
Fine-Tuning Homology-Directed Repair (HDR) for Precision Genome Editing: Current Strategies and Future Directions

Abstract
:1. Introduction
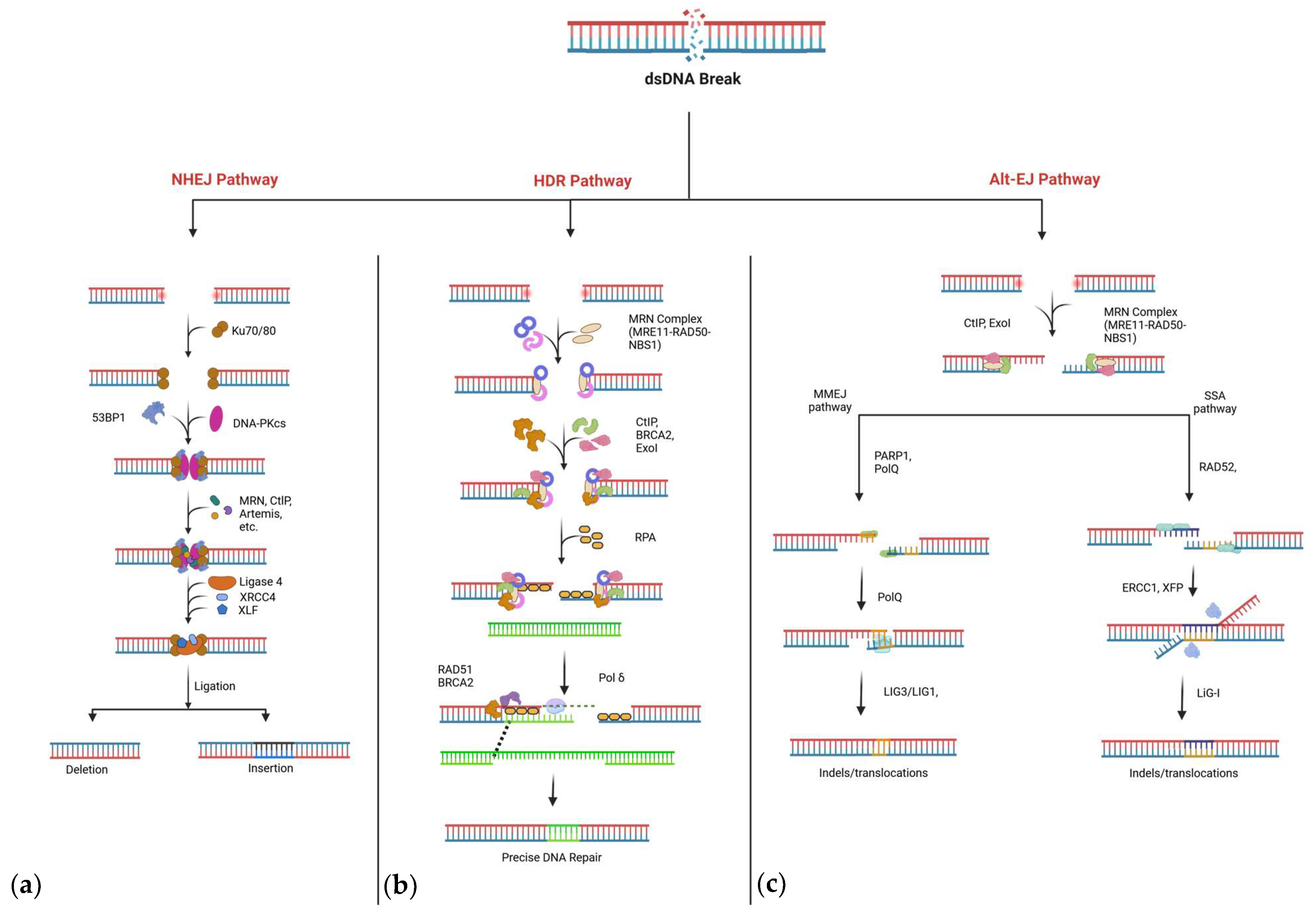
2. DNA Repair Pathways
2.1. Non-Homologous End-Joining (NHEJ)
2.2. Homology-Directed Repair (HDR)
2.3. Alternative Repair Pathways: MMEJ and SSA
Competition Between DNA Repair Pathways
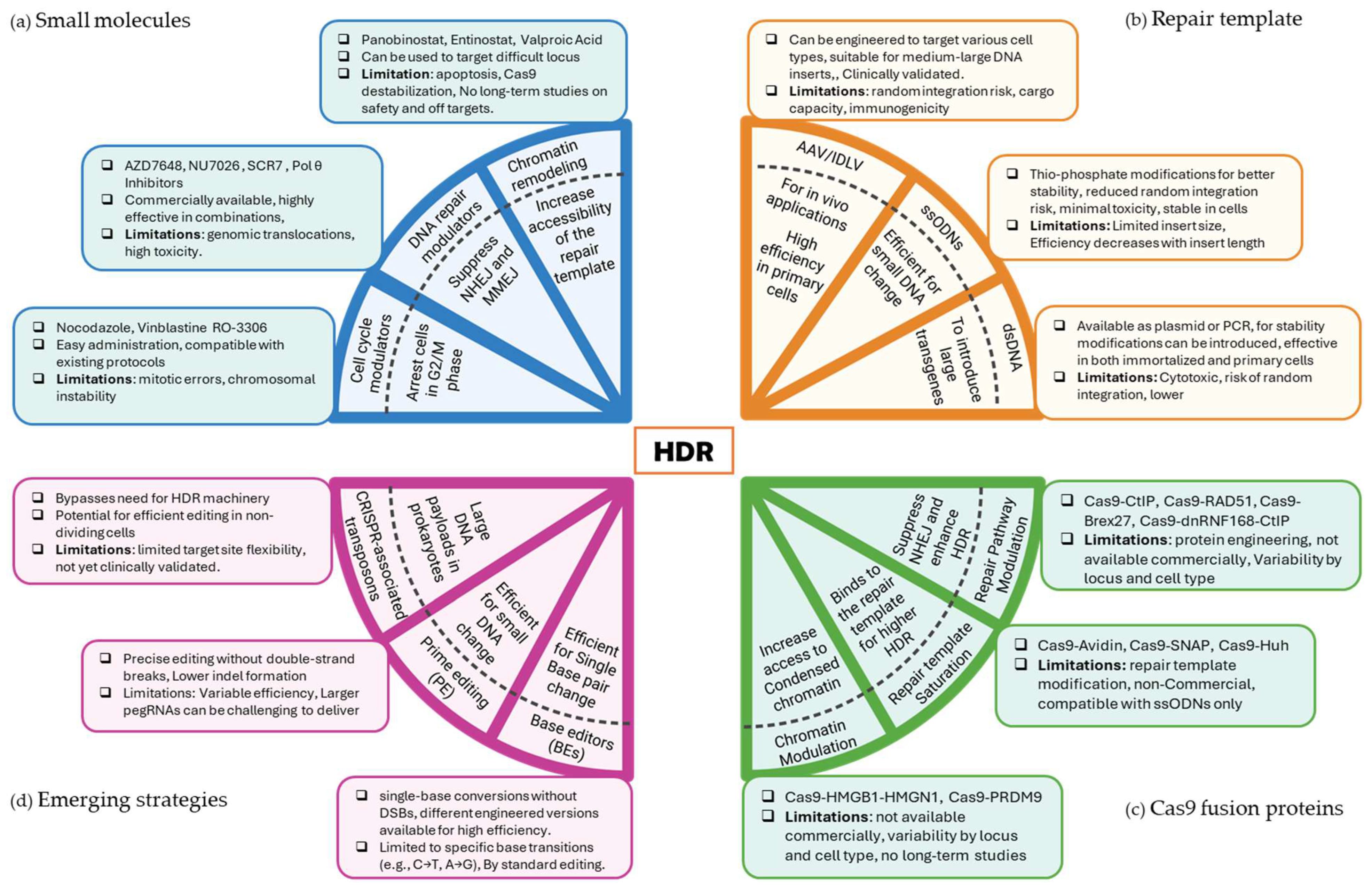
3. Strategies to Enhance HDR
3.1. Enhancing HDR Efficiency with Small Molecules

{kind=link}
{kind=link}
| Target Pathway or Mechanism | Small Molecules | Cell Lines | HDR Increase | Adverse Effects |
|---|---|---|---|---|
| Cell Cycle Modulation | Nocodazole [43] | HEK293T, HDFn, and H9 ESCs | Up to 3-fold | Mitotic errors and chromosomal instability [57] |
| Vinblastine [42] | HEK293T, HeLa, and HepG2 | Up to 6-fold | ||
| RO-3306 and XL413 [44] | K562 and T cells | Up to ~5-fold | ||
| NHEJ Inhibition | AZD7648 [45] | HSPCs and fibroblasts | >90% HDR | Genomic instability and inconsistent effectiveness [58,59] |
| NU7026 [46,47] | HEK293, K562, and hiPSCs | Between ~3- and ~7-fold | ||
| SCR7 [48] | MelJuSo, HEK293T, and DC2.4 | Up to 19-fold | ||
| DP308 [60] | ||||
| MMEJ Suppression | Pol θ inhibitors ART558 [49] | mESC, HEK293T, HeLa, U2OS, and RPE1 | Up to 2-fold | Genomic instability and lethality in HR-deficient cells [50] |
| PolQi1 and PolQi2 [51] | HEK293T, Jurkat HepG2, hiPSCs, T cells, and PHH | ~80% HDR | ||
| RAD51 Activation | RS-1 [52] | HEK-293 and U2OS | Between ~3- and ~6-fold | Limited validation; long-term effects unknown |
| Farrerol [61] | HEK 293FT, mESCs, and HCA2-hTERT | Up to ~3-fold | ||
| RPA-ssDNA Interaction | NSC15520 [46] | HEK293, K562, and h_iPSCs | Between ~3- and ~7-fold | Limited studies |
| Chromatin Remodeling | Panobinostat and entinostat [53] | H27, HEK293t, and HeLa | Up to ~3-fold | Cytotoxicity and apoptosis [53] |
| Valproic acid [55] | mESCs | Significant | Cas9 destabilization (valproic acid) [56] | |
| DNA Damage Response | VE-822 and AZD-77 [54] | hPSCs | Up to 6-fold | Limited studies; potential off-target effects [54] |
Adverse Effects of HDR-Enhancing Small Molecules
3.2. Optimizing Repair Templates to Enhance HDR
3.2.1. Viral-Based Repair Templates
3.2.2. Single-Stranded DNA (ssDNA) Templates
| Template Type | Key Modifications | Suitable Applications | HDR Increase | Limitations |
|---|---|---|---|---|
| Viral Templates | Capsid engineering [67] | T cells, HSCs, and in vivo editing | Between ~3 and ~25-fold | Integration risk; immune response [70,71] |
| Synthetic RNA-targeting sequences [69] | ||||
| ssODNs | Chemically modified (phosphorothioate) [82,83] | Point or small mutations; T cells, HEK293T, K562, and HSCs | ~21% HDR | Limited capacity for large insertions |
| Retron systems and transcription-coupled systems [85,86] | Between ~15% and ~60% HDR | |||
| cssDNA | High stability; reduced degradation; minimizes off-target integration [91,92] | Precise small edits; iPSCs and T cells | Between ~20% and ~70% HDR | Limited capacity for large insertions |
| Plasmid Templates | Synthetic RNA-targeting sequences [93,94,95] | Large insertions in immortalized cell lines | Between ~10% and ~30% HDR | Cytotoxicity at high concentrations [96,97] |
| Linear dsDNA | TEG or RNA::DNA hybrids [98] | Large insertions with homology arms (200–800 bp), can be used in primary cells, γδ-T cells, and NK cells | ~80% HDR | Cytotoxicity; random integration risks [96,97] |
| Doggybone DNA [99] | ||||
| Target sequences (tCTS) [100] | Between ~15% and ~30% HDR | |||
| Biotinylation [101] | ~80% |
3.2.3. Double-Stranded DNA (dsDNA) Templates
3.3. Enhancing HDR Using CRISPR-Cas9 Fusions Proteins
4. Emerging Strategies for Highly Precise Genome Editing
5. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kim, Y.-G.; Cha, J.; Chandrasegaran, S. Hybrid Restriction Enzymes: Zinc Finger Fusions to Fok I Cleavage Domain. Proc. Natl. Acad. Sci. USA 1996, 93, 1156–1160. [Google Scholar] [CrossRef] [PubMed]
- Christian, M.; Cermak, T.; Doyle, E.L.; Schmidt, C.; Zhang, F.; Hummel, A.; Bogdanove, A.J.; Voytas, D.F. Targeting DNA double-strand breaks with TAL effector nucleases. Genetics 2010, 186, 757–761. [Google Scholar] [CrossRef] [PubMed]
- Jinek, M.; Chylinski, K.; Fonfara, I.; Hauer, M.; Doudna, J.A.; Charpentier, E. A Programmable Dual-Rna–Guided DNA Endonuclease in Adaptive Bacterial Immunity. Science 2012, 337, 816–821. [Google Scholar] [CrossRef]
- Zhang, F. Development of Crispr-Cas Systems for Genome Editing and Beyond. Q. Rev. Biophys. 2019, 52, e6. [Google Scholar] [CrossRef]
- Dudáš, A.; Chovanec, M. DNA Double-Strand Break Repair by Homologous Recombination. Mutat. Res./Rev. Mutat. Res. 2004, 566, 131–167. [Google Scholar] [CrossRef]
- Kanaar, R.; Hoeijmakers, J.H.; van Gent, D.C. Molecular Mechanisms of DNA Double-Strand Break Repair. Trends Cell Biol. 1998, 8, 483–489. [Google Scholar] [CrossRef]
- Hefferin, M.L.; Tomkinson, A.E. Mechanism of DNA Double-Strand Break Repair by Non-Homologous End Joining. DNA Repair 2005, 4, 639–648. [Google Scholar] [CrossRef]
- Sfeir, A.; Symington, L.S. Microhomology-Mediated End Joining: A Back-up Survival Mechanism or Dedicated Pathway? Trends Biochem. Sci. 2015, 40, 701–714. [Google Scholar] [CrossRef]
- Seol, J.-H.; Shim, E.Y.; Lee, S.E. Microhomology-Mediated End Joining: Good, Bad and Ugly. Mutat. Res./Fundam. Mol. Mech. Mutagen. 2018, 809, 81–87. [Google Scholar] [CrossRef]
- Komor, A.C.; Kim, Y.B.; Packer, M.S.; Zuris, J.A.; Liu, D.R. Programmable Editing of a Target Base in Genomic DNA without Double-Stranded DNA Cleavage. Nature 2016, 533, 420–424. [Google Scholar] [CrossRef]
- Chen, P.J.; Liu, D.R. Prime Editing for Precise and Highly Versatile Genome Manipulation. Nat. Rev. Genet. 2023, 24, 161–177. [Google Scholar] [CrossRef]
- Vogt, A.; He, Y. Structure and Mechanism in Non-Homologous End Joining. DNA Repair 2023, 130, 103547. [Google Scholar] [CrossRef] [PubMed]
- Yue, X.; Bai, C.; Xie, D.; Ma, T.; Zhou, P.-K. DNA-Pkcs: A Multi-Faceted Player in DNA Damage Response. Front. Genet. 2020, 11, 607428. [Google Scholar] [CrossRef]
- Carusillo, A.; Mussolino, C. DNA Damage: From Threat to Treatment. Cells 2020, 9, 1665. [Google Scholar] [CrossRef] [PubMed]
- Liao, H.; Wu, J.; VanDusen, N.J.; Li, Y.; Zheng, Y. Crispr-Cas9-Mediated Homology-Directed Repair for Precise Gene Editing. Mol. Ther. Nucleic Acids 2024, 35, 102344. [Google Scholar] [CrossRef]
- Xue, C.; Greene, E.C. DNA Repair Pathway Choices in Crispr-Cas9-Mediated Genome Editing. Trends Genet. 2021, 37, 639–656. [Google Scholar] [CrossRef] [PubMed]
- Hepburn, M.R. Integrative Structural Model of DNA-Pkcs in the Initial Steps of Non-Homologous End Joining. Doctoral Thesis, University of Calgary, Calgary, AB, Canada, 2020. [Google Scholar]
- Lei, T.; Du, S.; Peng, Z.; Chen, L. Multifaceted Regulation and Functions of 53bp1 in Nhej-Mediated Dsb Repair. Int. J. Mol. Med. 2022, 50, 90. [Google Scholar] [CrossRef]
- Shibata, A.; Jeggo, P.A. Roles for the DNA-Pk Complex and 53bp1 in Protecting Ends from Resection During DNA Double-Strand Break Repair. J. Radiat. Res. 2020, 61, 718–726. [Google Scholar] [CrossRef]
- Ma, Y.; Pannicke, U.; Schwarz, K.; Lieber, M.R. Hairpin Opening and Overhang Processing by an Artemis/DNA-Dependent Protein Kinase Complex in Nonhomologous End Joining and V (D) J Recombination. Cell 2002, 108, 781–794. [Google Scholar] [CrossRef]
- Chang, H.H.; Pannunzio, N.R.; Adachi, N.; Lieber, M.R. Non-Homologous DNA End Joining and Alternative Pathways to Double-Strand Break Repair. Nat. Rev. Mol. Cell Biol. 2017, 18, 495–506. [Google Scholar] [CrossRef]
- Goodarzi, A.A.; Yu, Y.; Riballo, E.; Douglas, P.; Walker, S.A.; Ye, R.; Härer, C.; Marchetti, C.; Morrice, N.; Jeggo, P.A. DNA-Pk Autophosphorylation Facilitates Artemis Endonuclease Activity. EMBO J. 2006, 25, 3880–3889. [Google Scholar] [CrossRef] [PubMed]
- Ahnesorg, P.; Smith, P.; Jackson, S.P. Xlf Interacts with the Xrcc4-DNA Ligase Iv Complex to Promote DNA Nonhomologous End-Joining. Cell 2006, 124, 301–313. [Google Scholar] [CrossRef] [PubMed]
- Ochi, T.; Blackford, A.N.; Coates, J.; Jhujh, S.; Mehmood, S.; Tamura, N.; Travers, J.; Wu, Q.; Draviam, V.M.; Robinson, C.V. Paxx, a Paralog of Xrcc4 and Xlf, Interacts with Ku to Promote DNA Double-Strand Break Repair. Science 2015, 347, 185–188. [Google Scholar] [CrossRef]
- Bétermier, M.; Bertrand, P.; Lopez, B.S. Is Non-Homologous End-Joining Really an Inherently Error-Prone Process? PLoS Genet. 2014, 10, e1004086. [Google Scholar] [CrossRef]
- Gallagher, D.N.; Haber, J.E. Repair of a Site-Specific DNA Cleavage: Old-School Lessons for Cas9-Mediated Gene Editing. ACS Chem. Biol. 2018, 13, 397–405. [Google Scholar] [CrossRef] [PubMed]
- Heyer, W.-D.; Ehmsen, K.T.; Liu, J. Regulation of Homologous Recombination in Eukaryotes. Annu. Rev. Genet. 2010, 44, 113–139. [Google Scholar] [CrossRef]
- Williams, R.S.; Dodson, G.E.; Limbo, O.; Yamada, Y.; Williams, J.S.; Guenther, G.; Classen, S.; Glover, J.M.; Iwasaki, H.; Russell, P. Nbs1 Flexibly Tethers Ctp1 and Mre11-Rad50 to Coordinate DNA Double-Strand Break Processing and Repair. Cell 2009, 139, 87–99. [Google Scholar] [CrossRef]
- Yu, X.; Chen, J. DNA Damage-Induced Cell Cycle Checkpoint Control Requires Ctip, a Phosphorylation-Dependent Binding Partner of Brca1 C-Terminal Domains. Mol. Cell. Biol. 2004, 24, 9478–9486. [Google Scholar] [CrossRef]
- Chen, R.; Wold, M.S. Replication Protein A: Single-Stranded Dna’s First Responder: Dynamic DNA-Interactions Allow Replication Protein a to Direct Single-Strand DNA Intermediates into Different Pathways for Synthesis or Repair. Bioessays 2014, 36, 1156–1161. [Google Scholar] [CrossRef]
- Yang, H.; Li, Q.; Fan, J.; Holloman, W.K.; Pavletich, N.P. The Brca2 Homologue Brh2 Nucleates Rad51 Filament Formation at a Dsdna–Ssdna Junction. Nature 2005, 433, 653–657. [Google Scholar] [CrossRef]
- Miyabe, I.; Mizuno, K.I.; Keszthelyi, A.; Daigaku, Y.; Skouteri, M.; Mohebi, S.; Kunkel, T.A.; Murray, J.M.; Carr, A.M. Polymerase Δ Replicates Both Strands after Homologous Recombination–Dependent Fork Restart. Nat. Struct. Mol. Biol. 2015, 22, 932–938. [Google Scholar] [CrossRef] [PubMed]
- Roy, U.; Greene, E.C. Demystifying the D-Loop During DNA Recombination; Nature Publishing Group: London, UK, 2020. [Google Scholar]
- San Filippo, J.; Sung, P.; Klein, H. Mechanism of Eukaryotic Homologous Recombination. Annu. Rev. Biochem. 2008, 77, 229–257. [Google Scholar] [CrossRef]
- Ramsden, D.A.; Carvajal-Garcia, J.; Gupta, G.P. Mechanism, Cellular Functions and Cancer Roles of Polymerase-Theta-Mediated DNA End Joining. Nat. Rev. Mol. Cell Biol. 2022, 23, 125–140. [Google Scholar] [CrossRef]
- Kent, T.; Mateos-Gomez, P.A.; Sfeir, A.; Pomerantz, R.T. Polymerase Θ Is a Robust Terminal Transferase That Oscillates between Three Different Mechanisms During End-Joining. eLife 2016, 5, e13740. [Google Scholar] [CrossRef]
- McVey, M.; Lee, S.E. Mmej Repair of Double-Strand Breaks (Director’s Cut): Deleted Sequences and Alternative Endings. Trends Genet. 2008, 24, 529–538. [Google Scholar] [CrossRef] [PubMed]
- Mateos-Gomez, P.A.; Gong, F.; Nair, N.; Miller, K.M.; Lazzerini-Denchi, E.; Sfeir, A. Mammalian Polymerase Θ Promotes Alternative Nhej and Suppresses Recombination. Nature 2015, 518, 254–257. [Google Scholar] [CrossRef]
- Bhargava, R.; Onyango, D.O.; Stark, J.M. Regulation of Single-Strand Annealing and Its Role in Genome Maintenance. Trends Genet. 2016, 32, 566–575. [Google Scholar] [CrossRef] [PubMed]
- Sung, P.; Krejci, L.; Van Komen, S.; Sehorn, M.G. Rad51 Recombinase and Recombination Mediators. J. Biol. Chem. 2003, 278, 42729–42732. [Google Scholar] [CrossRef]
- Mimitou, E.P.; Symington, L.S. Sae2, Exo1 and Sgs1 Collaborate in DNA Double-Strand Break Processing. Nature 2008, 455, 770–774. [Google Scholar] [CrossRef]
- Li, G.; Wang, H.; Zhang, X.; Wu, Z.; Yang, H. A Cas9–Transcription Factor Fusion Protein Enhances Homology-Directed Repair Efficiency. J. Biol. Chem. 2021, 296, 100525. [Google Scholar] [CrossRef]
- Lin, S.; Staahl, B.T.; Alla, R.K.; Doudna, J.A. Enhanced Homology-Directed Human Genome Engineering by Controlled Timing of Crispr/Cas9 Delivery. eLife 2014, 3, e04766. [Google Scholar] [CrossRef] [PubMed]
- Wienert, B.; Nguyen, D.N.; Guenther, A.; Feng, S.J.; Locke, M.N.; Wyman, S.K.; Shin, J.; Kazane, K.R.; Gregory, G.L.; Carter, M.A. Timed Inhibition of Cdc7 Increases Crispr-Cas9 Mediated Templated Repair. Nat. Commun. 2020, 11, 2109. [Google Scholar] [CrossRef] [PubMed]
- Cloarec-Ung, F.-M.; Beaulieu, J.; Suthananthan, A.; Lehnertz, B.; Sauvageau, G.; Sheppard, H.M.; Knapp, D.J. Near-Perfect Precise on-Target Editing of Human Hematopoietic Stem and Progenitor Cells. eLife 2024, 12, RP91288. [Google Scholar] [CrossRef]
- Riesenberg, S.; Maricic, T. Targeting Repair Pathways with Small Molecules Increases Precise Genome Editing in Pluripotent Stem Cells. Nat. Commun. 2018, 9, 2164. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, K.; Tsunekawa, Y.; Hernandez-Benitez, R.; Wu, J.; Zhu, J.; Kim, E.J.; Hatanaka, F.; Yamamoto, M.; Araoka, T.; Li, Z. In Vivo Genome Editing Via Crispr/Cas9 Mediated Homology-Independent Targeted Integration. Nature 2016, 540, 144–149. [Google Scholar] [CrossRef]
- Maruyama, T.; Dougan, S.K.; Truttmann, M.C.; Bilate, A.M.; Ingram, J.R.; Ploegh, H.L. Increasing the Efficiency of Precise Genome Editing with Crispr-Cas9 by Inhibition of Nonhomologous End Joining. Nat. Biotechnol. 2015, 33, 538–542. [Google Scholar] [CrossRef]
- Schimmel, J.; Muñoz-Subirana, N.; Kool, H.; van Schendel, R.; van der Vlies, S.; Kamp, J.A.; de Vrij, F.M.; Kushner, S.A.; Smith, G.C.; Boulton, S.J. Modulating Mutational Outcomes and Improving Precise Gene Editing at Crispr-Cas9-Induced Breaks by Chemical Inhibition of End-Joining Pathways. Cell Rep. 2023, 42, 112019. [Google Scholar] [CrossRef]
- Zatreanu, D.; Robinson, H.M.; Alkhatib, O.; Boursier, M.; Finch, H.; Geo, L.; Grande, D.; Grinkevich, V.; Heald, R.A.; Langdon, S. Polθ Inhibitors Elicit Brca-Gene Synthetic Lethality and Target Parp Inhibitor Resistance. Nat. Commun. 2021, 12, 3636. [Google Scholar] [CrossRef]
- Wimberger, S.; Akrap, N.; Firth, M.; Brengdahl, J.; Engberg, S.; Schwinn, M.K.; Slater, M.R.; Lundin, A.; Hsieh, P.-P.; Li, S. Simultaneous Inhibition of DNA-Pk and Polϴ Improves Integration Efficiency and Precision of Genome Editing. Nat. Commun. 2023, 14, 4761. [Google Scholar] [CrossRef]
- Pinder, J.; Salsman, J.; Dellaire, G. Nuclear Domain ‘Knock-In’screen for the Evaluation and Identification of Small Molecule Enhancers of Crispr-Based Genome Editing. Nucleic Acids Res. 2015, 43, 9379–9392. [Google Scholar] [CrossRef]
- Liu, B.; Chen, S.; Rose, A.L.; Chen, D.; Cao, F.; Zwinderman, M.; Kiemel, D.; Aïssi, M.; Dekker, F.J.; Haisma, H.J. Inhibition of Histone Deacetylase 1 (Hdac1) and Hdac2 Enhances Crispr/Cas9 Genome Editing. Nucleic Acids Res. 2020, 48, 517–532. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.; Chen, X.; Jin, Y.; Ge, W.; Wang, W.; Kong, L.; Ji, J.; Guo, X.; Huang, J.; Feng, X.-H. Small Molecules Promote Crispr-Cpf1-Mediated Genome Editing in Human Pluripotent Stem Cells. Nat. Commun. 2018, 9, 1303. [Google Scholar] [CrossRef]
- Park, H.; Shin, J.; Choi, H.; Cho, B.; Kim, J. Valproic Acid Significantly Improves Crispr/Cas9-Mediated Gene Editing. Cells 2020, 9, 1447. [Google Scholar] [CrossRef] [PubMed]
- Cheng, X. Valproic Acid Thermally Destabilizes and Inhibits Spycas9 Activity. Mol. Ther. 2020, 28, 2635–2641. [Google Scholar] [CrossRef] [PubMed]
- Vasquez, R.J.; Howell, B.; Yvon, A.; Wadsworth, P.; Cassimeris, L. Nanomolar Concentrations of Nocodazole Alter Microtubule Dynamic Instability In Vivo and In Vitro. Mol. Biol. Cell 1997, 8, 973–985. [Google Scholar] [CrossRef]
- Cullot, G.; Aird, E.J.; Schlapansky, M.F.; Yeh, C.D.; van de Venn, L.; Vykhlyantseva, I.; Kreutzer, S.; Mailänder, D.; Lewków, B.; Klermund, J. Genome Editing with the Hdr-Enhancing DNA-Pkcs Inhibitor Azd7648 Causes Large-Scale Genomic Alterations. Nat. Biotechnol. 2024, 1–5. [Google Scholar] [CrossRef]
- Gerlach, M.; Kraft, T.; Brenner, B.; Petersen, B.; Niemann, H.; Montag, J. Efficient Knock-in of a Point Mutation in Porcine Fibroblasts Using the Crispr/Cas9-Gmnn Fusion Gene. Genes 2018, 9, 296. [Google Scholar] [CrossRef]
- Sun, Y.; Lu, H.; Fang, X.; Xiao, S.; Yang, F.; Chen, Y.; Wang, H.; Li, X.; Lu, J.; Lin, H. Discovery of a Novel 53bp1 Inhibitor through Alphascreen-Based High-Throughput Screening. Bioorg. Med. Chem. 2021, 34, 116054. [Google Scholar] [CrossRef]
- Zhang, W.; Chen, Y.; Yang, J.; Zhang, J.; Yu, J.; Wang, M.; Zhao, X.; Wei, K.; Wan, X.; Xu, X. A High-Throughput Small Molecule Screen Identifies Farrerol as a Potentiator of Crispr/Cas9-Mediated Genome Editing. eLife 2020, 9, e56008. [Google Scholar] [CrossRef]
- Eichenlaub-Ritter, U.; Boll, I. Nocodazole Sensitivity, Age-Related Aneuploidy, and Alterations in the Cell Cycle During Maturation of Mouse Oocytes. Cytogenet. Genome Res. 1989, 52, 170–176. [Google Scholar] [CrossRef]
- Hande, K.R.; Hagey, A.; Berlin, J.; Cai, Y.; Meek, K.; Kobayashi, H.; Lockhart, A.C.; Medina, D.; Sosman, J.; Gordon, G.B. The Pharmacokinetics and Safety of Abt-751, a Novel, Orally Bioavailable Sulfonamide Antimitotic Agent: Results of a Phase 1 Study. Clin. Cancer Res. 2006, 12, 2834–2840. [Google Scholar] [CrossRef] [PubMed]
- Gangopadhyay, S.A.; Cox, K.J.; Manna, D.; Lim, D.; Maji, B.; Zhou, Q.; Choudhary, A. Precision Control of Crispr-Cas9 Using Small Molecules and Light. Biochemistry 2019, 58, 234–244. [Google Scholar] [CrossRef]
- Moço, P.D.; Aharony, N.; Kamen, A. Adeno-Associated Viral Vectors for Homology-Directed Generation of Car-T Cells. Biotechnol. J. 2020, 15, 1900286. [Google Scholar] [CrossRef] [PubMed]
- Duddy, G.; Courtis, K.; Horwood, J.; Olsen, J.; Horsler, H.; Hodgson, T.; Varsani-Brown, S.; Abdullah, A.; Denti, L.; Lane, H. Donor Template Delivery by Recombinant Adeno-Associated Virus for the Production of Knock-in Mice. BMC Biol. 2024, 22, 26. [Google Scholar] [CrossRef]
- Ling, C.; Yu, C.; Wang, C.; Yang, M.; Yang, H.; Yang, K.; He, Y.; Shen, Y.; Tang, S.; Yu, X. Raav Capsid Mutants Eliminate Leaky Expression from DNA Donor Template for Homologous Recombination. Nucleic Acids Res. 2024, 52, 6518–6531. [Google Scholar] [CrossRef]
- Gwiazda, K.S.; Grier, A.E.; Sahni, J.; Burleigh, S.M.; Martin, U.; Yang, J.G.; Popp, N.A.; Krutein, M.C.; Khan, I.F.; Jacoby, K. High Efficiency Crispr/Cas9-Mediated Gene Editing in Primary Human T-Cells Using Mutant Adenoviral E4orf6/E1b55k “Helper” Proteins. Mol. Ther. 2016, 24, 1570–1580. [Google Scholar] [CrossRef] [PubMed]
- Karnan, S.; Ota, A.; Konishi, Y.; Wahiduzzaman, M.; Hosokawa, Y.; Konishi, H. Improved Methods of Aav-Mediated Gene Targeting for Human Cell Lines Using Ribosome-Skipping 2A Peptide. Nucleic Acids Res. 2016, 44, e54. [Google Scholar] [CrossRef]
- Suoranta, T.; Laham-Karam, N.; Ylä-Herttuala, S. Strategies to Improve Safety Profile of AAV Vectors. Front. Mol. Med. 2022, 2, 1054069. [Google Scholar] [CrossRef]
- Hanlon, K.S.; Kleinstiver, B.P.; Garcia, S.P.; Zaborowski, M.P.; Volak, A.; Spirig, S.E.; Muller, A.; Sousa, A.A.; Tsai, S.Q.; Bengtsson, N.E. High Levels of AAV Vector Integration into CRISPR-Induced DNA Breaks. Nat. Commun. 2019, 10, 4439. [Google Scholar] [CrossRef]
- Cornu, T.I.; Cathomen, T. Targeted Genome Modifications Using Integrase-Deficient Lentiviral Vectors. Mol. Ther. 2007, 15, 2107–2113. [Google Scholar] [CrossRef]
- Uchida, N.; Drysdale, C.M.; Nassehi, T.; Gamer, J.; Yapundich, M.; DiNicola, J.; Shibata, Y.; Hinds, M.; Gudmundsdottir, B.; Haro-Mora, J.J.; et al. Cas9 Protein Delivery Non-Integrating Lentiviral Vectors for Gene Correction in Sickle Cell Disease. Mol. Ther. Methods Clin. Dev. 2021, 21, 121–132. [Google Scholar] [CrossRef] [PubMed]
- Lombardo, A.; Genovese, P.; Beausejour, C.M.; Colleoni, S.; Lee, Y.L.; Kim, K.A.; Ando, D.; Urnov, F.D.; Galli, C.; Gregory, P.D.; et al. Gene Editing in Human Stem Cells Using Zinc Finger Nucleases and Integrase-Defective Lentiviral Vector Delivery. Nat. Biotechnol. 2007, 25, 1298–1306. [Google Scholar] [CrossRef]
- Asperti, C.; Canarutto, D.; Porcellini, S.; Sanvito, F.; Cecere, F.; Vavassori, V.; Ferrari, S.; Rovelli, E.; Albano, L.; Jacob, A. Scalable Gmp-Compliant Gene Correction of Cd4+ T Cells with Idlv Template Functionally Validated In Vitro and In Vivo. Mol. Ther. Methods Clin. Dev. 2023, 30, 546–557. [Google Scholar] [CrossRef]
- Joglekar, A.V.; Hollis, R.P.; Kuftinec, G.; Senadheera, S.; Chan, R.; Kohn, D.B. Integrase-Defective lentiviral Vectors as a Delivery Platform for Targeted Modification of Adenosine Deaminase Locus. Mol. Ther. 2013, 21, 1705–1717. [Google Scholar] [CrossRef] [PubMed]
- Gutierrez-Guerrero, A.; Sanchez-Hernandez, S.; Galvani, G.; Pinedo-Gomez, J.; Martin-Guerra, R.; Sanchez-Gilabert, A.; Aguilar-González, A.; Cobo, M.; Gregory, P.; Holmes, M.; et al. Comparison of Zinc Finger Nucleases Versus Crispr-Specific Nucleases for Genome Editing of the Wiskott-Aldrich Syndrome Locus. Hum. Gene Ther. 2018, 29, 366–380. [Google Scholar] [CrossRef] [PubMed]
- Ferrari, S.; Jacob, A.; Cesana, D.; Laugel, M.; Beretta, S.; Varesi, A.; Unali, G.; Conti, A.; Canarutto, D.; Albano, L.; et al. Choice of Template Delivery Mitigates the Genotoxic Risk and Adverse Impact of Editing in Human Hematopoietic Stem Cells. Cell Stem Cell 2022, 29, 1428–1444.e9. [Google Scholar] [CrossRef]
- Okamoto, S.; Amaishi, Y.; Maki, I.; Enoki, T.; Mineno, J. Highly Efficient Genome Editing for Single-Base Substitutions Using Optimized ssODNs with Cas9-RNPs. Sci. Rep. 2019, 9, 4811. [Google Scholar] [CrossRef]
- Wang, K.; Tang, X.; Liu, Y.; Xie, Z.; Zou, X.; Li, M.; Yuan, H.; Ouyang, H.; Jiao, H.; Pang, D. Efficient Generation of Orthologous Point Mutations in Pigs via CRISPR-Assisted ssODN-Mediated Homology-Directed Repair. Mol. Ther. Nucleic Acids 2016, 5, e396. [Google Scholar] [CrossRef]
- Foschi, N.; Athanasakis, E.; Gasparini, P.; Di Stazio, M.; d’Adamo, A.P. Systematic Analysis of Factors That Improve Hdr Efficiency in Crispr/Cas9 Technique. Res. Square 2020. [Google Scholar] [CrossRef]
- Renaud, J.-B.; Boix, C.; Charpentier, M.; De Cian, A.; Cochennec, J.; Duvernois-Berthet, E.; Perrouault, L.; Tesson, L.; Edouard, J.; Thinard, R. Improved Genome Editing Efficiency and Flexibility Using Modified Oligonucleotides with Talen and Crispr-Cas9 Nucleases. Cell Rep. 2016, 14, 2263–2272. [Google Scholar] [CrossRef]
- Clavé, G.; Reverte, M.; Vasseur, J.-J.; Smietana, M. Modified Internucleoside Linkages for Nuclease-Resistant Oligonucleotides. RSC Chem. Biol. 2021, 2, 94–150. [Google Scholar] [CrossRef] [PubMed]
- Jin, Y.-Y.; Zhang, P.; Liu, L.-L.; Zhao, X.; Hu, X.-Q.; Liu, S.-Z.; Li, Z.-K.; Liu, Q.; Wang, J.-Q.; Hao, D.-L. Enhancing Homology-Directed Repair Efficiency with HDR-Boosting Modular ssDNA Donor. Nat. Commun. 2024, 15, 6843. [Google Scholar] [CrossRef]
- Zhao, B.; Chen, S.-A.A.; Lee, J.; Fraser, H.B. Bacterial Retrons Enable Precise Gene Editing in Human Cells. Cris. J. 2022, 5, 31–39. [Google Scholar] [CrossRef]
- Lopez, S.C.; Crawford, K.D.; Lear, S.K.; Bhattarai-Kline, S.; Shipman, S.L. Precise Genome Editing Across Kingdoms of Life Using Retron-Derived DNA. Nat. Chem. Biol. 2022, 18, 199–206. [Google Scholar] [CrossRef] [PubMed]
- Aird, E.J.; Lovendahl, K.N.; St. Martin, A.; Harris, R.S.; Gordon, W.R. Increasing Cas9-Mediated Homology-Directed Repair Efficiency Through Covalent Tethering of DNA Repair Template. Commun. Biol. 2018, 1, 54. [Google Scholar] [CrossRef] [PubMed]
- Ling, X.; Xie, B.; Gao, X.; Chang, L.; Zheng, W.; Chen, H.; Huang, Y.; Tan, L.; Li, M.; Liu, T. Improving the Efficiency of Precise Genome Editing with Site-Specific Cas9-Oligonucleotide Conjugates. Sci. Adv. 2020, 6, eaaz0051. [Google Scholar] [CrossRef]
- Kang, Y.K.; Lee, J.; Lee, J.H.; Jeong, J.; Kim, D.K.; Yang, S.Y.; Jung, K.; Kim, S.-G.; Chung, H.J. Cas9 Conjugate Complex Delivering Donor DNA for Efficient Gene Editing by Homology-Directed Repair. J. Ind. Eng. Chem. 2021, 102, 241–250. [Google Scholar] [CrossRef]
- Gao, K.; Zhang, X.; Zhang, Z.; Wu, X.; Guo, Y.; Fu, P.; Sun, A.; Peng, J.; Zheng, J.; Yu, P. Transcription-Coupled Donor DNA Expression Increases Homologous Recombination for Efficient Genome Editing. Nucleic Acids Res. 2022, 50, e109. [Google Scholar] [CrossRef]
- Iyer, S.; Mir, A.; Vega-Badillo, J.; Roscoe, B.P.; Ibraheim, R.; Zhu, L.J.; Lee, J.; Liu, P.; Luk, K.; Mintzer, E. Efficient Homology-Directed Repair with Circular Single-Stranded DNA Donors. Cris. J. 2022, 5, 685–701. [Google Scholar] [CrossRef]
- Xie, K.; Starzyk, J.; Majumdar, I.; Rincones, K.; Tran, T.; Lee, D.; Niemi, S.; Famiglietti, J.; Suter, B.; Shan, R. Circular Single-Stranded DNA Is a Superior Homology-Directed Repair Donor Template for Efficient Genome Engineering. bioRxiv 2022. [Google Scholar] [CrossRef]
- Shepelev, M.; Komkov, D.; Golubev, D.; Borovikova, S.; Mazurov, D.; Kruglova, N. Donor DNA Modification with Cas9 Targeting Sites Improves the Efficiency of MTC34 Knock-in into the CXCR4 Locus. Mol. Biol. 2024, 58, 672–682. [Google Scholar] [CrossRef]
- Shepelev, M.; Komkov, D.; Golubev, D.; Borovikova, S.; Mazurov, D.; Kruglova, N. Increasing the Level of Knock-In of the MT-C34-Encoding Construct into the CXCR4 Locus by Modifying Donor DNA with Cas9 Target Sites. Mol. Biol. 2024, 58, 590–600. [Google Scholar] [CrossRef]
- Ishibashi, R.; Maki, R.; Toyoshima, F. Gene Targeting in Adult Organs Using in Vivo Cleavable Donor Plasmids for CRISPR-Cas9 and CRISPR-Cas12a. Sci. Rep. 2024, 14, 7615. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Li, T.; Ou, J.; Huang, J.; Liang, P. Homology-Based Repair Induced by CRISPR-Cas Nucleases in Mammalian Embryo Genome Editing. Protein Cell 2022, 13, 316–335. [Google Scholar] [CrossRef] [PubMed]
- Agudelo, D.; Duringer, A.; Bozoyan, L.; Huard, C.C.; Carter, S.; Loehr, J.; Synodinou, D.; Drouin, M.; Salsman, J.; Dellaire, G. Marker-Free Coselection for CRISPR-Driven Genome Editing in Human Cells. Nat. Methods 2017, 14, 615–620. [Google Scholar] [CrossRef]
- Ghanta, K.S.; Chen, Z.; Mir, A.; Dokshin, G.A.; Krishnamurthy, P.M.; Yoon, Y.; Gallant, J.; Xu, P.; Zhang, X.-O.; Ozturk, A.R. 5′-Modifications Improve Potency and Efficacy of DNA Donors for Precision Genome Editing. eLife 2021, 10, e72216. [Google Scholar] [CrossRef]
- Moretti, A.; Ponzo, M.; Nicolette, C.A.; Tcherepanova, I.Y.; Biondi, A.; Magnani, C.F. The Past, Present, and Future of Non-Viral CAR T Cells. Front. Immunol. 2022, 13, 867013. [Google Scholar] [CrossRef]
- Nguyen, D.N.; Roth, T.L.; Li, J.; Chen, P.A.; Mamedov, M.R.; Vo, L.T.; Tobin, V.; Apathy, R.; Goodman, D.; Shifrut, E. A Cas9 Nanoparticle System with Truncated Cas9 Target Sequences on Dna Repair Templates Enhances Genome Targeting in Diverse Human Immune Cell Types. bioRxiv 2019. [Google Scholar] [CrossRef]
- Roche, P.J.; Gytz, H.; Hussain, F.; Cameron, C.J.; Paquette, D.; Blanchette, M.; Dostie, J.; Nagar, B.; Akavia, U.D. Double-Stranded Biotinylated Donor Enhances Homology-Directed Repair In Combination with Cas9 Monoavidin in Mammalian Cells. Cris. J. 2018, 1, 414–430. [Google Scholar] [CrossRef]
- Kim, D.; Kim, S.M.; Lee, J.; Kim, J.; Lee, J.S. Knockout of the Lysosomal Membrane Protein, LAMP2C, Improves Transient Gene Expression in HEK293 Cells via Increased Intracellular Plasmid Availability. Biotechnol. J. 2024, 19, 2300017. [Google Scholar] [CrossRef]
- Seki, A.; Rutz, S. Optimized RNP Transfection for Highly Efficient CRISPR/Cas9-Mediated Gene Knockout in Primary T Cells. J. Exp. Med. 2018, 215, 985–997. [Google Scholar] [CrossRef] [PubMed]
- Hendel, A.; Kildebeck, E.J.; Fine, E.J.; Clark, J.T.; Punjya, N.; Sebastiano, V.; Bao, G.; Porteus, M.H. Quantifying Genome-Editing Outcomes at Endogenous Loci with SMRT Sequencing. Cell Rep. 2014, 7, 293–305. [Google Scholar] [CrossRef] [PubMed]
- Roth, T.L.; Puig-Saus, C.; Yu, R.; Shifrut, E.; Carnevale, J.; Li, P.J.; Hiatt, J.; Saco, J.; Krystofinski, P.; Li, H. Reprogramming Human T Cell Function and Specificity with Non-Viral Genome Targeting. Nature 2018, 559, 405–409. [Google Scholar] [CrossRef]
- Yang, M.; Tkach, D.; Boyne, A.; Kazancioglu, S.; Duclert, A.; Poirot, L.; Duchateau, P.; Juillerat, A. Optimized Two-Step Electroporation Process to Achieve Efficient Nonviral-Mediated Gene Insertion into Primary T Cells. FEBS Open Bio 2022, 12, 38–50. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Guo, Y.; Tian, Q.; Lan, Y.; Yeh, H.; Zhang, M.; Tasan, I.; Jain, S.; Zhao, H. An Efficient Gene Knock-In Strategy Using 5′-Modified Double-Stranded DNA Donors with Short Homology Arms. Nat. Chem. Biol. 2020, 16, 387–390. [Google Scholar] [CrossRef]
- Pawłowska, R.; Guga, P. Phosphorothioate Nucleic Acids: Artificial Modification Envisaged by Nature. In Handbook of Chemical Biology of Nucleic Acids; Springer: Berlin/Heidelberg, Germany, 2023; pp. 1–26. [Google Scholar]
- Lin, Y.; Wagner, E.; Lächelt, U. Non-Viral Delivery of the CRISPR/Cas System: DNA Versus RNA Versus RNP. Biomater. Sci. 2022, 10, 1166–1192. [Google Scholar] [CrossRef]
- An, J.; Zhang, C.-P.; Qiu, H.-Y.; Zhang, H.-X.; Chen, Q.-B.; Zhang, Y.-M.; Lei, X.-L.; Zhang, C.-X.; Yin, H.; Zhang, Y. Enhancement of the Viability of T Cells Electroporated with DNA via Osmotic Dampening of the DNA-Sensing cGAS–STING Pathway. Nat. Biomed. Eng. 2024, 8, 149–164. [Google Scholar] [CrossRef]
- Charpentier, M.; Khedher, A.; Menoret, S.; Brion, A.; Lamribet, K.; Dardillac, E.; Boix, C.; Perrouault, L.; Tesson, L.; Geny, S. CtIP FUSION to Cas9 enhances Transgene Integration by Homology-Dependent Repair. Nat. Commun. 2018, 9, 1133. [Google Scholar] [CrossRef]
- Carusillo, A.; Haider, S.; Schäfer, R.; Rhiel, M.; Türk, D.; Chmielewski, K.O.; Klermund, J.; Mosti, L.; Andrieux, G.; Schäfer, R. A Novel Cas9 Fusion Protein Promotes Targeted Genome Editing with Reduced Mutational Burden in Primary Human Cells. Nucleic Acids Res. 2023, 51, 4660–4673. [Google Scholar] [CrossRef]
- Rees, H.A.; Yeh, W.-H.; Liu, D.R. Development of hRad51–Cas9 Nickase Fusions that Mediate HDR Without Double-Stranded Breaks. Nat. Commun. 2019, 10, 2212. [Google Scholar] [CrossRef]
- Ma, L.; Ruan, J.; Song, J.; Wen, L.; Yang, D.; Zhao, J.; Xia, X.; Chen, Y.E.; Zhang, J.; Xu, J. MiCas9 Increases Large Size Gene Knock-In Rates and Reduces Undesirable On-Target and Off-Target Indel Edits. Nat. Commun. 2020, 11, 6082. [Google Scholar] [CrossRef] [PubMed]
- Ma, M.; Zhuang, F.; Hu, X.; Wang, B.; Wen, X.-Z.; Ji, J.-F.; Xi, J.J. Efficient Generation of Mice Carrying Homozygous Double-Floxp Alleles Using the Cas9-Avidin/Biotin-Donor DNA System. Cell Res. 2017, 27, 578–581. [Google Scholar] [CrossRef] [PubMed]
- Savic, N.; Ringnalda, F.C.; Lindsay, H.; Berk, C.; Bargsten, K.; Li, Y.; Neri, D.; Robinson, M.D.; Ciaudo, C.; Hall, J. Covalent Linkage of the DNA Repair Template to the CRISPR-Cas9 Nuclease Enhances Homology-Directed Repair. eLife 2018, 7, e33761. [Google Scholar] [CrossRef]
- Ding, X.; Seebeck, T.; Feng, Y.; Jiang, Y.; Davis, G.D.; Chen, F. Improving CRISPR-Cas9 Genome EDITING efficiency by Fusion with Chromatin-Modulating Peptides. Cris. J. 2019, 2, 51–63. [Google Scholar] [CrossRef]
- Tabassum, T.; Pietrogrande, G.; Healy, M.; Wolvetang, E.J. CRISPR-Cas9 Direct Fusions for Improved Genome Editing via Enhanced Homologous Recombination. Int. J. Mol. Sci. 2023, 24, 14701. [Google Scholar] [CrossRef]
- Chen, E.; Lin-Shiao, E.; Trinidad, M.; Saffari Doost, M.; Colognori, D.; Doudna, J.A. Decorating Chromatin for Enhanced Genome Editing Using CRISPR-Cas9. Proc. Natl. Acad. Sci. USA 2022, 119, e2204259119. [Google Scholar] [CrossRef] [PubMed]
- Gutschner, T.; Haemmerle, M.; Genovese, G.; Draetta, G.F.; Chin, L. Post-Translational Regulation of Cas9 During G1 Enhances Homology-Directed Repair. Cell Rep. 2016, 14, 1555–1566. [Google Scholar] [CrossRef]
- Jayavaradhan, R.; Pillis, D.M.; Goodman, M.; Zhang, F.; Zhang, Y.; Andreassen, P.R.; Malik, P. CRISPR-Cas9 Fusion to Dominant-Negative 53BP1 Enhances HDR and Inhibits NHEJ Specifically at Cas9 Target Sites. Nat. Commun. 2019, 10, 2866. [Google Scholar] [CrossRef]
- Macarrón Palacios, A.; Korus, P.; Wilkens, B.G.; Heshmatpour, N.; Patnaik, S.R. Revolutionizing In Vivo Therapy with CRISPR/Cas Genome Editing: Breakthroughs, Opportunities and Challenges. Front. Genome Ed. 2024, 6, 1342193. [Google Scholar] [CrossRef]
- Anzalone, A.V.; Koblan, L.W.; Liu, D.R. Genome Editing with CRISPR–Cas Nucleases, BASE editors, Transposases and Prime Editors. Nat. Biotechnol. 2020, 38, 824–844. [Google Scholar] [CrossRef]
- Nishida, K.; Arazoe, T.; Yachie, N.; Banno, S.; Kakimoto, M.; Tabata, M.; Mochizuki, M.; Miyabe, A.; Araki, M.; Hara, K.Y. Targeted Nucleotide Editing Using Hybrid Prokaryotic and Vertebrate Adaptive Immune Systems. Science 2016, 353, aaf8729. [Google Scholar] [CrossRef]
- Gaudelli, N.M.; Komor, A.C.; Rees, H.A.; Packer, M.S.; Badran, A.H.; Bryson, D.I.; Liu, D.R. Programmable Base Editing of A• T to G• C in genomic DNA without DNA Cleavage. Nature 2017, 551, 464–471. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Wang, Y.; Liu, Y.; Yang, B.; Wang, X.; Wei, J.; Lu, Z.; Zhang, Y.; Wu, J.; Huang, X. Base Editing with a Cpf1–Cytidine Deaminase Fusion. Nat. Biotechnol. 2018, 36, 324–327. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Qin, W.; Lu, X.; Xu, J.; Huang, H.; Bai, H.; Li, S.; Lin, S. Programmable Base Editing of Zebrafish Genome Using a Modified CRISPR-Cas9 System. Nat. Commun. 2017, 8, 118. [Google Scholar] [CrossRef]
- Kurt, I.C.; Zhou, R.; Iyer, S.; Garcia, S.P.; Miller, B.R.; Langner, L.M.; Grünewald, J.; Joung, J.K. CRISPR C-to-G Base Editors for Inducing Targeted DNA Transversions in Human Cells. Nat. Biotechnol. 2021, 39, 41–46. [Google Scholar] [CrossRef]
- Rees, H.A.; Komor, A.C.; Yeh, W.-H.; Caetano-Lopes, J.; Warman, M.; Edge, A.S.; Liu, D.R. Improving the DNA Specificity and Applicability of Base Editing Through Protein Engineering and Protein Delivery. Nat. Commun. 2017, 8, 15790. [Google Scholar] [CrossRef] [PubMed]
- Anzalone, A.V.; Randolph, P.B.; Davis, J.R.; Sousa, A.A.; Koblan, L.W.; Levy, J.M.; Chen, P.J.; Wilson, C.; Newby, G.A.; Raguram, A. Search-and-Replace Genome Editing without Double-Strand Breaks or Donor DNA. Nature 2019, 576, 149–157. [Google Scholar] [CrossRef]
- Chen, P.J.; Hussmann, J.A.; Yan, J.; Knipping, F.; Ravisankar, P.; Chen, P.-F.; Chen, C.; Nelson, J.W.; Newby, G.A.; Sahin, M. Enhanced Prime Editing Systems by Manipulating Cellular Determinants of Editing Outcomes. Cell 2021, 184, 5635–5652.e29. [Google Scholar] [CrossRef]
- Liu, P.; Liang, S.-Q.; Zheng, C.; Mintzer, E.; Zhao, Y.G.; Ponnienselvan, K.; Mir, A.; Sontheimer, E.J.; Gao, G.; Flotte, T.R. Improved Prime Editors Enable Pathogenic Allele Correction and Cancer Modelling in Adult Mice. Nat. Commun. 2021, 12, 2121. [Google Scholar] [CrossRef]
- Nelson, J.W.; Randolph, P.B.; Shen, S.P.; Everette, K.A.; Chen, P.J.; Anzalone, A.V.; An, M.; Newby, G.A.; Chen, J.C.; Hsu, A. Engineered pegRNAs Improve Prime Editing Efficiency. Nat. Biotechnol. 2022, 40, 402–410. [Google Scholar] [CrossRef]
- Gaidukov, L.; Wroblewska, L.; Teague, B.; Nelson, T.; Zhang, X.; Liu, Y.; Jagtap, K.; Mamo, S.; Tseng, W.A.; Lowe, A. A Multi-Landing Pad DNA Integration Platform for Mammalian Cell Engineering. Nucleic Acids Res. 2018, 46, 4072–4086. [Google Scholar] [CrossRef] [PubMed]
- Davis, R.P.; Costa, M.; Grandela, C.; Holland, A.M.; Hatzistavrou, T.; Micallef, S.J.; Li, X.; Goulburn, A.L.; Azzola, L.; Elefanty, A.G. A Protocol for Removal of Antibiotic Resistance Cassettes From Human Embryonic Stem Cells Genetically Modified by Homologous Recombination or Transgenesis. Nat. Protoc. 2008, 3, 1550–1558. [Google Scholar] [CrossRef] [PubMed]
- Tate, P.H.; Skarnes, W.C. Bi-Allelic Gene Targeting in Mouse Embryonic Stem Cells. Methods 2011, 53, 331–338. [Google Scholar] [CrossRef] [PubMed]
- Blanch-Asensio, A.; Grandela, C.; Brandao, K.O.; de Korte, T.; Mei, H.; Ariyurek, Y.; Yiangou, L.; Mol, M.P.; van Meer, B.J.; Kloet, S.L. STRAIGHT-IN Enables High-Throughput Targeting of Large DNA Payloads in Human Pluripotent Stem Cells. Cell Rep. Methods 2022, 2, 100300. [Google Scholar] [CrossRef]
- Peters, J.E.; Makarova, K.S.; Shmakov, S.; Koonin, E.V. Recruitment of CRISPR-Cas Systems by Tn7-like Transposons. Proc. Natl. Acad. Sci. USA 2017, 114, E7358–E7366. [Google Scholar] [CrossRef]
- Hoffmann, F.T.; Kim, M.; Beh, L.Y.; Wang, J.; Vo, P.L.H.; Gelsinger, D.R.; George, J.T.; Acree, C.; Mohabir, J.T.; Fernández, I.S. Selective TnsC Recruitment Enhances the Fidelity of RNA-Guided Transposition. Nature 2022, 609, 384–393. [Google Scholar] [CrossRef]
| Category | Fusion Protein | Mechanism | Cell Lines | HDR Increase |
|---|---|---|---|---|
| Repair Pathway Modulation | Cas9-CtIP | Promotes DNA resection | Fibroblasts and iPSCs | Between ~1- and ~2-fold [111] |
| Cas9-dnRNF168 | Blocks NHEJ by inhibiting 53BP1 recruitment | HSPCs and T cells | Not specified [112] | |
| Cas9-CtIP-dnRNF168 | Synergistically promotes HDR and inhibits NHEJ | K562, Jurkat, and HSPCs | Up to 7-fold [112] | |
| Cas9-RAD51 | Promotes strand invasion | Multiple cell lines | ~1-fold [113] | |
| Cas9-Brex27 | BRCA2 domain recruiting Rad51 | iPSCs and fibroblasts | Between ~2- and ~3-fold [114] | |
| Repair Template Saturation | Cas9-Avidin | Binds biotinylated templates | HEK293T and K562 | ~2-fold [115] |
| Cas9-SNAP | Covalently attaches donor DNA | HEK293T and K562 | Between ~3- and ~24-fold [116] | |
| Cas9-Huh | Tethers ssDNA donors via phospho-tyrosine bonds | HEK293T and U2OS | Between ~15- and ~30-fold [87] | |
| Chromatin Modulation | Cas9-HMGB1 | Increases chromatin accessibility | K562 and Jurkat | Between ~2- and ~3-fold [117,118] |
| Cas9-HMGN1 | Increases chromatin accessibility | K562 and Jurkat | ~2-fold [117,118] | |
| Cas9-HMGB1-HMGN1 | Dual fusion for synergistic chromatin effects | K562 and Jurkat | ~3-fold [117,118] | |
| Cas9-PRDM9 | Histone methyltransferase-promoting HDR | HEK293T | ~2-fold [119] | |
| Cell Cycle Control | Cas9-Geminin | Restricts Cas9 activity to S/G2 phases | HEK293T and iPSCs | ~2-fold [120] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Haider, S.; Mussolino, C. Fine-Tuning Homology-Directed Repair (HDR) for Precision Genome Editing: Current Strategies and Future Directions. Int. J. Mol. Sci. 2025, 26, 4067. https://doi.org/10.3390/ijms26094067
Haider S, Mussolino C. Fine-Tuning Homology-Directed Repair (HDR) for Precision Genome Editing: Current Strategies and Future Directions. International Journal of Molecular Sciences. 2025; 26(9):4067. https://doi.org/10.3390/ijms26094067
Chicago/Turabian StyleHaider, Sibtain, and Claudio Mussolino. 2025. "Fine-Tuning Homology-Directed Repair (HDR) for Precision Genome Editing: Current Strategies and Future Directions" International Journal of Molecular Sciences 26, no. 9: 4067. https://doi.org/10.3390/ijms26094067
APA StyleHaider, S., & Mussolino, C. (2025). Fine-Tuning Homology-Directed Repair (HDR) for Precision Genome Editing: Current Strategies and Future Directions. International Journal of Molecular Sciences, 26(9), 4067. https://doi.org/10.3390/ijms26094067