
GPR75: Advances, Challenges in Deorphanization, and Potential as a Novel Drug Target for Disease Treatment
Abstract
:1. Introduction
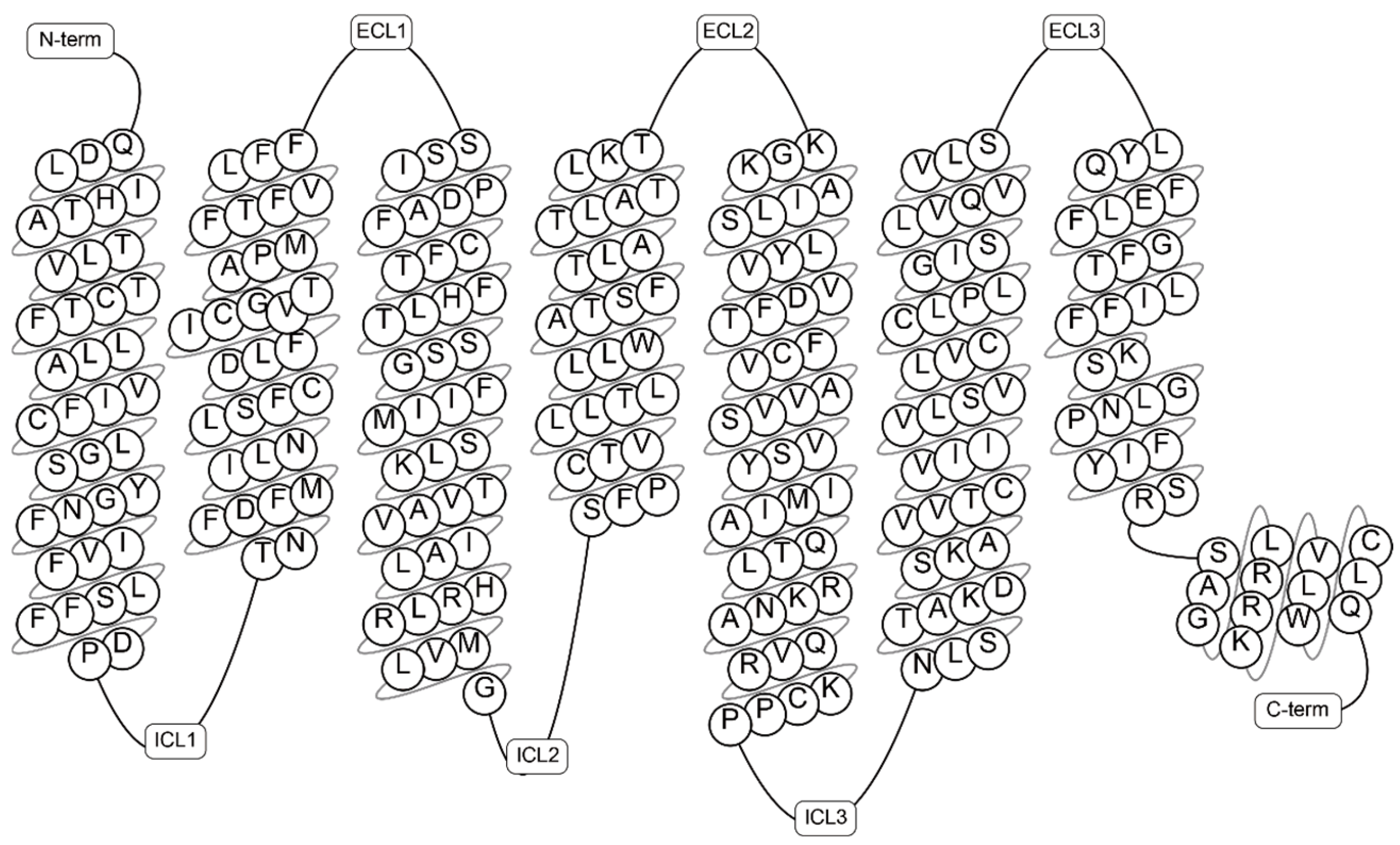
2. Overview of G Protein-Coupled Receptor 75 (GPR75)
3. Deorphanization Research of GPR75
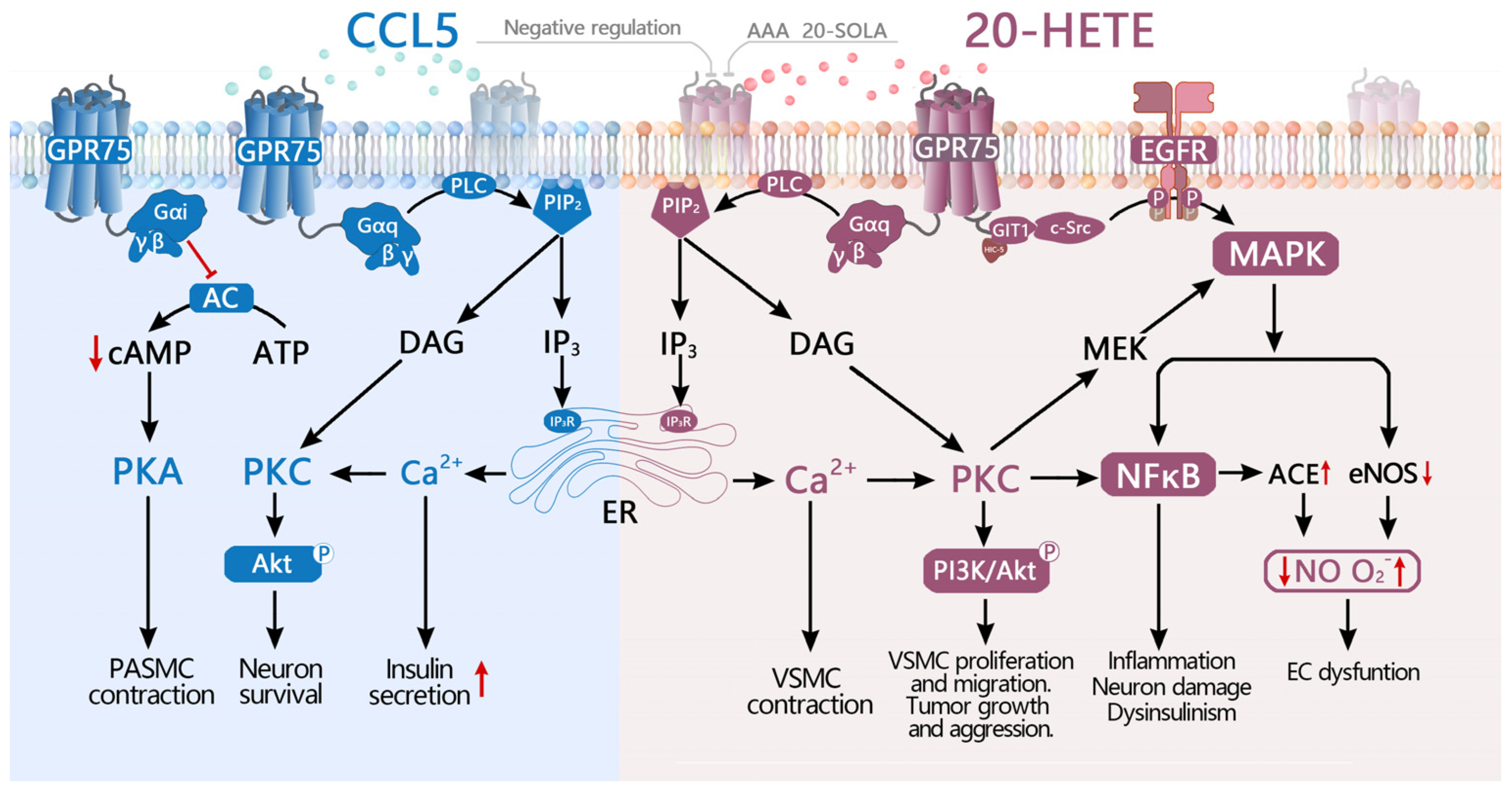
3.1. Identification of CCL5 as the First Potential Ligand for GPR75
3.2. 20-HETE Is a High-Affinity Potential Ligand for GPR75
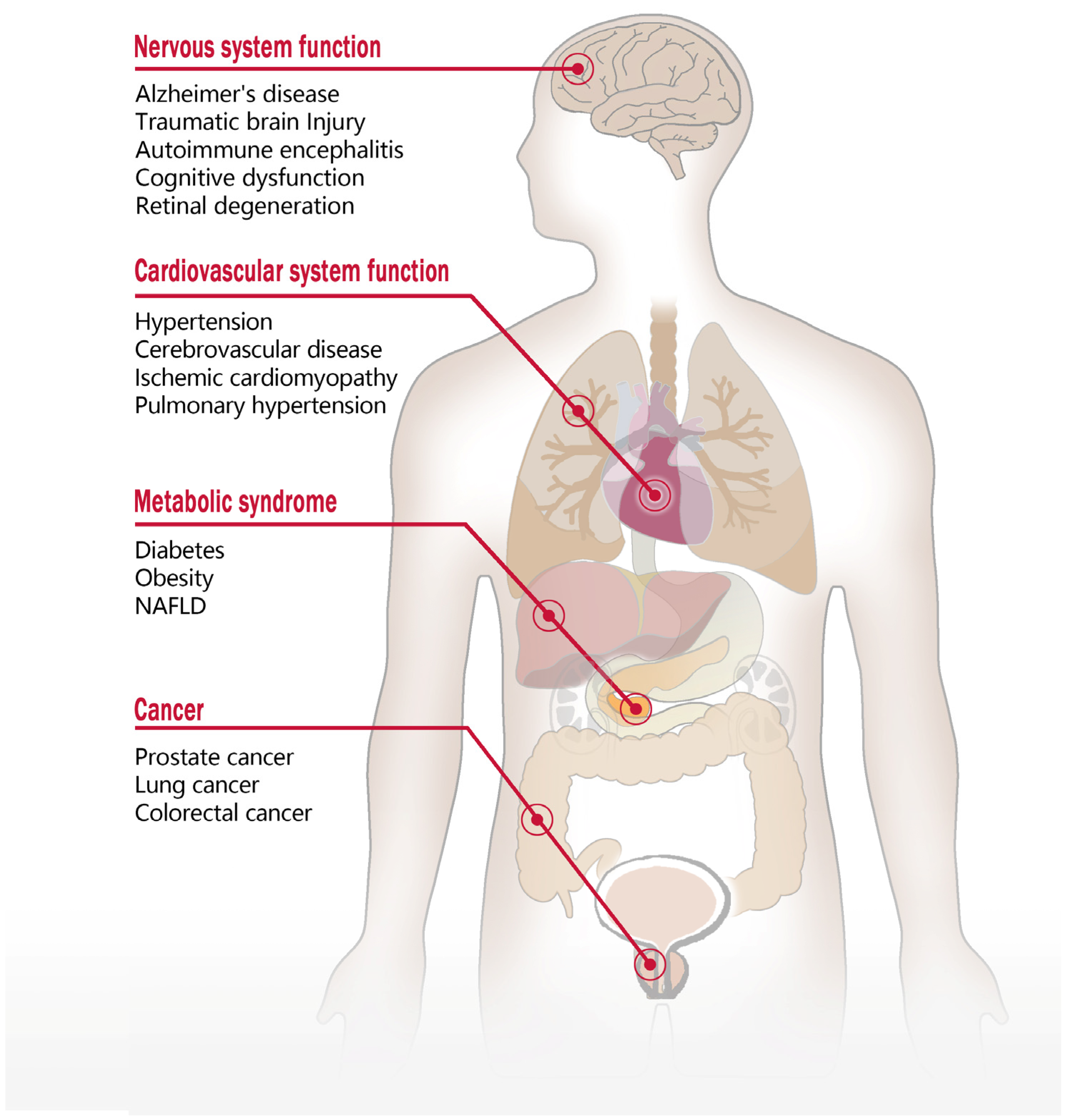
4. Function of GPR75 and Involvement in Diseases
4.1. The Role of GPR75 in the Nervous System
4.2. The Impact of GPR75 on Cardiovascular System Function
4.3. The Role of GPR75 in Metabolic Disorders
4.4. The Role of GPR75 in Tumor Development and Progression
5. Current Issues and Perspectives
Author Contributions
Funding
Conflicts of Interest
References
- Venkatakrishnan, A.J.; Deupi, X.; Lebon, G.; Tate, C.G.; Schertler, G.F.; Babu, M.M. Molecular Signatures of G-Protein-Coupled Receptors. Nature 2013, 494, 185–194. [Google Scholar] [CrossRef] [PubMed]
- Schöneberg, T.; Liebscher, I. Mutations in G Protein-Coupled Receptors: Mechanisms, Pathophysiology and Potential Therapeutic Approaches. Pharmacol. Rev. 2021, 73, 89–119. [Google Scholar] [CrossRef] [PubMed]
- Yang, D.; Zhou, Q.; Labroska, V.; Qin, S.; Darbalaei, S.; Wu, Y.; Yuliantie, E.; Xie, L.; Tao, H.; Cheng, J.; et al. G Protein-Coupled Receptors: Structure- and Function-Based Drug Discovery. Signal Transduct. Target. Ther. 2021, 6, 7. [Google Scholar] [CrossRef]
- Alexander, S.P.H.; Christopoulos, A.; Davenport, A.P.; Kelly, E.; Mathie, A.A.; Peters, J.A.; Veale, E.L.; Armstrong, J.F.; Faccenda, E.; Harding, S.D.; et al. The Concise Guide to PHARMACOLOGY 2023/24: G Protein-Coupled Receptors. Br. J. Pharmacol. 2023, 180 (Suppl. S2), S23–S144. [Google Scholar] [CrossRef]
- Ignatov, A.; Robert, J.; Gregory-Evans, C.; Schaller, H.C. RANTES Stimulates Ca2+ Mobilization and Inositol Trisphosphate (IP3) Formation in Cells Transfected with G Protein-Coupled Receptor 75: GPR75 Is a New Receptor for the Chemokine RANTES. Br. J. Pharmacol. 2006, 149, 490–497. [Google Scholar] [CrossRef]
- Garcia, V.; Gilani, A.; Shkolnik, B.; Pandey, V.; Zhang, F.F.; Dakarapu, R.; Gandham, S.K.; Reddy, N.R.; Graves, J.P.; Gruzdev, A.; et al. 20-HETE Signals Through G-Protein-Coupled Receptor GPR75 (Gq) to Affect Vascular Function and Trigger Hypertension. Circ. Res. 2017, 120, 1776–1788. [Google Scholar] [CrossRef]
- Bonafini, S.; Fava, C. Omega-3 Fatty Acids and Cytochrome P450-Derived Eicosanoids in Cardiovascular Diseases: Which Actions and Interactions Modulate Hemodynamics? Prostaglandins Other Lipid Mediat. 2017, 128, 34–42. [Google Scholar] [CrossRef]
- Tarttelin, E.E.; Kirschner, L.S.; Bellingham, J.; Baffi, J.; Taymans, S.E.; Gregory-Evans, K.; Csaky, K.; Stratakis, C.A.; Gregory-Evans, C.Y. Cloning and Characterization of a Novel Orphan G-Protein-Coupled Receptor Localized to Human Chromosome 2p16. Biochem. Biophys. Res. Commun. 1999, 260, 174–180. [Google Scholar] [CrossRef]
- Kakarala, K.K.; Jamil, K. Sequence-Structure Based Phylogeny of GPCR Class A Rhodopsin Receptors. Mol. Phylogenet. Evol. 2014, 74, 66–96. [Google Scholar] [CrossRef]
- Sauer, C.G.; White, K.; Stöhr, H.; Grimm, T.; Hutchinson, A.; Bernstein, P.S.; Lewis, R.A.; Simonelli, F.; Pauleikhoff, D.; Allikmets, R.; et al. Evaluation of the G Protein Coupled Receptor-75 (GPR75) in Age Related Macular Degeneration. Br. J. Ophthalmol. 2001, 85, 969–975. [Google Scholar] [CrossRef]
- Voogdt, C.G.P.; Merchant, M.E.; Wagenaar, J.A.; Van Putten, J.P.M. Evolutionary Regression and Species-Specific Codon Usage of TLR15. Front. Immunol. 2018, 9, 2626. [Google Scholar] [CrossRef] [PubMed]
- Dedoni, S.; Campbell, L.A.; Harvey, B.K.; Avdoshina, V.; Mocchetti, I. The Orphan G-protein-coupled Receptor 75 Signaling Is Activated by the Chemokine CCL 5. J. Neurochem. 2018, 146, 526–539. [Google Scholar] [CrossRef] [PubMed]
- Isaikina, P.; Tsai, C.-J.; Petrovic, I.; Rogowski, M.; Dürr, A.M.; Grzesiek, S. Preparation of a Stable CCL5·CCR5·Gi Signaling Complex for Cryo-EM Analysis. Methods Cell Biol. 2022, 169, 115–141. [Google Scholar] [CrossRef]
- Lah Turnšek, T.; Jiao, X.; Novak, M.; Jammula, S.; Cicero, G.; Ashton, A.W.; Joyce, D.; Pestell, R.G. An Update on Glioblastoma Biology, Genetics, and Current Therapies: Novel Inhibitors of the G Protein-Coupled Receptor CCR5. Int. J. Mol. Sci. 2021, 22, 4464. [Google Scholar] [CrossRef]
- Zeng, Z.; Lan, T.; Wei, Y.; Wei, X. CCL5/CCR5 Axis in Human Diseases and Related Treatments. Genes Dis. 2022, 9, 12–27. [Google Scholar] [CrossRef]
- Decrock, E.; De Bock, M.; Wang, N.; Gadicherla, A.K.; Bol, M.; Delvaeye, T.; Vandenabeele, P.; Vinken, M.; Bultynck, G.; Krysko, D.V.; et al. IP3, a Small Molecule with a Powerful Message. Biochim. Biophys. Acta 2013, 1833, 1772–1786. [Google Scholar] [CrossRef]
- Liu, B.; Hassan, Z.; Amisten, S.; King, A.J.; Bowe, J.E.; Huang, G.C.; Jones, P.M.; Persaud, S.J. The Novel Chemokine Receptor, G-Protein-Coupled Receptor 75, Is Expressed by Islets and Is Coupled to Stimulation of Insulin Secretion and Improved Glucose Homeostasis. Diabetologia 2013, 56, 2467–2476. [Google Scholar] [CrossRef]
- Gençoğlu, H.; Şahin, K.; Jones, P.M. Determining the Insulin Secretion Potential for Certain Specific G-Protein Coupled Receptors in MIN6 Pancreatic Beta Cells. Turk. J. Med. Sci. 2019, 49, 403–411. [Google Scholar] [CrossRef]
- D’Addario, C.A.; Matsumura, S.; Kitagawa, A.; Lainer, G.M.; Zhang, F.; D’silva, M.; Khan, M.Y.; Froogh, G.; Gruzdev, A.; Zeldin, D.C.; et al. Global and Endothelial G-Protein Coupled Receptor 75 (GPR75) Knockout Relaxes Pulmonary Artery and Mitigates Hypoxia-Induced Pulmonary Hypertension. Vasc. Pharmacol. 2023, 153, 107235. [Google Scholar] [CrossRef]
- Rajagopal, S.; Kim, J.; Ahn, S.; Craig, S.; Lam, C.M.; Gerard, N.P.; Gerard, C.; Lefkowitz, R.J. Beta-Arrestin- but Not G Protein-Mediated Signaling by the “Decoy” Receptor CXCR7. Proc. Natl. Acad. Sci. USA 2010, 107, 628–632. [Google Scholar] [CrossRef]
- Duan, J.; Liu, H.; Zhao, F.; Yuan, Q.; Ji, Y.; Cai, X.; He, X.; Li, X.; Li, J.; Wu, K.; et al. GPCR Activation and GRK2 Assembly by a Biased Intracellular Agonist. Nature 2023, 620, 676–681. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Patterson, B.; Zhu, L. Biased Signaling in GPCRs: Structural Insights and Implications for Drug Development. Pharmacol. Ther. 2025, 266, 108786. [Google Scholar] [CrossRef] [PubMed]
- Bernhard, S.M.; Han, J.; Che, T. GPCR-G Protein Selectivity Revealed by Structural Pharmacology. FEBS J. 2024, 291, 2784–2791. [Google Scholar] [CrossRef]
- Ham, D.; Shihoya, W.; Nureki, O.; Inoue, A.; Chung, K.Y. Molecular Mechanism of the Endothelin Receptor Type B Interactions with Gs, Gi, and Gq. Structure 2024, 32, 1632–1639.e4. [Google Scholar] [CrossRef]
- Southern, C.; Cook, J.M.; Neetoo-Isseljee, Z.; Taylor, D.L.; Kettleborough, C.A.; Merritt, A.; Bassoni, D.L.; Raab, W.J.; Quinn, E.; Wehrman, T.S.; et al. Screening β-Arrestin Recruitment for the Identification of Natural Ligands for Orphan G-Protein–Coupled Receptors. J. Biomol. Screen. 2013, 18, 599–609. [Google Scholar] [CrossRef]
- Pascale, J.V.; Wolf, A.; Kadish, Y.; Diegisser, D.; Kulaprathazhe, M.-M.; Yemane, D.; Ali, S.; Kim, N.; Baruch, D.E.; Yahaya, M.A.F.; et al. 20-Hydroxyeicosatetraenoic Acid (20-HETE): Bioactions, Receptors, Vascular Function, Cardiometabolic Disease and Beyond. Adv. Pharmacol. 2023, 97, 229–255. [Google Scholar] [CrossRef]
- Nithipatikom, K.; DiCamelli, R.F.; Kohler, S.; Gumina, R.J.; Falck, J.R.; Campbell, W.B.; Gross, G.J. Determination of Cytochrome P450 Metabolites of Arachidonic Acid in Coronary Venous Plasma during Ischemia and Reperfusion in Dogs. Anal. Biochem. 2001, 292, 115–124. [Google Scholar] [CrossRef]
- Nithipatikom, K.; Gross, E.R.; Endsley, M.P.; Moore, J.M.; Isbell, M.A.; Falck, J.R.; Campbell, W.B.; Gross, G.J. Inhibition of Cytochrome P450ω-Hydroxylase: A Novel Endogenous Cardioprotective Pathway. Circ. Res. 2004, 95, e65–e71. [Google Scholar] [CrossRef]
- Han, Y.; Zhao, H.; Tang, H.; Li, X.; Tan, J.; Zeng, Q.; Sun, C. 20-Hydroxyeicosatetraenoic Acid Mediates Isolated Heart Ischemia/Reperfusion Injury by Increasing NADPH Oxidase-Derived Reactive Oxygen Species Production. Circ. J. 2013, 77, 1807–1816. [Google Scholar] [CrossRef]
- Bao, Y.; Wang, X.; Li, W.; Huo, D.; Shen, X.; Han, Y.; Tan, J.; Zeng, Q.; Sun, C. 20-Hydroxyeicosatetraenoic Acid Induces Apoptosis in Neonatal Rat Cardiomyocytes through Mitochondrial-Dependent Pathways. J. Cardiovasc. Pharm. 2011, 57, 294–301. [Google Scholar] [CrossRef]
- Roman, R.J. 20-HETE and Hypertension. Hypertension 2024, 81, 2012–2015. [Google Scholar] [CrossRef] [PubMed]
- Garcia, V.; Cheng, J.; Weidenhammer, A.; Ding, Y.; Wu, C.; Zhang, F.; Gotlinger, K.; Falck, J.; Schwartzman, M. Androgen-Induced Hypertension in Angiotensinogen Deficient Mice: Role of 20-HETE and EETS. Prostaglandins Other Lipid Mediat. 2015, 116, 124–130. [Google Scholar] [CrossRef] [PubMed]
- Yu, M.; Cambj-Sapunar, L.; Kehl, F.; Maier, K.G.; Takeuchi, K.; Miyata, N.; Ishimoto, T.; Reddy, L.M.; Falck, J.R.; Gebremedhin, D.; et al. Effects of a 20-HETE Antagonist and Agonists on Cerebral Vascular Tone. Eur. J. Pharmacol. 2004, 486, 297–306. [Google Scholar] [CrossRef]
- Chen, L.; Ackerman, R.; Guo, A.M. 20-HETE in Neovascularization. Prostaglandins Other Lipid Mediat. 2012, 98, 63–68. [Google Scholar] [CrossRef]
- Palanisamy, S.; Xue, C.; Ishiyama, S.; Naga Prasad, S.V.; Gabrielson, K. GPCR-ErbB Transactivation Pathways and Clinical Implications. Cell. Signal. 2021, 86, 110092. [Google Scholar] [CrossRef]
- Han, J.; Li, J.; Liu, L.; Li, K.; Zhang, C.; Han, Y. 20-HETE Mediates Ang II-Induced Cardiac Hypertrophy via ROS and Ca2+ Signaling in H9c2 Cells. Sci. Rep. 2025, 15, 2342. [Google Scholar] [CrossRef]
- Cárdenas, S.; Colombero, C.; Panelo, L.; Dakarapu, R.; Falck, J.R.; Costas, M.A.; Nowicki, S. GPR75 Receptor Mediates 20-HETE-Signaling and Metastatic Features of Androgen-Insensitive Prostate Cancer Cells. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2020, 1865, 158573. [Google Scholar] [CrossRef]
- Gilani, A.; Agostinucci, K.; Hossain, S.; Pascale, J.V.; Garcia, V.; Adebesin, A.M.; Falck, J.R.; Schwartzman, M.L. 20-HETE Interferes with Insulin Signaling and Contributes to Obesity-Driven Insulin Resistance. Prostaglandins Other Lipid Mediat. 2021, 152, 106485. [Google Scholar] [CrossRef]
- Chen, X.; Liu, Y.; Wang, Y.; Wang, C.; Chen, X.; Xiong, Y.; Liu, L.; Yuan, X.; Tang, H.; Shu, C.; et al. CYP4F2-Catalyzed Metabolism of Arachidonic Acid Promotes Stromal Cell-Mediated Immunosuppression in Non–Small Cell Lung Cancer. Cancer Res. 2022, 82, 4016–4030. [Google Scholar] [CrossRef]
- Pascale, J.V.; Wolf, A.; Kulaprathazhe, M.-M.; Ali, S.; Kim, N.; Froogh, G.; Adebesin, A.M.; Falck, J.R.; Schwartzman, M.L.; Garcia, V. Structure-Function Relationship of the 20-HETE-GPR75 Pairing: Development and Characterization of Agonist, Partial Agonists, and Receptor Blockers. FASEB J. 2022, 36. [Google Scholar] [CrossRef]
- Pascale, J.V.; Park, E.J.; Adebesin, A.M.; Falck, J.R.; Schwartzman, M.L.; Garcia, V. Uncovering the Signalling, Structure and Function of the 20-HETE-GPR75 Pairing: Identifying the Chemokine CCL5 as a Negative Regulator of GPR75. Br. J. Pharmacol. 2021, 178, 3813–3828. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Xun, Y.; Zhang, Z. Central Regulation of Feeding and Body Weight by Ciliary GPR75. J. Clin. Investig. 2024, 134, e182121. [Google Scholar] [CrossRef] [PubMed]
- Agostinucci, K.; Hutcheson, R.; Hossain, S.; Pascale, J.V.; Villegas, E.; Zhang, F.; Adebesin, A.M.; Falck, J.R.; Gupte, S.; Garcia, V.; et al. Blockade of 20-Hydroxyeicosatetraenoic Acid Receptor Lowers Blood Pressure and Alters Vascular Function in Mice with Smooth Muscle-Specific Overexpression of CYP4A12-20-HETE Synthase. J. Hypertens. 2022, 40, 498. [Google Scholar] [CrossRef] [PubMed]
- Tunctan, B.; Senol, S.P.; Temiz-Resitoglu, M.; Yilmaz, D.E.; Guden, D.S.; Bahceli, O.; Horat, M.F.; Sahan-Firat, S.; Sari, A.N.; Falck, J.R.; et al. Activation of GPR75 Signaling Pathway Contributes to the Effect of a 20-HETE Mimetic, 5,14-HEDGE, to Prevent Hypotensive and Tachycardic Responses to Lipopolysaccharide in a Rat Model of Septic Shock. J. Cardiovasc. Pharm. 2022, 80, 276–293. [Google Scholar] [CrossRef]
- Liu, J.; Ji, Y.; Mao, L.; Guo, L.; Han, Y. Role of G-Protein-Coupled Receptor 75 Signaling Pathway in Apoptosis of Neonatal Rat Cardiomyocytes Induced by 20-Hydroxyeicosatetraenoic Acid. Chin. J. Pathophysiol. 2022, 38, 65–73. [Google Scholar] [CrossRef]
- Mao, L.; Lu, G.; Tang, Q.; Li, X.; Han, Y.; Chen, Y. Knockdown of G-protein-coupled receptor 75 expression inhibits 20-HETE-induced apoptosis in H9c2 cardiomyocytes. Chin. J. Pathophysiol. 2020, 36, 421–426. [Google Scholar] [CrossRef]
- Gonzalez-Fernandez, E.; Staursky, D.; Lucas, K.; Nguyen, B.V.; Li, M.; Liu, Y.; Washington, C.; Coolen, L.M.; Fan, F.; Roman, R.J. 20-HETE Enzymes and Receptors in the Neurovascular Unit: Implications in Cerebrovascular Disease. Front. Neurol. 2020, 11, 983. [Google Scholar] [CrossRef]
- Weijiang, M.; Aihua, L.; Xinya, W.; Li, G.; Jingjing, C.; Hanxin, W.; Meixiao, L.; Yuxin, F.; Li, P.; Jiaru, Y.; et al. The Intricate Role of CCL5/CCR5 Axis in Alzheimer Disease. J. Neuropathol. Exp. Neurol. 2023, 82, 894–900. [Google Scholar] [CrossRef]
- Vacinova, G.; Vejražkova, D.; Rusina, R.; Holmerová, I.; Vaňková, H.; Jarolímová, E.; Včelák, J.; Bendlová, B.; Vaňková, M. Regulated upon Activation, Normal T Cell Expressed and Secreted (RANTES) Levels in the Peripheral Blood of Patients with Alzheimer’s Disease. Neural Regen. Res. 2021, 16, 796–800. [Google Scholar] [CrossRef]
- Wee, J.J.; Kumar, S. Prediction of Hub Genes of Alzheimer’s Disease Using a Protein Interaction Network and Functional Enrichment Analysis. Genom. Inform. 2020, 18, e39. [Google Scholar] [CrossRef]
- Jorda, A.; Cauli, O.; Santonja, J.M.; Aldasoro, M.; Aldasoro, C.; Obrador, E.; Vila, J.M.; Mauricio, M.D.; Iradi, A.; Guerra-Ojeda, S.; et al. Changes in Chemokines and Chemokine Receptors Expression in a Mouse Model of Alzheimer’s Disease. Int. J. Biol. Sci. 2019, 15, 453–463. [Google Scholar] [CrossRef] [PubMed]
- Kester, M.I.; van der Flier, W.M.; Visser, A.; Blankenstein, M.A.; Scheltens, P.; Oudejans, C.B. Decreased mRNA Expression of CCL5 [RANTES] in Alzheimer’s Disease Blood Samples. Clin. Chem. Lab. Med. 2011, 50, 61–65. [Google Scholar] [CrossRef] [PubMed]
- Tripathy, D.; Thirumangalakudi, L.; Grammas, P. RANTES Upregulation in the Alzheimer’s Disease Brain: A Possible Neuroprotective Role. Neurobiol. Aging 2010, 31, 8–16. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Q.; Wang, X. Alzheimer’s Disease: Insights into Pathology, Molecular Mechanisms, and Therapy. Protein Cell 2024, 16, 83–120. [Google Scholar] [CrossRef]
- Gonzalez-Fernandez, E.; Liu, Y.; Auchus, A.P.; Fan, F.; Roman, R.J. Vascular Contributions to Cognitive Impairment and Dementia: The Emerging Role of 20-HETE. Clin. Sci. 2021, 135, 1929–1944. [Google Scholar] [CrossRef]
- Kaplan, L.; Chow, B.W.; Gu, C. Neuronal Regulation of the Blood-Brain Barrier and Neurovascular Coupling. Nat. Rev. Neurosci. 2020, 21, 416–432. [Google Scholar] [CrossRef]
- Yi, X.; Han, Z.; Zhou, Q.; Lin, J.; Liu, P. 20-Hydroxyeicosatetraenoic Acid as a Predictor of Neurological Deterioration in Acute Minor Ischemic Stroke. Stroke 2016, 47, 3045–3047. [Google Scholar] [CrossRef]
- Li, Z.; McConnell, H.L.; Stackhouse, T.L.; Pike, M.M.; Zhang, W.; Mishra, A. Increased 20-HETE Signaling Suppresses Capillary Neurovascular Coupling After Ischemic Stroke in Regions Beyond the Infarct. Front. Cell. Neurosci. 2021, 15, 762843. [Google Scholar] [CrossRef]
- Ma, Z.; Ning, Y.; Chen, X.; Zhao, S.; Yan, J.; Wang, B.; Li, C.; Gao, R.; Chen, X.; Yang, N.; et al. 20-Hydroxyeicosatetraenoic Acid Regulates the Src/EGFR/NF-κB Signaling Pathway Via GPR75 to Activate Microglia and Promote TBI in the Immature Brain. Neurochem. Res. 2024, 50, 7. [Google Scholar] [CrossRef]
- Jamieson, K.; Endo, T.; Darwesh, A.; Samokhvalov, V.; Seubert, J. Cytochrome P450-Derived Eicosanoids and Heart Function. Pharmacol. Ther. 2017, 179, 47–83. [Google Scholar] [CrossRef]
- Imig, J.D. Epoxyeicosatrienoic Acids and 20-Hydroxyeicosatetraenoic Acid on Endothelial and Vascular Function. Adv. Pharmacol. 2016, 77, 105–141. [Google Scholar] [CrossRef] [PubMed]
- Horat, M.F.; Senol, S.P.; Bahceli, O.; Temiz-Resitoglu, M.; Sahan-Firat, S.; Sevim, S.; Tunctan, B. Pro-Inflammatory GPR75 and Anti-Apoptotic Phospholipase Signaling Pathways Contribute to the Ameliorating Effect of Soluble Epoxide Hydrolase Inhibition on Chronic Experimental Autoimmune Encephalomyelitis in Mice. Cell Mol. Biol. 2023, 69, 9–16. [Google Scholar] [CrossRef] [PubMed]
- Speidell, A.; Walton, S.; Campbell, L.A.; Tomassoni-Ardori, F.; Tessarollo, L.; Corbo, C.; Taraballi, F.; Mocchetti, I. Mice Deficient for G-Protein-Coupled Receptor 75 Display Altered Presynaptic Structural Protein Expression and Disrupted Fear Conditioning Recall. J. Neurochem. 2023, 165, 827–841. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.; Liu, T.; Li, J.; Yang, J.; Du, Q.; Wang, J.; Yang, Y.; Liu, X.; Fan, Y.; Lu, F.; et al. A Genome-Wide Scan Maps a Novel Autosomal Dominant Juvenile- Onset Open-Angle Glaucoma Locus to 2p15-16. Mol. Vis. 2008, 14, 739–744. [Google Scholar]
- Vasudevan, S.; Samuels, I.S.; Park, P.S.-H. Gpr75 Knockout Mice Display Age-Dependent Cone Photoreceptor Cell Loss. J. Neurochem. 2023, 167, 538–555. [Google Scholar] [CrossRef]
- Sedláková, L.; Kikerlová, S.; Husková, Z.; Červenková, L.; Chábová, V.Č.; Zicha, J.; Falck, J.R.; Imig, J.D.; Kompanowska-Jezierska, E.; Sadowski, J.; et al. 20-Hydroxyeicosatetraenoic Acid Antagonist Attenuates the Development of Malignant Hypertension and Reverses It Once Established: A Study in Cyp1a1-Ren-2 Transgenic Rats. Biosci. Rep. 2018, 38, BSR20171496. [Google Scholar] [CrossRef]
- Gawrys, O.; Husková, Z.; Baranowska, I.; Walkowska, A.; Sadowski, J.; Kikerlová, S.; Vaňourková, Z.; Honetschlägerová, Z.; Škaroupková, P.; Červenka, L.; et al. Combined Treatment with Epoxyeicosatrienoic Acid Analog and 20-Hydroxyeicosatetraenoic Acid Antagonist Provides Substantial Hypotensive Effect in Spontaneously Hypertensive Rats. J. Hypertens. 2020, 38, 1802–1810. [Google Scholar] [CrossRef]
- Baranowska, I.; Gawrys, O.; Walkowska, A.; Olszynski, K.H.; Červenka, L.; Falck, J.R.; Adebesin, A.M.; Imig, J.D.; Kompanowska-Jezierska, E. Epoxyeicosatrienoic Acid Analog and 20-HETE Antagonist Combination Prevent Hypertension Development in Spontaneously Hypertensive Rats. Front. Pharmacol. 2021, 12, 798642. [Google Scholar] [CrossRef]
- Pais, R.; Zietek, T.; Hauner, H.; Daniel, H.; Skurk, T. RANTES (CCL5) Reduces Glucose-Dependent Secretion of Glucagon-like Peptides 1 and 2 and Impairs Glucose-Induced Insulin Secretion in Mice. Am. J. Physiol. Gastrointest. Liver Physiol. 2014, 307, G330–G337. [Google Scholar] [CrossRef]
- Lopes, L.R.; Ribeiro, S.M.L.T.; Figueiredo, V.P.; Leite, A.L.J.; Nicolato, R.L.d.C.; Gomes, J.A.E.; de Oliveira, F.L.P.; Talvani, A. The Overweight Increases Circulating Inflammatory Mediators Commonly Associated with Obesity in Young Individuals. Cytokine 2018, 110, 169–173. [Google Scholar] [CrossRef]
- van der Weerd, K.; Dik, W.A.; Schrijver, B.; Schweitzer, D.H.; Langerak, A.W.; Drexhage, H.A.; Kiewiet, R.M.; van Aken, M.O.; van Huisstede, A.; van Dongen, J.J.M.; et al. Morbidly Obese Human Subjects Have Increased Peripheral Blood CD4+ T Cells with Skewing toward a Treg- and Th2-Dominated Phenotype. Diabetes 2012, 61, 401–408. [Google Scholar] [CrossRef] [PubMed]
- Gilani, A.; Pandey, V.; Garcia, V.; Agostinucci, K.; Singh, S.P.; Schragenheim, J.; Bellner, L.; Falck, J.R.; Paudyal, M.P.; Capdevila, J.H.; et al. High-Fat Diet-Induced Obesity and Insulin Resistance in CYP4a14-/- Mice Is Mediated by 20-HETE. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2018, 315, R934–R944. [Google Scholar] [CrossRef] [PubMed]
- Hardwick, J.P.; Song, B.-J.; Rote, P.; Leahy, C.; Lee, Y.K.; Wolf, A.R.; Diegisser, D.; Garcia, V. The CYP4/20-HETE/GPR75 Axis in the Progression Metabolic Dysfunction-Associated Steatosis Liver Disease (MASLD) to Chronic Liver Disease. Front. Physiol. 2024, 15, 1497297. [Google Scholar] [CrossRef]
- Akbari, P.; Gilani, A.; Sosina, O.; Kosmicki, J.A.; Khrimian, L.; Fang, Y.-Y.; Persaud, T.; Garcia, V.; Sun, D.; Li, A.; et al. Sequencing of 640,000 Exomes Identifies GPR75 Variants Associated with Protection from Obesity. Science 2021, 373, eabf8683. [Google Scholar] [CrossRef]
- Yeo, G.S.H.; O’Rahilly, S. Finding Genes That Control Body Weight. Science 2021, 373, 30–31. [Google Scholar] [CrossRef]
- Powell, D.R.; Doree, D.D.; DaCosta, C.M.; Platt, K.A.; Brommage, R.; Buhring, L.; Revelli, J.-P.; Shadoan, M.K. Mice Lacking Gpr75 Are Hypophagic and Thin. Diabetes Metab. Syndr. Obes. 2022, 15, 45–58. [Google Scholar] [CrossRef]
- Hossain, S.; Gilani, A.; Pascale, J.; Villegas, E.; Diegisser, D.; Agostinucci, K.; Kulaprathazhe, M.-M.; Dirice, E.; Garcia, V.; Schwartzman, M.L. Gpr75-Deficient Mice Are Protected from High-Fat Diet-Induced Obesity. Obesity 2023, 31, 1024–1037. [Google Scholar] [CrossRef]
- Zhong, Z.; Fan, J.; Tian, Y.; Lin, M.; Zhu, H.; Ma, D. Whole-Genome Resequencing and RNA-Seq Analysis Implicates GPR75 as a Potential Genetic Basis Related to Retarded Growth in South China Carp (Cyprinus carpio rubrofuscus). Genomics 2024, 116, 110934. [Google Scholar] [CrossRef]
- Brewer, K.M.; Brewer, K.K.; Richardson, N.C.; Berbari, N.F. Neuronal Cilia in Energy Homeostasis. Front. Cell Dev. Biol. 2022, 10, 1082141. [Google Scholar] [CrossRef]
- Leeson-Payne, A.; Iyinikkel, J.; Malcolm, C.; Lam, B.Y.H.; Sommer, N.; Dowsett, G.K.C.; Martinez de Morentin, P.B.; Thompson, D.; Mackenzie, A.; Chianese, R.; et al. Loss of GPR75 Protects against Non-Alcoholic Fatty Liver Disease and Body Fat Accumulation. Cell Metab. 2024, 36, 1076–1087.e4. [Google Scholar] [CrossRef]
- Wang, B.; Wu, L.; Chen, J.; Dong, L.; Chen, C.; Wen, Z.; Hu, J.; Fleming, I.; Wang, D.W. Metabolism Pathways of Arachidonic Acids: Mechanisms and Potential Therapeutic Targets. Signal Transduct. Target. Ther. 2021, 6, 94. [Google Scholar] [CrossRef] [PubMed]
- Cárdenas, S.; Colombero, C.; Cruz, M.; Mormandi, E.; Adebesin, A.M.; Falck, J.R.; Nowicki, S. 20-HETE/GPR75 Pairing Modulates the Expression and Transcriptional Activity of the Androgen Receptor in Androgen-Sensitive Prostate Cancer Cells. Mol. Cell Endocrinol. 2023, 559, 111784. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Gu, J.; Xu, F.; Zhu, Q.; Ge, D.; Lu, C. Novel Methylation-Driven Genes Identified as Prognostic Indicators for Lung Squamous Cell Carcinoma. Am. J. Transl. Res. 2019, 11, 1997–2012. [Google Scholar]
- Ghorbanzadeh, F.; Jafari-Gharabaghlou, D.; Dashti, M.R.; Hashemi, M.; Zarghami, N. Advanced Nano-Therapeutic Delivery of Metformin: Potential Anti-Cancer Effect against Human Colon Cancer Cells through Inhibition of GPR75 Expression. Med. Oncol. 2023, 40, 255. [Google Scholar] [CrossRef]
- Trauelsen, M.; Lückmann, M.; Frimurer, T.M.; Schwartz, T.W. The HETE Is on FFAR1 and Pancreatic Islet Cells. Cell Metab. 2018, 27, 273–275. [Google Scholar] [CrossRef]
- Yu, J.; Waresi, M.; Zhong, H.; Wu, H.; Ge, J. 20-HETE Induced Platelet Activation via a GPR75-Independent Pathway. Thromb. Res. 2025, 247, 109277. [Google Scholar] [CrossRef]
- Zhang, C.; Booz, G.W.; Yu, Q.; He, X.; Wang, S.; Fan, F. Conflicting Roles of 20-HETE in Hypertension and Renal End Organ Damage. Eur. J. Pharmacol. 2018, 833, 190–200. [Google Scholar] [CrossRef]
- Liu, J.; Tang, H.; Xu, C.; Zhou, S.; Zhu, X.; Li, Y.; Prézeau, L.; Xu, T.; Pin, J.-P.; Rondard, P.; et al. Biased Signaling Due to Oligomerization of the G Protein-Coupled Platelet-Activating Factor Receptor. Nat. Commun. 2022, 13, 6365. [Google Scholar] [CrossRef]
- Li, W.; Li, T.; Ali, T.; Mou, S.; Gong, Q.; Yu, Z.-J.; Li, S. Uncoupling Serotonin (2C) and Dopamine (D2) Receptor Heterodimers Ameliorate PTSD-like Behaviors. J. Affect. Disord. 2025, 380, 63–77. [Google Scholar] [CrossRef]
- Varney, M.J.; Benovic, J.L. The Role of G Protein-Coupled Receptors and Receptor Kinases in Pancreatic β-Cell Function and Diabetes. Pharmacol. Rev. 2024, 76, 267–299. [Google Scholar] [CrossRef]
- Zhang, X.; Yu, W.; Li, Y.; Wang, A.; Cao, H.; Fu, Y. Drug Development Advances in Human Genetics-based Targets. MedComm 2024, 5, e481. [Google Scholar] [CrossRef]
- Lv, Z.; He, Y.; Xiang, Y.; Li, J.; Zhang, S.; Meng, F.; Lan, B.; Guo, H.; He, D.; Wang, Y.; et al. Cryo-EM Complex Structure of Active GPR75 with a Nanobody. bioRxiv 2022. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
| Year | Reference | Putative Ligands | Related Disease | Major Findings |
|---|---|---|---|---|
| 1999 | Tarttelin [8] | - | - | The GPR75 gene was first identified in the human genome. |
| 2001 | Sauer [10] | - | - | GPR75, primarily expressed in the CNS and retina, shows no link to age-related macular degeneration. |
| 2006 | Ignatov [5] | CCL5 | Alzheimer’s disease | CCL5, an endogenous ligand for GPR75, activates the MAPK/AKT pathway to inhibit Aβ-induced neuronal apoptosis. |
| 2008 | Lin [64] | - | Glaucoma | The 2p16 region containing GPR75 is linked to POAG, though GPR75 may not be a direct cause. |
| 2013 | Liu [17] | CCL5 | Diabetes | GPR75 is expressed in pancreatic β cells, where CCL5 enhances insulin secretion and glucose homeostasis by this receptor. |
| 2013 | Southern [25] | - | - | CCL5/GPR75 pairing was not confirmed by β-arrestin recruitment assays. |
| 2014 | Kakarala [9] | - | - | GPR75 shares transmembrane domain homology with several receptors across species, including neuropeptide Y and galanin receptors. |
| 2017 | Garcia [6] | 20-HETE | Hypertension | Gpr75 is expressed in the cardiovascular system, where 20-HETE activates it to induce endothelial dysfunction and VSMC contraction, contributing to hypertension. |
| 2018 | Dedoni [12] | CCL5 | - | CCL5 activates GPR75 via internalization, while Gpr75 knockdown blocks CCL5-induced pERK activation in SH-SY5Y cells. |
| 2018 | Sedláková [66] | 20-HETE | Hypertension | AAA, a 20-HETE receptor antagonist, reverses malignant hypertension by blocking GPR75 in transgenic rats. |
| 2018 | Voogdt [11] | - | - | In chickens, Gpr75 is adjacent to Tlr15, suggesting similar biological functions. |
| 2019 | Gençoğlu [18] | CCL5 | Diabetes | CCL5 promotes insulin secretion in MIN6 cells through GPR75. |
| 2019 | Li [83] | - | Lung cancer | In lung SCC, GPR75 hypermethylation serves as a prognostic marker. |
| 2020 | Cárdenas [37] | 20-HETE | Prostate cancer | 20-HETE activates EGFR/AKT signaling via GPR75, enhancing prostate cancer metastasis. |
| 2020 | Gonzalez-Fernandez [47] | 20-HETE | Cerebrovascular disease | Cyp4a and Gpr75 are co-expressed in the brain, where 20-HETE regulates VSMCs and pericytes via GPR75. |
| 2020 | Mao [46] | 20-HETE | Ischemic cardiomyopathy | In H9c2 cardiomyocytes, GPR75 mediates 20-HETE-induced apoptosis through mitochondrial damage. |
| 2020 | Gawrys [67] | 20-HETE | Hypertension | AAA and EET-A improve endothelial function and lower blood pressure in hypertensive rats. |
| 2021 | Gilani [38] | 20-HETE | Diabetes | In 3T3-L1 adipocytes, 20-HETE activates GPR75 to inhibit insulin receptor phosphorylation, leading to insulin resistance. |
| 2021 | Pascale [41] | 20-HETE | - | 20-HETE is a high-affinity ligand, while CCL5 is a low-affinity negative regulator of GPR75. |
| 2021 | Akbari [74] | - | Obesity | GPR75 truncating variants are linked to BMI, and its knockout in mice prevents weight gain and improves glycemic control on a high-fat diet. |
| 2022 | Agostinucci [43] | 20-HETE | Hypertension | AAA reduces 20-HETE-dependent hypertension in Cyp4a12 transgenic mice. |
| 2022 | Tunctan [44] | 20-HETE | Hypotension and tachycardia | The 20-HETE mimetic 5,14-HEDGE protects against LPS-induced hypotension via GPR75, reversed by AAA. |
| 2022 | Chen [39] | 20-HETE | Lung cancer | 20-HETE upregulates GPR75 and activates fibroblasts, promoting immune evasion and invasiveness in lung cancer. |
| 2022 | Powell [76] | - | Obesity | Gpr75 knockout mice show reduced weight and improved glucose tolerance and insulin sensitivity. |
| 2022 | Liu [45] | 20-HETE | Ischemic cardiomyopathy | GPR75 mediates 20-HETE-induced cardiomyocyte apoptosis via Ca2+ overload and ROS overproduction in NRCMs. |
| 2022 | Lv [92] | - | - | Cryo-EM revealed the 3.6 Å structure of GPR75, detailing its ligand-binding pocket for drug design. |
| 2022 | Pascale [40] | 20-HETE | - | 20-HETE and agonists activate GPR75, while 20-SOLA, AAA, and antagonists inhibit its activity. |
| 2023 | Hossain [77] | - | Obesity | Gpr75-deficient mice resist weight gain on a high-fat diet without affecting insulin signaling. |
| 2023 | Speidell [63] | - | Cognitive dysfunction | Gpr75 knockout mice show anxiety and memory deficits linked to hippocampal protein dysregulation. |
| 2023 | Vasudevan [65] | - | Retinal degeneration | Gpr75 knockout causes age-related cone photoreceptor loss and dysfunction. |
| 2023 | D’Addario [19] | CCL5 | Pulmonary hypertension | In hypoxia-induced PH, CCL5 causes IPA contraction by decreasing cAMP levels via the GPR75/Gαi pathway. |
| 2023 | Cárdenas [82] | 20-HETE | Prostate cancer | In prostate cancer, Gpr75 expression correlates with AR, and 20-HETE/GPR75 axis increases AR activity. |
| 2023 | Ghorbanzadeh [84] | - | Colorectal cancer | Metformin reduces Gpr75 expression, potentially suppressing colon cancer growth. |
| 2024 | Leeson [80] | - | NAFLD | Gpr75 co-expressed in hypothalamic neurons regulates appetite and contributes to fatty liver disease. |
| 2024 | Jiang [42] | - | Obesity | In hypothalamic cilia, Gpr75 regulates food intake and energy, with its loss promoting a lean phenotype. |
| 2024 | Ma [59] | 20-HETE | Traumatic brain injury | The 20-HETE/GPR75 axis drives TBI-induced inflammation, reversed by Gpr75 knockdown or HET0016. |
| 2024 | Zhong [78] | - | Obesity | In growth-retarded carp, reduced Gpr75 expression impairs insulin metabolism, affecting body weight. |
| 2024 | Hardwick [73] | 20-HETE | MASLD | The 20-HETE/GPR75 axis is involved in regulating the progression of MASLD. |
| 2025 | Han [36] | 20-HETE | Myocardial hypertrophy | The 20-HETE/GPR75 axis is involved in Ang II-induced cardiomyocyte hypertrophy. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Han, J.; Li, J.; Yao, S.; Wei, Z.; Jiang, H.; Xu, T.; Zeng, J.; Xu, L.; Han, Y. GPR75: Advances, Challenges in Deorphanization, and Potential as a Novel Drug Target for Disease Treatment. Int. J. Mol. Sci. 2025, 26, 4084. https://doi.org/10.3390/ijms26094084
Han J, Li J, Yao S, Wei Z, Jiang H, Xu T, Zeng J, Xu L, Han Y. GPR75: Advances, Challenges in Deorphanization, and Potential as a Novel Drug Target for Disease Treatment. International Journal of Molecular Sciences. 2025; 26(9):4084. https://doi.org/10.3390/ijms26094084
Chicago/Turabian StyleHan, Jingyi, Jiaojiao Li, Sirui Yao, Zao Wei, Hui Jiang, Tao Xu, Junwei Zeng, Lin Xu, and Yong Han. 2025. "GPR75: Advances, Challenges in Deorphanization, and Potential as a Novel Drug Target for Disease Treatment" International Journal of Molecular Sciences 26, no. 9: 4084. https://doi.org/10.3390/ijms26094084
APA StyleHan, J., Li, J., Yao, S., Wei, Z., Jiang, H., Xu, T., Zeng, J., Xu, L., & Han, Y. (2025). GPR75: Advances, Challenges in Deorphanization, and Potential as a Novel Drug Target for Disease Treatment. International Journal of Molecular Sciences, 26(9), 4084. https://doi.org/10.3390/ijms26094084