [CrossRef]
- Kawahara, Y.; Mieda-Sato, A. TDP-43 promotes microRNA biogenesis as a component of the Drosha and Dicer complexes. Proc. Natl. Acad. Sci. USA 2012, 109, 3347–3352. [Google Scholar] [CrossRef]
- McDonald, K.K.; Aulas, A.; Destroismaisons, L.; Pickles, S.; Beleac, E.; Camu, W.; Rouleau, G.A.; Vande Velde, C. TAR DNA-binding protein 43 (TDP-43) regulates stress granule dynamics via differential regulation of G3BP and TIA-1. Hum. Mol. Genet. 2011, 20, 1400–1410. [Google Scholar] [CrossRef]
- Lukavsky, P.J.; Daujotyte, D.; Tollervey, J.R.; Ule, J.; Stuani, C.; Buratti, E.; Baralle, F.E.; Damberger, F.F.; Allain, F.H.-T. Molecular basis of UG-rich RNA recognition by the human splicing factor TDP-43. Nat. Struct. Mol. Biol. 2013, 20, 1443–1449. [Google Scholar] [CrossRef]
- Ayala, Y.M.; De Conti, L.; Avendaño-Vázquez, S.E.; Dhir, A.; Romano, M.; D’ambrogio, A.; Tollervey, J.; Ule, J.; Baralle, M.; Buratti, E.; et al. TDP-43 regulates its mRNA levels through a negative feedback loop. EMBO J. 2011, 30, 277–288. [Google Scholar] [CrossRef]
- Russo, A.; Scardigli, R.; La Regina, F.; Murray, M.E.; Romano, N.; Dickson, D.W.; Wolozin, B.; Cattaneo, A.; Ceci, M. Increased cytoplasmic TDP-43 reduces global protein synthesis by interacting with RACK1 on polyribosomes. Hum. Mol. Genet. 2017, 26, 1407–1418. [Google Scholar] [CrossRef] [PubMed]
- Casafont, I.; Bengoechea, R.; Tapia, O.; Berciano, M.T.; Lafarga, M. TDP-43 localizes in mRNA transcription and processing sites in mammalian neurons. J. Struct. Biol. 2009, 167, 235–241. [Google Scholar] [CrossRef] [PubMed]
- Osaka, M.; Ito, D.; Suzuki, N. Disturbance of proteasomal and autophagic protein degradation pathways by amyotrophic lateral sclerosis-linked mutations in ubiquilin 2. Biochem. Biophys. Res. Commun. 2016, 472, 324–331. [Google Scholar] [CrossRef]
- Buratti, E.; De Conti, L.; Stuani, C.; Romano, M.; Baralle, M.; Baralle, F. Nuclear factor TDP-43 can affect selected microRNA levels. FEBS J. 2010, 277, 2268–2281. [Google Scholar] [CrossRef]
- Avendaño-Vázquez, S.E.; Dhir, A.; Bembich, S.; Buratti, E.; Proudfoot, N.; Baralle, F.E. Autoregulation of TDP-43 mRNA levels involves interplay between transcription, splicing, and alternative polyA site selection. Genes Dev. 2012, 26, 1679–1684. [Google Scholar] [CrossRef]
- Baughn, M.W.; Melamed, Z.; López-Erauskin, J.; Beccari, M.S.; Ling, K.; Zuberi, A.; Presa, M.; Gil, E.G.; Maimon, R.; Vazquez-Sanchez, S.; et al. Mechanism of STMN2 cryptic splice/polyadenylation and its correction for TDP-43 proteinopathies. Science 2023, 379, 1140–1149. [Google Scholar] [CrossRef]
- Guerra San Juan, I.; Nash, L.A.; Smith, K.S.; Leyton-Jaimes, M.F.; Qian, M.; Klim, J.R.; Limone, F.; Dorr, A.B.; Couto, A.; Pintacuda, G.; et al. Loss of mouse Stmn2 function causes motor neuropathy. Neuron 2022, 110, 1671–1688.e6. [Google Scholar] [CrossRef]
- Deng, H.; Gao, K.; Jankovic, J. The role of FUS gene variants in neurodegenerative diseases. Nat. Rev. Neurol. 2014, 10, 337–348. [Google Scholar] [CrossRef]
- Vance, C.; Rogelj, B.; Hortobágyi, T.; De Vos, K.J.; Nishimura, A.L.; Sreedharan, J.; Hu, X.; Smith, B.; Ruddy, D.; Wright, P.; et al. Mutations in FUS, an RNA processing protein, cause familial amyotrophic lateral sclerosis type 6. Science 2009, 323, 1208–1211. [Google Scholar] [CrossRef]
- Andersson, M.K.; Ståhlberg, A.; Arvidsson, Y.; Olofsson, A.; Semb, H.; Stenman, G.; Nilsson, O.; Åman, P. The multifunctional FUS, EWS and TAF15 proto-oncoproteins show cell type-specific expression patterns and involvement in cell spreading and stress response. BMC Cell Biol. 2008, 9, 37. [Google Scholar] [CrossRef]
- Nakaya, T.; Maragkakis, M. Amyotrophic Lateral Sclerosis associated FUS mutation shortens mitochondria and induces neurotoxicity. Sci. Rep. 2018, 8, 15575. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.-Y.; Pan, L.; Su, S.C.; Quinn, E.J.; Sasaki, M.; Jimenez, J.C.; Mackenzie, I.R.A.; Huang, E.J.; Tsai, L.-H. Interaction of FUS and HDAC1 Regulates DNA Damage Response and Repair in Neurons. Nat. Neurosci. 2013, 16, 1383–1391. [Google Scholar] [CrossRef] [PubMed]
- Protter, D.S.W.; Parker, R. Principles and Properties of Stress Granules. Trends Cell Biol. 2016, 26, 668–679. [Google Scholar] [CrossRef] [PubMed]
- Tan, A.Y.; Manley, J.L. TLS inhibits RNA polymerase III transcription. Mol. Cell. Biol. 2010, 30, 186–196. [Google Scholar] [CrossRef]
- Lagier-Tourenne, C.; Polymenidou, M.; Hutt, K.R.; Vu, A.Q.; Baughn, M.; Huelga, S.C.; Clutario, K.M.; Ling, S.-C.; Liang, T.Y.; Mazur, C.; et al. Divergent roles of ALS-linked proteins FUS/TLS and TDP-43 intersect in processing long pre-mRNAs. Nat. Neurosci. 2012, 15, 1488–1497. [Google Scholar] [CrossRef]
- Huang, E.J.; Zhang, J.; Geser, F.; Trojanowski, J.Q.; Strober, J.B.; Dickson, D.W.; Brown, R.H., Jr.; Shapiro, B.E.; Lomen-Hoerth, C. Extensive FUS-Immunoreactive Pathology in Juvenile Amyotrophic Lateral Sclerosis with Basophilic Inclusions. Brain Pathol. 2010, 20, 1069–1076. [Google Scholar] [CrossRef]
- Stoica, R.; Paillusson, S.; Gomez-Suaga, P.; Mitchell, J.C.; Lau, D.H.; Gray, E.H.; Sancho, R.M.; Vizcay-Barrena, G.; De Vos, K.J.; Shaw, C.E.; et al. ALS/FTD-associated FUS activates GSK-3β to disrupt the VAPB–PTPIP51 interaction and ER–mitochondria associations. EMBO Rep. 2016, 17, 1326–1342. [Google Scholar] [CrossRef]
- Tsai, Y.-L.; Coady, T.H.; Lu, L.; Zheng, D.; Alland, I.; Tian, B.; Shneider, N.A.; Manley, J.L. ALS/FTD-associated protein FUS induces mitochondrial dysfunction by preferentially sequestering respiratory chain complex mRNAs. Genes Dev. 2020, 34, 785–805. [Google Scholar] [CrossRef]
- DeJesus-Hernandez, M.; Mackenzie, I.R.; Boeve, B.F.; Boxer, A.L.; Baker, M.; Rutherford, N.J.; Nicholson, A.M.; Finch, N.A.; Gilmer, H.F.; Adamson, J.; et al. Expanded GGGGCC hexanucleotide repeat in non-coding region of C9ORF72 causes chromosome 9p-linked frontotemporal dementia and amyotrophic lateral sclerosis. Neuron 2011, 72, 245–256. [Google Scholar] [CrossRef]
- Renton, A.E.; Majounie, E.; Waite, A.; Simón-Sánchez, J.; Rollinson, S.; Gibbs, J.R.; Schymick, J.C.; Laaksovirta, H.; van Swieten, J.C.; Myllykangas, L.; et al. A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron 2011, 72, 257–268. [Google Scholar] [CrossRef]
- Lall, D.; Baloh, R.H. Microglia and C9orf72 in neuroinflammation and ALS and frontotemporal dementia. J. Clin. Investig. 2017, 127, 3250–3258. [Google Scholar] [CrossRef] [PubMed]
- Trageser, K.J.; Smith, C.; Herman, F.J.; Ono, K.; Pasinetti, G.M. Mechanisms of Immune Activation by c9orf72-Expansions in Amyotrophic Lateral Sclerosis and Frontotemporal Dementia. Front. Neurosci. 2019, 13, 1298. [Google Scholar] [CrossRef] [PubMed]
- Moumné, L.; Marie, A.-C.; Crouvezier, N. Oligonucleotide Therapeutics: From Discovery and Development to Patentability. Pharmaceutics 2022, 14, 260. [Google Scholar] [CrossRef] [PubMed]
- Bennett, C.F. Therapeutic Antisense Oligonucleotides Are Coming of Age. Annu. Rev. Med. 2019, 70, 307–321. [Google Scholar] [CrossRef]
- Smith, R.A.; Miller, T.M.; Yamanaka, K.; Monia, B.P.; Condon, T.P.; Hung, G.; Lobsiger, C.S.; Ward, C.M.; McAlonis-Downes, M.; Wei, H.; et al. Antisense oligonucleotide therapy for neurodegenerative disease. J. Clin. Investig. 2006, 116, 2290. [Google Scholar] [CrossRef]
- McCampbell, A.; Cole, T.; Wegener, A.J.; Tomassy, G.S.; Setnicka, A.; Farley, B.J.; Schoch, K.M.; Hoye, M.L.; Shabsovich, M.; Sun, L.; et al. Antisense oligonucleotides extend survival and reverse decrement in muscle response in ALS models. J. Clin. Investig. 2018, 128, 3558–3567. [Google Scholar] [CrossRef]
- Donnelly, C.J.; Zhang, P.-W.; Pham, J.T.; Heusler, A.R.; Mistry, N.A.; Vidensky, S.; Daley, E.L.; Poth, E.M.; Hoover, B.; Fines, D.M.; et al. RNA Toxicity from the ALS/FTD C9ORF72 Expansion Is Mitigated by Antisense Intervention. Neuron 2013, 80, 415–428. [Google Scholar] [CrossRef]
- Sareen, D.; O’Rourke, J.G.; Meera, P.; Muhammad, A.K.M.G.; Grant, S.; Simpkinson, M.; Bell, S.; Carmona, S.; Ornelas, L.; Sahabian, A.; et al. Targeting RNA foci in iPSC-derived motor neurons from ALS patients with C9ORF72 repeat expansion. Sci. Transl. Med. 2013, 5, 208ra149. [Google Scholar] [CrossRef]
- Lagier-Tourenne, C.; Baughn, M.; Rigo, F.; Sun, S.; Liu, P.; Li, H.-R.; Jiang, J.; Watt, A.T.; Chun, S.; Katz, M.; et al. Targeted degradation of sense and antisense C9orf72 RNA foci as therapy for ALS and frontotemporal degeneration. Proc. Natl. Acad. Sci. USA 2013, 110, E4530–E4539. [Google Scholar] [CrossRef]
- Jiang, J.; Zhu, Q.; Gendron, T.F.; Saberi, S.; McAlonis-Downes, M.; Seelman, A.; Stauffer, J.E.; Jafar-nejad, P.; Drenner, K.; Schulte, D.; et al. Gain of toxicity from ALS/FTD-linked repeat expansions in C9ORF72 is alleviated by antisense oligonucleotides targeting GGGGCC-containing RNAs. Neuron 2016, 90, 535–550. [Google Scholar] [CrossRef]
- Liu, Y.; Dodart, J.-C.; Tran, H.; Berkovitch, S.; Braun, M.; Byrne, M.; Durbin, A.F.; Hu, X.S.; Iwamoto, N.; Jang, H.G.; et al. Variant-selective stereopure oligonucleotides protect against pathologies associated with C9orf72-repeat expansion in preclinical models. Nat. Commun. 2021, 12, 847. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Andreucci, A.; Iwamoto, N.; Yin, Y.; Yang, H.; Liu, F.; Bulychev, A.; Hu, X.S.; Lin, X.; Lamore, S.; et al. Preclinical evaluation of WVE-004, aninvestigational stereopure oligonucleotide forthe treatment of C9orf72-associated ALS or FTD. Mol. Ther. Nucleic Acids 2022, 28, 558–570. [Google Scholar] [CrossRef] [PubMed]
- Mejzini, R.; Caruthers, M.H.; Schafer, B.; Kostov, O.; Sudheendran, K.; Ciba, M.; Danielsen, M.; Wilton, S.; Akkari, P.A.; Flynn, L.L. Allele-Selective Thiomorpholino Antisense Oligonucleotides as a Therapeutic Approach for Fused-in-Sarcoma Amyotrophic Lateral Sclerosis. Int. J. Mol. Sci. 2024, 25, 8495. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.-Y.; Liu, Y.-J.; Zhang, X.-L.; Liu, Y.-H.; Jiang, L.-L.; Hu, H.-Y. PolyQ-expanded ataxin-2 aggregation impairs cellular processing-body homeostasis via sequestering the RNA helicase DDX6. J. Biol. Chem. 2024, 300, 107413. [Google Scholar] [CrossRef]
- Elden, A.C.; Kim, H.-J.; Hart, M.P.; Chen-Plotkin, A.S.; Johnson, B.S.; Fang, X.; Armakola, M.; Geser, F.; Greene, R.; Lu, M.M.; et al. Ataxin-2 intermediate-length polyglutamine expansions are associated with increased risk for ALS. Nature 2010, 466, 1069–1075. [Google Scholar] [CrossRef]
- Becker, L.A.; Huang, B.; Bieri, G.; Ma, R.; Knowles, D.A.; Jafar-Nejad, P.; Messing, J.; Kim, H.J.; Soriano, A.; Auburger, G.; et al. Therapeutic reduction of ataxin 2 extends lifespan and reduces pathology in TDP-43 mice. Nature 2017, 544, 367–371. [Google Scholar] [CrossRef]
- Zhang, M.M.; Bahal, R.; Rasmussen, T.P.; Manautou, J.E.; Zhong, X.B. The growth of siRNA-based therapeutics: Updated clinical studies. Biochem. Pharmacol. 2021, 189, 114432. [Google Scholar] [CrossRef]
- Kilikevicius, A.; Meister, G.; Corey, D.R. Reexamining assumptions about miRNA-guided gene silencing. Nucleic Acids Res. 2022, 50, 617. [Google Scholar] [CrossRef]
- Ding, H.; Schwarz, D.S.; Keene, A.; Affar, E.B.; Fenton, L.; Xia, X.; Shi, Y.; Zamore, P.D.; Xu, Z. Selective silencing by RNAi of a dominant allele that causes amyotrophic lateral sclerosis. Aging Cell 2003, 2, 209–217. [Google Scholar] [CrossRef]
- Miller, T.M.; Kaspar, B.K.; Kops, G.J.; Yamanaka, K.; Christian, L.J.; Gage, F.H.; Cleveland, D.W. Virus-delivered small RNA silencing sustains strength in amyotrophic lateral sclerosis. Ann. Neurol. 2005, 57, 773–776. [Google Scholar] [CrossRef]
- Ralph, G.S.; Radcliffe, P.A.; Day, D.M.; Carthy, J.M.; Leroux, M.A.; Lee, D.C.P.; Wong, L.-F.; Bilsland, L.G.; Greensmith, L.; Kingsman, S.M.; et al. Silencing mutant SOD1 using RNAi protects against neurodegeneration and extends survival in an ALS model. Nat. Med. 2005, 11, 429–433. [Google Scholar] [CrossRef] [PubMed]
- Foust, K.D.; Salazar, D.L.; Likhite, S.; Ferraiuolo, L.; Ditsworth, D.; Ilieva, H.; Meyer, K.; Schmelzer, L.; Braun, L.; Cleveland, D.W.; et al. Therapeutic AAV9-mediated suppression of mutant SOD1 slows disease progression and extends survival in models of inherited ALS. Mol. Ther. J. Am. Soc. Gene Ther. 2013, 21, 2148–2159. [Google Scholar] [CrossRef] [PubMed]
- Thomsen, G.M.; Gowing, G.; Latter, J.; Chen, M.; Vit, J.-P.; Staggenborg, K.; Avalos, P.; Alkaslasi, M.; Ferraiuolo, L.; Likhite, S.; et al. Delayed Disease Onset and Extended Survival in the SOD1G93A Rat Model of Amyotrophic Lateral Sclerosis after Suppression of Mutant SOD1 in the Motor Cortex. J. Neurosci. 2014, 34, 15587–15600. [Google Scholar] [CrossRef] [PubMed]
- Duan, C.; Kang, M.; Pan, X.; Gan, Z.; Huang, V.; Li, G.; Place, R.F.; Li, L.-C. Intrathecal administration of a novel siRNA modality extends survival and improves motor function in the SOD1G93A ALS mouse model. Mol. Ther. Nucleic Acids 2024, 35, 102147. [Google Scholar] [CrossRef]
- Wang, H.; Yang, B.; Qiu, L.; Yang, C.; Kramer, J.; Su, Q.; Guo, Y.; Brown, R.H., Jr.; Gao, G.; Xu, Z. Widespread spinal cord transduction by intrathecal injection of rAAV delivers efficacious RNAi therapy for amyotrophic lateral sclerosis. Hum. Mol. Genet. 2014, 23, 668–681. [Google Scholar] [CrossRef]
- Stoica, L.; Todeasa, S.H.; Cabrera, G.T.; Salameh, J.S.; ElMallah, M.K.; Mueller, C.; Brown, R.H.; Miguel, S.-E. AAV delivered artificial microRNA extends survival and delays paralysis in an Amyotrophic Lateral Sclerosis mouse model. Ann. Neurol. 2016, 79, 687–700. [Google Scholar] [CrossRef]
- Martier, R.; Liefhebber, J.M.; García-Osta, A.; Miniarikova, J.; Cuadrado-Tejedor, M.; Espelosin, M.; Ursua, S.; Petry, H.; van Deventer, S.J.; Evers, M.M.; et al. Targeting RNA-Mediated Toxicity in C9orf72 ALS and/or FTD by RNAi-Based Gene Therapy. Mol. Ther.-Nucleic Acids 2019, 16, 26–37. [Google Scholar] [CrossRef]
- Nishimura, A.L.; Shum, C.; Scotter, E.L.; Abdelgany, A.; Sardone, V.; Wright, J.; Lee, Y.-B.; Chen, H.-J.; Bilican, B.; Carrasco, M.; et al. Allele-Specific Knockdown of ALS-Associated Mutant TDP-43 in Neural Stem Cells Derived from Induced Pluripotent Stem Cells. PLoS ONE 2014, 9, e91269. [Google Scholar] [CrossRef]
- Romano, R.; De Luca, M.; Del Fiore, V.S.; Pecoraro, M.; Lattante, S.; Sabatelli, M.; La Bella, V.; Bucci, C. Allele-specific silencing as therapy for familial amyotrophic lateral sclerosis caused by the p.G376D TARDBP mutation. Brain Commun. 2022, 4, fcac315. [Google Scholar] [CrossRef]
- Duan, C.; Kang, M.; Liu, K.; Gan, Z.; Li, G.; Chen, J.; Schacht, I.; Place, R.F.; Li, L.-C. RAG-17: A Novel siRNA Conjugate Demonstrating Efficacy in Late-Stage Treatment of SOD1G93A ALS mice. bioRxiv 2023. [Google Scholar] [CrossRef]
- Mueller, C.; Berry, J.D.; McKenna-Yasek, D.M.; Gernoux, G.; Owegi, M.A.; Pothier, L.M.; Douthwright, C.L.; Gelevski, D.; Luppino, S.D.; Blackwood, M.; et al. SOD1 Suppression with Adeno-Associated Virus and MicroRNA in Familial ALS. N. Engl. J. Med. 2020, 383, 151–158. [Google Scholar] [CrossRef] [PubMed]
- Jinek, M.; Chylinski, K.; Fonfara, I.; Hauer, M.; Doudna, J.A.; Charpentier, E. A programmable dual RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 2012, 337, 816–821. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.; Chang, R.; Yang, H.; Zhao, T.; Hong, Y.; Kong, H.E.; Sun, X.; Qin, Z.; Jin, P.; Li, S.; et al. CRISPR/Cas9-mediated gene editing ameliorates neurotoxicity in mouse model of Huntington’s disease. J. Clin. Investig. 2017, 127, 2719–2724. [Google Scholar] [CrossRef]
- Ortiz-Virumbrales, M.; Moreno, C.L.; Kruglikov, I.; Marazuela, P.; Sproul, A.; Jacob, S.; Zimmer, M.; Paull, D.; Zhang, B.; Schadt, E.E.; et al. CRISPR/Cas9-Correctable mutation-related molecular and physiological phenotypes in iPSC-derived Alzheimer’s PSEN2N141Ineurons. Acta Neuropathol. Commun. 2017, 5, 77. [Google Scholar] [CrossRef]
- Nelson, C.E.; Hakim, C.H.; Ousterout, D.G.; Thakore, P.I.; Moreb, E.A.; Rivera, R.M.C.; Madhavan, S.; Pan, X.; Ran, F.A.; Yan, W.X.; et al. In Vivo Genome Editing Improves Muscle Function in a Mouse Model of Duchenne Muscular Dystrophy. Science 2006, 351, 403–407. [Google Scholar] [CrossRef]
- Meijboom, K.E.; Abdallah, A.; Fordham, N.P.; Nagase, H.; Rodriguez, T.; Kraus, C.; Gendron, T.F.; Krishnan, G.; Esanov, R.; Andrade, N.S.; et al. CRISPR/Cas9-mediated excision of ALS/FTD-causing hexanucleotide repeat expansion in C9ORF72 rescues major disease mechanisms in vivo and in vitro. Nat. Commun. 2022, 13, 6286. [Google Scholar] [CrossRef]
- Gaj, T.; Ojala, D.S.; Ekman, F.K.; Byrne, L.C.; Limsirichai, P.; Schaffer, D.V. In vivo genome editing improves motor function and extends survival in a mouse model of ALS. Sci. Adv. 2017, 3, eaar3952. [Google Scholar] [CrossRef]
- Doench, J.G.; Hartenian, E.; Graham, D.B.; Tothova, Z.; Hegde, M.; Smith, I.; Sullender, M.; Ebert, B.L.; Xavier, R.J.; Root, D.E. Rational design of highly active sgRNAs for CRISPR-Cas9-mediated gene inactivation. Nat. Biotechnol. 2014, 32, 1262–1267. [Google Scholar] [CrossRef]
- Zetsche, B.; Gootenberg, J.S.; Abudayyeh, O.O.; Slaymaker, I.M.; Makarova, K.S.; Essletzbichler, P.; Volz, S.E.; Joung, J.; van der Oost, J.; Regev, A.; et al. Cpf1 is a single RNA-guided endonuclease of a class 2 CRISPR-Cas system. Cell 2015, 163, 759–771. [Google Scholar] [CrossRef]
- Cox, D.B.; Gootenberg, J.S.; Abudayyeh, O.O.; Franklin, B.; Kellner, M.J.; Joung, J.; Zhang, F. RNA editing with CRISPR-Cas13. Science 2017, 358, 1019–1027. [Google Scholar] [CrossRef]
- Duan, W.; Guo, M.; Yi, L.; Liu, Y.; Li, Z.; Ma, Y.; Zhang, G.; Liu, Y.; Bu, H.; Song, X.; et al. The deletion of mutant SOD1 via CRISPR/Cas9/sgRNA prolongs survival in an amyotrophic lateral sclerosis mouse model. Gene Ther. 2020, 27, 157–169. [Google Scholar] [CrossRef] [PubMed]
- Ababneh, N.A.; Scaber, J.; Flynn, R.; Douglas, A.; Barbagallo, P.; Candalija, A.; Turner, M.R.; Sims, D.; Dafinca, R.; Cowley, S.A.; et al. Correction of amyotrophic lateral sclerosis related phenotypes in induced pluripotent stem cell-derived motor neurons carrying a hexanucleotide expansion mutation in C9orf72 by CRISPR/Cas9 genome editing using homology-directed repair. Hum. Mol. Genet. 2020, 29, 2200–2217. [Google Scholar] [CrossRef] [PubMed]
- Krishnan, G.; Zhang, Y.; Gu, Y.; Kankel, M.W.; Gao, F.-B.; Almeida, S. CRISPR deletion of the C9ORF72 promoter in ALS/FTD patient motor neurons abolishes production of dipeptide repeat proteins and rescues neurodegeneration. Acta Neuropathol. 2020, 140, 81–84. [Google Scholar] [CrossRef]
- Wang, H.; Guo, W.; Mitra, J.; Hegde, P.M.; Vandoorne, T.; Eckelmann, B.J.; Mitra, S.; Tomkinson, A.E.; Van Den Bosch, L.; Hegde, M.L. Mutant FUS causes DNA ligation defects to inhibit oxidative damage repair in Amyotrophic Lateral Sclerosis. Nat. Commun. 2018, 9, 3683. [Google Scholar] [CrossRef]
- Tann, J.; Wong, L.; Sajikumar, S.; Ibáñez, C. Abnormal TDP-43 function impairs activity-dependent BDNF secretion, synaptic plasticity, and cognitive behavior through altered Sortilin splicing. EMBO J. 2019, 38, e100989. [Google Scholar] [CrossRef]
- Hermonat, P.L.; Muzyczka, N. Use of adeno-associated virus as a mammalian DNA cloning vector: Transduction of neomycin resistance into mammalian tissue culture cells. Proc. Natl. Acad. Sci. USA 1984, 81, 6466–6470. [Google Scholar] [CrossRef]
- Rose, J.A.; Hoggan, M.D.; Shatkin, A.J. Nucleic acid from an adeno-associated virus: Chemical and physical studies. Proc. Natl. Acad. Sci. USA 1966, 56, 86–92. [Google Scholar] [CrossRef]
- Nidetz, N.F.; McGee, M.C.; Tse, L.V.; Li, C.; Cong, L.; Li, Y.; Huang, W. Adeno-associated viral vector-mediated immune responses: Understanding barriers to gene delivery. Pharmacol. Ther. 2020, 207, 107453. [Google Scholar] [CrossRef]
- Gao, G.; Alvira, M.; Wang, L.; Calcedo, R.; Johnston, J.; Wilson, J. Novel adeno-associated viruses from rhesus monkeys as vectors for human gene therapy. Proc. Natl. Acad. Sci. USA 2002, 99, 11854–11859. [Google Scholar] [CrossRef]
- Nakai, H.; Fuess, S.; Storm, T.; Muramatsu, S.; Nara, Y.; Kay, M. Unrestricted Hepatocyte Transduction with Adeno-Associated Virus Serotype 8 Vectors in Mice. J. Virol. 2005, 79, 214–224. [Google Scholar] [CrossRef]
- Xiao, W.; Chirmule, N.; Berta, S.; McCullough, B.; Gao, G.; Wilson, J. Gene Therapy Vectors Based on Adeno-Associated Virus Type 1. J. Virol. 1999, 73, 3994–4003. [Google Scholar] [CrossRef] [PubMed]
- Blankinship, M.J.; Gregorevic, P.; Allen, J.M.; Harper, S.Q.; Harper, H.; Halbert, C.L.; Miller, A.D.; Chamberlain, J.S. Efficient transduction of skeletal muscle using vectors based on adeno-associated virus serotype 6. Mol. Ther. J. Am. Soc. Gene Ther. 2004, 10, 671–678. [Google Scholar] [CrossRef] [PubMed]
- Alisky, J.M.; Hughes, S.M.; Sauter, S.L.; Jolly, D.; Dubensky, T.W.J.; Staber, P.D.; Chiorini, J.A.; Davidson, B.L. Transduction of murine cerebellar neurons with recombinant FIV and AAV5 vectors. NeuroReport 2000, 11, 2669. [Google Scholar] [CrossRef] [PubMed]
- Burger, C.; Gorbatyuk, O.S.; Velardo, M.J.; Peden, C.S.; Williams, P.; Zolotukhin, S.; Reier, P.J.; Mandel, R.J.; Muzyczka, N. Recombinant AAV viral vectors pseudotyped with viral capsids from serotypes 1, 2, and 5 display differential efficiency and cell tropism after delivery to different regions of the central nervous system. Mol. Ther. J. Am. Soc. Gene Ther. 2004, 10, 302–317. [Google Scholar] [CrossRef]
- Wang, C.; Wang, C.-M.; Clark, K.R.; Sferra, T.J. Recombinant AAV serotype 1 transduction efficiency and tropism in the murine brain. Gene Ther. 2003, 10, 1528–1534. [Google Scholar] [CrossRef]
- Guangping, G.; Vandenberghe, L.H.; Альвира, М.Р.; Лу, Ю.; Кальседo, Р.; Чжoу, С.; Уилсoн, Д.М. Clades of Adeno-Associated Viruses Are Widely Disseminated in Human Tissues. J. Virol. 2004, 78, 6381–6388. [Google Scholar] [CrossRef]
- Wang, Z.; Zhu, T.; Qiao, C.; Zhou, L.; Wang, B.; Zhang, J.; Chen, C.; Li, J.; Xiao, X. Adeno-associated virus serotype 8 efficiently delivers genes to muscle and heart. Nat. Biotechnol. 2005, 23, 321–328. [Google Scholar] [CrossRef]
- Cappella, M.; Ciotti, C.; Cohen-Tannoudji, M.; Biferi, M.G. Gene Therapy for ALS—A Perspective. Int. J. Mol. Sci. 2019, 20, 4388. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shiryaeva, O.; Tolochko, C.; Alekseeva, T.; Dyachuk, V. Targets and Gene Therapy of ALS (Part 1). Int. J. Mol. Sci. 2025, 26, 4063. https://doi.org/10.3390/ijms26094063
Shiryaeva O, Tolochko C, Alekseeva T, Dyachuk V. Targets and Gene Therapy of ALS (Part 1). International Journal of Molecular Sciences. 2025; 26(9):4063. https://doi.org/10.3390/ijms26094063
Chicago/Turabian StyleShiryaeva, Olga, Christina Tolochko, Tatiana Alekseeva, and Vyacheslav Dyachuk. 2025. "Targets and Gene Therapy of ALS (Part 1)" International Journal of Molecular Sciences 26, no. 9: 4063. https://doi.org/10.3390/ijms26094063
APA StyleShiryaeva, O., Tolochko, C., Alekseeva, T., & Dyachuk, V. (2025). Targets and Gene Therapy of ALS (Part 1). International Journal of Molecular Sciences, 26(9), 4063. https://doi.org/10.3390/ijms26094063