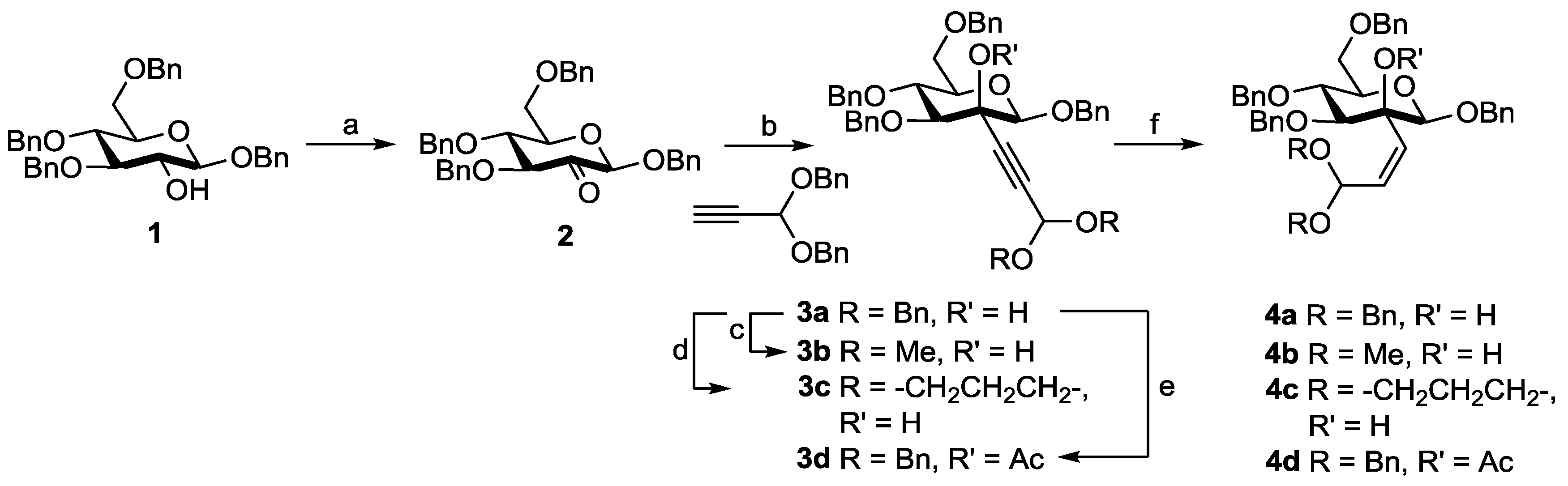
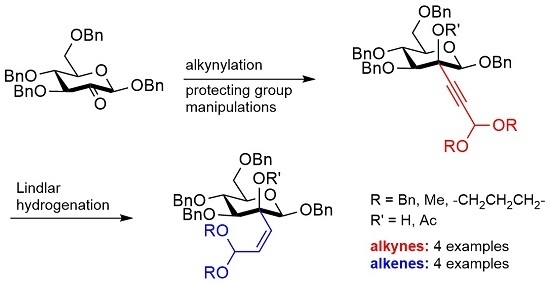
2. Results and Discussion
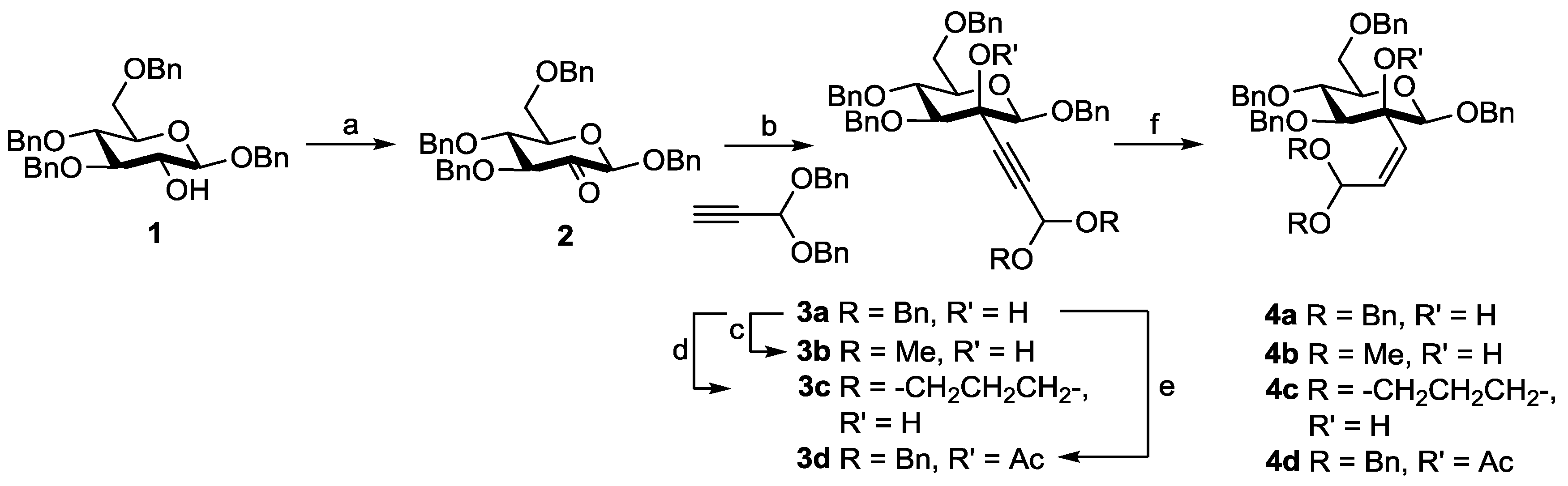
Our synthetic sequence started by subjecting benzyl β-glucoside
1 [
7] to Dess-Martin oxidation to afford the corresponding perbenzylated 2-uloside
2 (
Scheme 1). Previously, Fokt et al. [
8] described the synthesis of
2 via glycosylation reactions with a 2-ulosyl bromide. However, based on our previously encountered difficulties with comparable glycosylation reactions [
5] (i.e., low yields and the formation of side products) we anticipated our approach via Dess-Martin oxidation of
1 to be more efficient. Indeed, 2-uloside
2 was obtained in this way in an excellent 96% yield. Due to the high crystallinity of 2-uloside
2, X-ray structural analysis was performed in order to complete the analytical data of
2. Compound
2 crystallized as thin needles upon diffusion of
n-pentane into a solution of
2 in ethyl acetate/chloroform (2:1). The space group was found to be P2
1 and the asymmetric unit contained one molecule of
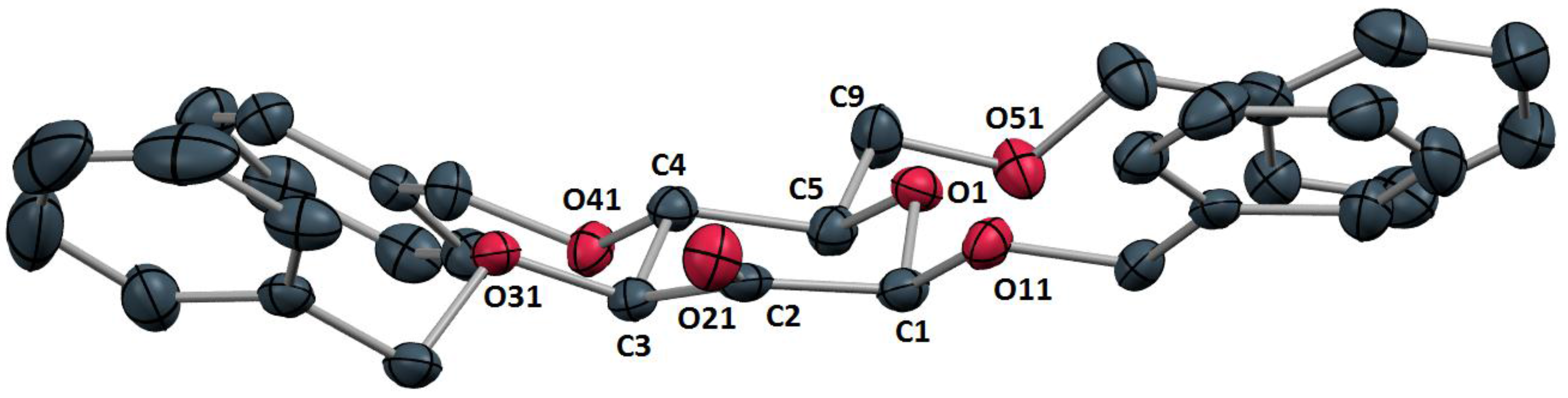
2. The molecular structure (
Figure 1) revealed a carbonyl bond length of 1.204 Å (C2-O21) as well as carbonyl angles of 115.3 Å (C1-C2-C3) and 122.3° (C1-C2-O21 and C3-C2-O21). The carbohydrate system was found to comprise a distorted
4C
1 conformation.
Subsequently, alkynylation of
2 with propiolaldehyde dibenzyl acetal [
9] and
n-butyllithium gave 2-
C-alkynyl mannoside
3a in 85% yield. The attack of the nucleophile solely occurred from the equatorial side and no diastereomeric byproduct could be detected. Similarly, high stereoselectivities were previously observed for a wide variety of nucleophilic addition reactions to β-2-ulosides [
5,
10,
11,
12]. In order to expand the synthetic applicability of the thus prepared branched mannosides we performed a series of transacetalizations affording products with variably orthogonally protected side chain formyl moieties. Thus, iodine-catalyzed [
13] and acid-catalyzed transacetalization of
3a with methanol and 1,3-dihydroxypropane, respectively, afforded dimethyl acetal
3b (88%) and 1,3-dioxane
3c (84%) (
Scheme 1). Acetylation of the tertiary 2-OH function under standard conditions [
14] gave acetate
3d in 81% yield. The subsequent partial hydrogenation of
3a employing Lindlar’s catalyst and H
2 [
15] was initially performed in ethyl acetate as well as methanol. However, only minor hydrogenation of the starting material was observed in these solvents over several days. We attributed this finding to the sterical crowdedness of the alkyne moiety in
3a caused by the bulky dibenzyl acetal and the other benzyl protecting groups in the glycoside. Changing the solvent to 1,4-dioxane had only a slightly accelerating effect. In contrast, using DMF as the solvent resulted in a smooth conversion of alkynes
3a–
3d to afford the corresponding
cis-alkenes
4a–
4d in typically 2–5 days and in good yields (81%–93%). Although the partial hydrogenation of alkynes with Lindlar catalyst has been used frequently, solvent effects as well as effects of sterical crowdedness seem to be hardly assessable, and the conditions often need rigorous optimization regarding the individual reaction system [
16,
17,
18,
19]. Therefore, we believe the protocol described above might be of further use for the partial hydrogenation of comparable branched alkyne systems. The vicinal alkene coupling constants
J7,8 of glycosides
4 were found to be in a region between the anticipated values for
cis- and
trans-alkenes (
J7,8 = 12.4 (
4a), 12.4 (
4b), 12.1 (
4c), 12.4 (
4d) Hz). Furthermore, 2D NMR experiments (H,H-NOESY) as well as X-ray structural analysis (
vide infra) were utilized for additional structural elucidation and final proof of the structures of
4a–
4d.
Originally we anticipated a synthetic route to 1,2-annulated glycosides based on the
syn-selective catalytic osmoylation of
cis-alkenes of type
4. However,
4a,
4c and
4d, respectively, appeared to be remarkably inert under various osmoylation conditions [
20]. Even stoichiometric amounts of osmium tetroxide at elevated temperatures did not result in dihydroxylation of the double bond. For example, dibenzyl acetal
4a did not react over several days when stirred at 70 °C in acetone/water/
t-butyl alcohol with 1.2 equivalents of osmium tetroxide and 5 vol % of pyridine as an additive. Dimethyl acetal
4b only resulted in acetal hydrolysis and subsequent decomposition.
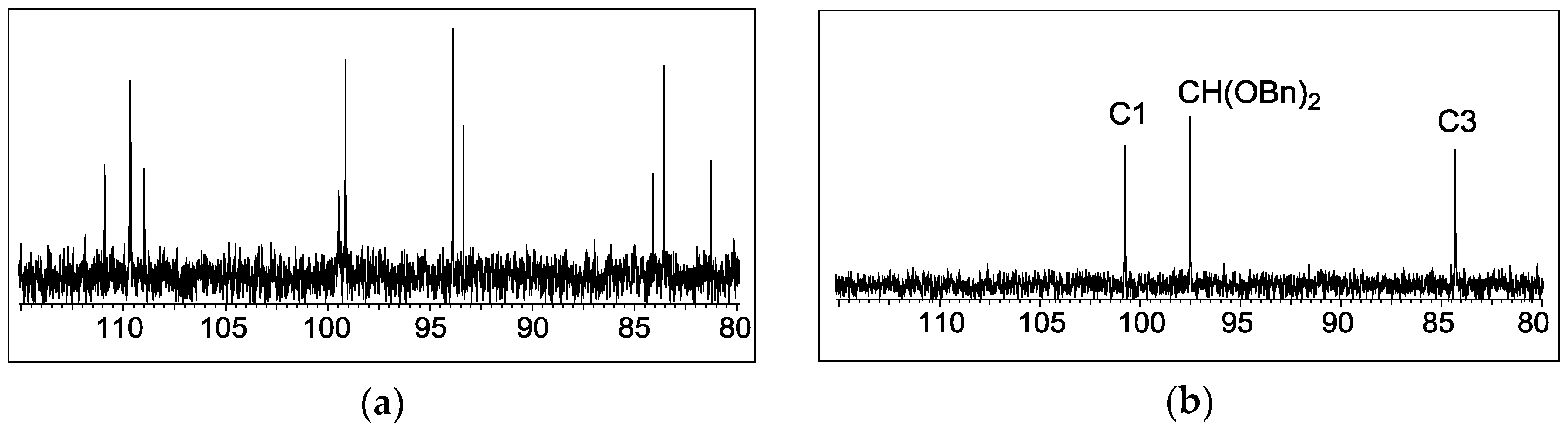
Both glycosides
4a and
4b but not glycosides
4c and
4d exhibited each remarkably complex
1H and
13C-NMR spectra in CDCl
3 (
Figure 2; for the full spectra see the Supporting Information). We attributed these unusually complex spectra to a conformational isomerism caused by constrained rotation of the single bonds adjacent to the bulky alkene moiety. In DMSO-
d6 as the solvent compound
4a showed a simpler NMR spectrum and no rotamers were observed. Similar distinct solvent dependencies of conformational isomerism have been reported in the literature [
21]. In the case of compound
4b, rotamers were still observable in DMSO-
d6, albeit to a much lesser extent. NMR measurements performed at elevated temperatures (50 °C) failed because of decomposition. The structure of glycoside
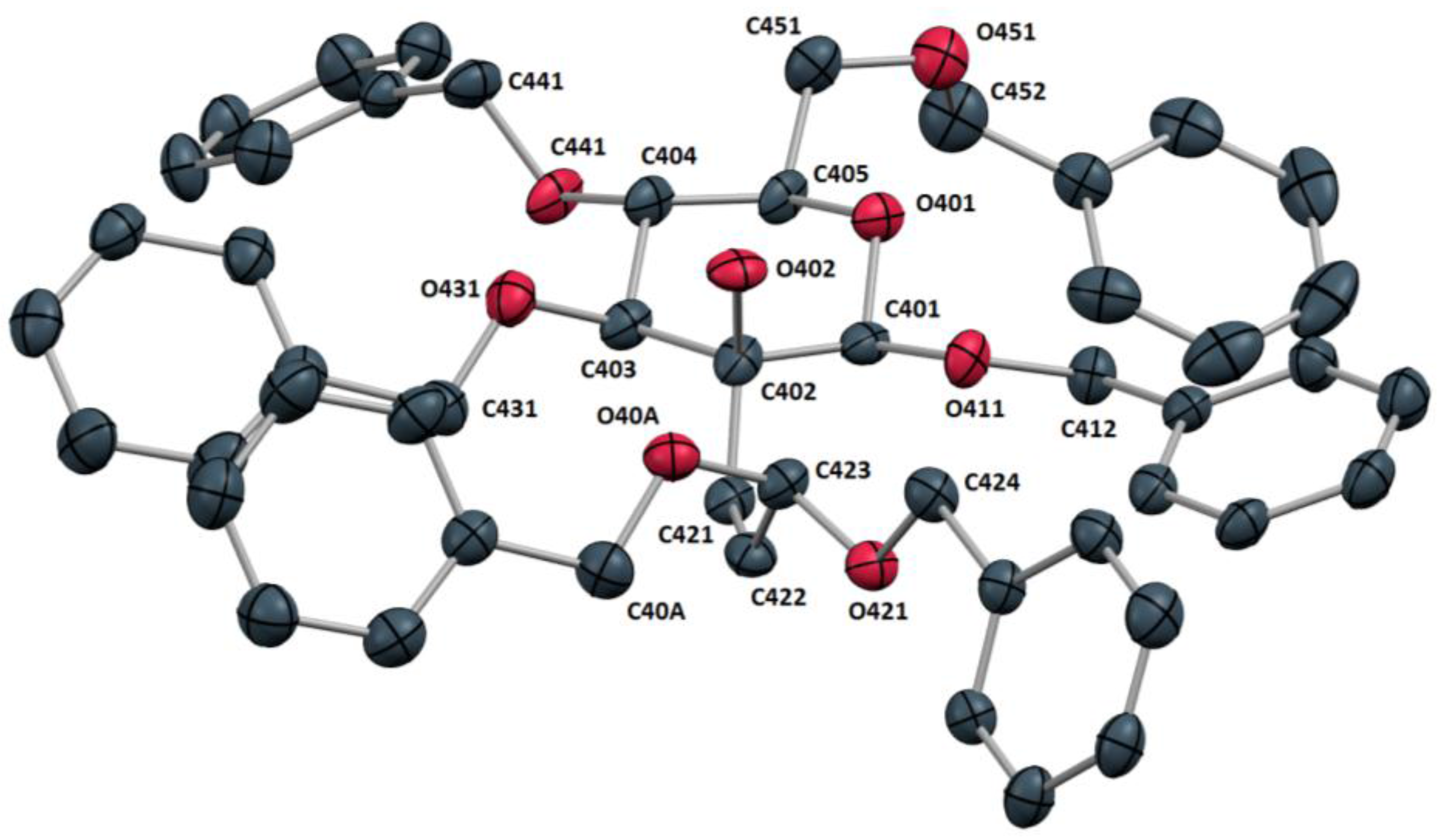
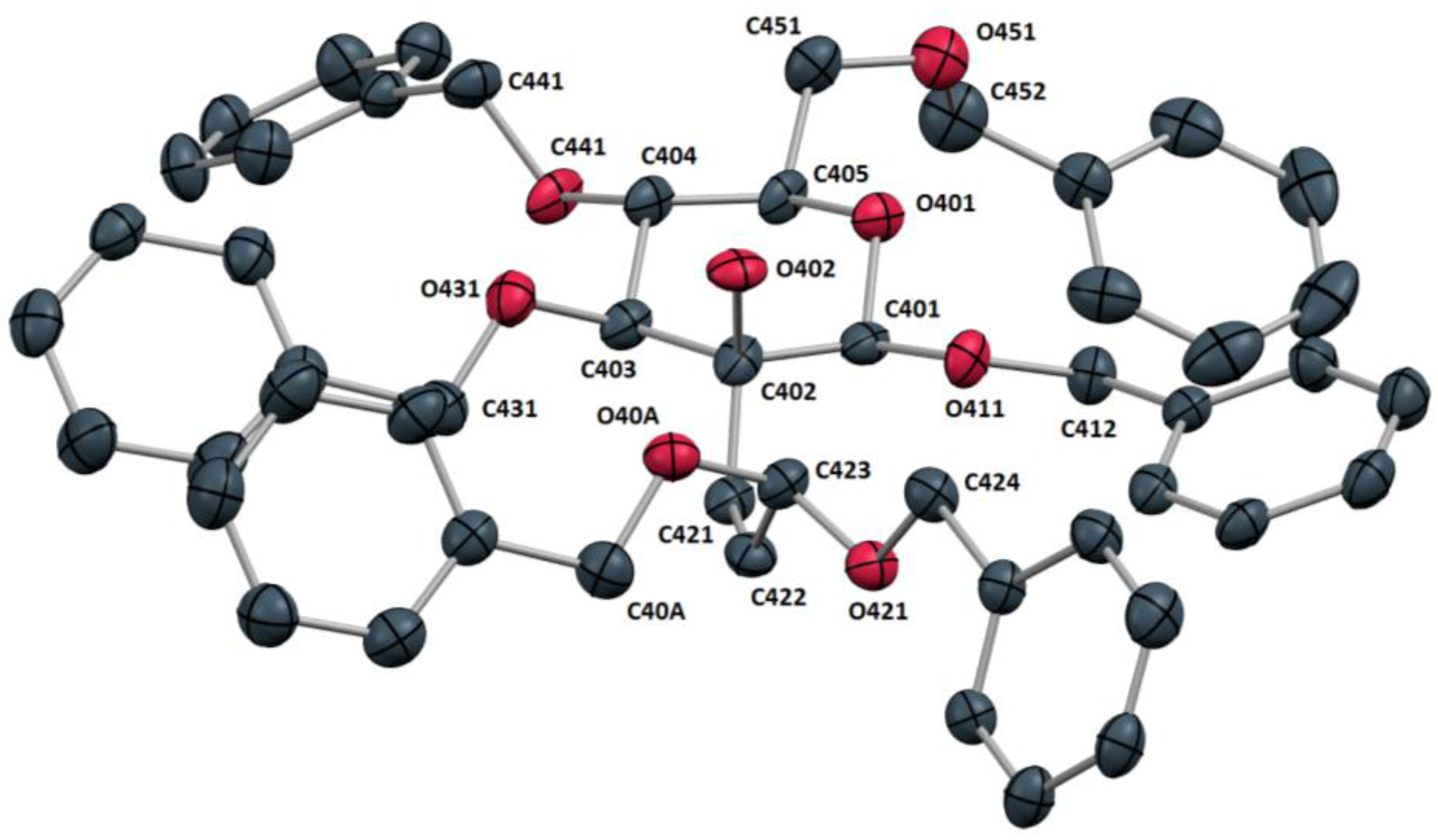
4a could finally be unambiguously confirmed by X-ray crystallography. Glycoside
4a formed thin needles suitable for X-ray analysis by slow diffusion of
n-pentane into a solution of the compound in ethyl acetate. The X-ray structure revealed compound
4a to crystallize in the triclinic space group P1. The asymmetric unit contained four molecules of
4a. An examination of the molecular structure (
Figure 3) confirmed the expected
4C
1 conformation of the carbohydrate system. The olefinic bond length (C421-C422: 1.325 Å) and torsion angle (C402-C421-C422-C423: 3.0°) were found to be as anticipated for an isolated
cis-alkene. The sterically crowded side chain dibenzyl acetal moiety was found to shield both sides of the alkene double bond as illustrated by the torsion angles involving the acetal oxygen atoms and the alkene carbon atoms (C421-C422-C423-O40A: 94.3° and C421-C422-C423-O421: −144.1°).
In conclusion, we have described a methodology for the straightforward synthesis of 2-C-alkynyl and 2-C-cis-alkenyl β-mannosides with an acetal-protected γ-aldehyde functionality. The synthesis comprises the manno-selective alkynylation of a benzylated 2-uloside and subsequent partial hydrogenation of the intermediate branched alkynes utilizing Lindlar’s catalyst. DMF as the solvent of choice was found to have a distinctly accelerating effect on the partial hydrogenation, and this finding could be of further use for the Lindlar hydrogenation of similar sterically crowded alkyne systems. The 2-C-branched carbohydrates reported here might be valuable building blocks for synthetic carbohydrate chemistry due to the potential orthogonality of the side chain carbonyl protection. For example, C-glycosyl-substituted (Z)-acrolein systems might be accessible from the described compounds.
3. Experimental Section
3.1. General Methods
NMR spectra were recorded with a Bruker Avance 400 spectrometer (Bruker BioSpin GmbH, Rheinstetten, Germany) and calibrated for the solvent signal (
1H: CDCl
3: 7.26 ppm; acetone-D
6: 2.05 ppm; DMSO-
d6: 2.50 ppm;
13C: CDCl
3: 77.16 ppm; acetone-
d6: 29.84 ppm; DMSO-
d6: 39.52 ppm). For diastereotopic methylene protons, the signal appearing at higher chemical shift was designated H
a, the one appearing at lower chemical shift values was designated H
b. NMR spectra of compounds
2-
4 are given in the Supporting Information. ESI-TOF-HRMS spectra were measured with a Bruker maXis 4 g spectrometer (Bruker Daltonik GmbH, Bremen, Germany), ESI-FTICR-HRMS spectra with a Bruker Apex II spectrometer and FAB-spectra with a Finnigan MAT spectrometer model TSQ 70 (Finnigan MAT, Bremen, Germany). Elemental analysis was performed with a HEKAtech Euro 3000 CHN analyzer (HEKAtech GmbH, Wegberg, Germany). Optical rotations were measured with a Perkin-Elmer Polarimeter 341 (Perkin Elmer Inc., Waltham, MA, USA) in a 10 cm cuvette at 20 °C. Melting points were determined with a Büchi Melting Point M-560 apparatus (BÜCHI Labortechnik GmbH, Essen, Germany). Reactions were monitored by TLC on Polygram Sil G/UV silica gel plates from Macherey-Nagel. Detection of spots was effected by charring with H
2SO
4 (5% in EtOH), staining by spraying the plates with (NH
4)
2MoO
4/Ce(SO
4)
2 in diluted sulfuric acid or by inspection of the TLC plates under UV light. Preparative chromatography was performed on silica gel (0.032–0.063 mm) from Macherey-Nagel (Macherey-Nagel GmbH & Co. KG, Düren, Germany). All solvents were purchased in technical grade and purified by distillation. For the reactions, the following solvents were dried according to standard methods and stored over molecular sieves 4 Å: CH
2Cl
2, THF and DMF: P
4O
10; toluene: Na/benzophenone. Dess-Martin periodinane was prepared via the potassium bromate method [
22]. Propiolaldehyde dibenzyl acetal was prepared according to the method published by Fliegel et al. [
9]. The following reagents were commercially available and used without further purification: Lindlar catalyst,
para-toluenesulfonic acid, acetic anhydride, 4-(dimethylamino)pyridine (Sigma-Aldrich Chemie, Munich, Germany), 1,3-dihydroxypropane (abcr, Karlsruhe, Germany),
n-butyllithium (Fisher Scientific, Nidderrau, Germany), triethylamine (Th. Geyer, Renningen, Germany). All yields given below are isolated yields.
3.2. X-ray Crystallography
Crystals of
2 were grown by slow diffusion of
n-pentane into a solution of the compound in ethyl acetate/chloroform (2:1; 20 mg, 750 µL) at ambient temperature. Crystals of
4a were grown by diffusion of
n-pentane into a solution of
4a in ethyl acetate (20 mg, 500 µL) at ambient temperature. X-ray data were collected on a Bruker SMART APEX II DUO diffractometer (Bruker AXS Advanced X-ray Solutions GmbH, Karlsruhe, Germany) using a Cu Kα radiation (λ = 1.54178 Å) or Mo Kα radiation (λ = 0.71073 Å). Corrections for absorption effects were applied using SADABS [
23]. All structures were solved by direct methods using SHELXS and SHELXL for structure solution and refinement [
24,
25,
26]. Complete crystallographic data for the structural analysis have been deposited with the Cambridge Crystallographic Data, CCDC 1505647 (for
2) and CCDC 1505648 (for
4a). Copies of this information may be obtained free of charge from The Cambridge Crystallographic Data Centre.
3.3. Benzyl 3,4,6-Tri-O-benzyl-β-d-arabino-hexos-2-ulo-1,5-pyranoside (2)
To a solution of glucoside
1 [
7] (1.65 g; 3.05 mmol) in CH
2Cl
2 (100 mL) Dess-Martin periodinane (2.85 g; 6.71 mmol) was added in portions over 15 min at r.t., and the mixture was stirred until TLC (toluene/ethyl acetate, 4:1) indicated complete transformation of the starting material (16 h). The solution was diluted with CH
2Cl
2 (100 mL) and washed with water, three times with a 1:1 mixture of satd. aqueous NaHCO
3 solution and an aqueous 0.2 M Na
2S
2O
3 solution and with water (80 mL each). The organic layer was dried with Na
2SO
4, filtered and concentrated in vacuo. The crude product was suspended in toluene/acetone (20 mL/15 mL), and the suspension was purified by column chromatography (SiO
2, toluene/acetone, 20:1) to afford pure ketone
2 (1.58 g; 2.92 mmol; 96%) as a colorless solid. Crystals suitable for X-ray structural analysis were obtained from
n-pentane/ethyl acetate/chloroform. Analytical data were found to be in accordance with ref. [
8]. Additional characterization data:
13C-NMR (100 MHz, CDCl
3) δ: 197.4 (C-2), 138.1, 137.8, 137.4, 136.2 (4 × C
Ar,quart), 128.6, 128.6, 128.5, 128.3, 128.2, 128.1, 128.0, 127.8 (20 × C
Ar), 97.4 (C-1), 85.8 (C-3), 80.1 (C-4), 75.9 (C-5), 75.0 ,73.6, 73.6, 70.3 (4 × CH
2Ph), 69.1 (C-6). Anal. calcd. for C
34H
34O
6: C, 75.82; H, 6.36. Found: C, 75.54; H, 6.35.
3.4. Benzyl 3,4,6-Tri-O-benzyl-2-C-(3,3-dibenzyloxypropyn-1-yl)-β-d-mannopyranoside (3a)
Propiolaldehyde dibenzyl acetal [
9] (1.02 g; 4.04 mmol) was dissolved in anhydrous THF (35 mL) and cooled to −80 °C.
n-Butyllithium (1.50 mL; 4.04 mmol; 2.7 M in
n-heptane) was added dropwise and stirring at −80 °C was continued for 2.5 h. Next, 2-uloside
2 (1.98 g; 3.67 mmol) in 30 mL THF was added dropwise over 15 min. After stirring for another 15 min, the reaction was quenched by the addition of satd. aqueous NaCl solution (25 mL), allowed to warm to ambient temperature, diluted with chloroform (200 mL) and washed with water, satd. aqueous NH
4Cl solution, satd. aqueous NaHCO
3 solution and water (100 mL each). Drying of the organic layer with Na
2SO
4, filtration, evaporation of the solvents and chromatographic purification (SiO
2, toluene/acetone, 20:1) afforded alkyne
3a (2.47 g; 3.12 mmol; 85%) as a colorless solid. An analytical sample was recrystallized from
n-hexane/ethyl acetate.
Rf = 0.65 (toluene/ethyl acetate, 4:1); [α]
+3.5 (
c 1, CHCl
3); m.p. 85 °C (
n-hexane/ethyl acetate);
1H-NMR (400 MHz, CDCl
3) δ: 7.50–7.21 (m, 30H, H
Ar), 5.57 (s, 1H, H-9), 5.14 (d, 1H,
Jgem = 10.6 Hz, CH
2Ph), 5.04 (d, 1H,
Jgem = 12.1 Hz, CH
2Ph), 4.95 (d, 1H,
Jgem = 10.6 Hz, CH
2Ph), 4.88 (d, 1H,
Jgem = 11.1 Hz, CH
2Ph), 4.85–4.78 (m, 3H, CH
2Ph), 4.74–4.63 (m, 4H, CH
2Ph), 4.59 (d, 1H,
Jgem = 10.9 Hz, CH
2Ph), 4.56 (s, 1H, H-1), 3.91–3.76 (m, 3H, H-4, H-6a, H-6b), 3.71 (d, 1H,
J3,4 = 9.1 Hz, H-3), 3.52 (ddd, 1H,
J4,5 = 9.9,
J5,6 = 5.1, 1.9 Hz, H-5), 3.10 (bs, 1H, OH);
13C-NMR (100 MHz, CDCl
3) δ: 138.2, 138.1, 137.8 137.4, 137.3, 136.7 (6 × C
Ar,quart), 128.5, 128.4, 128.4, 128.4, 128.3, 128.0, 128.0, 128.0, 127.9, 127.9, 127.8, 127.8, 127.7 (30 × C
Ar), 99.7 (C-1), 90.7 (C-9), 85.3 (C-3), 85.1 (C-7), 80.8 (C-8), 76.1 (CH
2Ph), 75.3 (C-5), 75.2 (CH
2Ph), 75.0 (C-4), 73.6 (CH
2Ph), 71.6 (C-2), 70.8 (CH
2Ph), 69.0 (C-6), 67.5 (C9-O-
CH
2Ph), 67.4 (C9-O-
CH2Ph); HR-ESI-TOF calcd. for C
51H
50O
8Na [M + Na]
+: 813.3398 Found: 813.3384; Anal. calcd. for C
51H
50O
8: C, 77.45; H, 6.37. Found: C, 77.21; H, 6.40.
3.5. Benzyl 3,4,6-Tri-O-benzyl-2-C-(3,3-dimethoxypropyn-1-yl)-β-d-mannopyranoside (3b)
Dibenzyl acetal 3a (301 mg; 0.38 mmol) was dissolved in anhydrous methanol (20 mL) and iodine (200 mg; 0.79 mmol) was added. The solution was stirred at 60 °C for 48 h until TLC (toluene/ethyl acetate, 4:1) indicated complete consumption of the starting material. The reaction mixture was cooled to 0 °C, treated with solid Na2S2O3 (1.0 g), stirred for 15 min and allowed to warm to ambient temperature. Next, chloroform (100 mL) was added and the organic layer was washed with water (2 × 30 mL) and brine (30 mL). The solution was dried with Na2SO4, filtered and concentrated in vacuo. Chromatographic purification of the residue (SiO2, toluene/acetone, 20:1) afforded 3b (213 mg; 0.33 mmol; 88%) as a colorless oil. Rf = 0.30 (toluene/ethyl acetate, 4:1); [α] −6.7 (c 1, CHCl3); 1H-NMR (400 MHz, CDCl3) δ: 7.48–7.27 (m, 18H, HAr), 7.22–7.15 (m, 2H, HAr), 5.18 (s, 1H, H-9), 5.08 (d, 1H, Jgem = 10.9 Hz, CH2Ph), 5.00 (d, 1H, Jgem = 12.1 Hz, CH2Ph), 4.90 (d, 1H, Jgem = 10.9 Hz, CH2Ph), 4.85–4.74 (m, 2H, CH2Ph), 4.70–4.51 (m, 4H, CH2Ph, H-1), 3.86–3.67 (m, 4H, H-4, H-6a, H-6b, H-3, visible coupling constants: J3,4 = 9.1 Hz), 3.52–3.45 (m, 1H, H-5), 3.36 (s, 6H, 2 × CH3), 3.04 (bs, 1H, OH); 13C-NMR (100 MHz, CDCl3) δ: 138.3, 138.1, 137.9 136.8 (4 × CAr,quart), 128.5, 128.4, 128.0, 128.0, 128.0, 127.9, 127.9, 127.7 (20 × CAr), 99.9 (C-1), 93.1 (C-9), 85.4 (C-3), 84.9 (C-7), 80.5 (C-8), 76.1 (CH2Ph), 75.4 (C-5), 75.2 (CH2Ph), 75.0 (C-4), 73.6 (CH2Ph), 71.7 (C-2), 70.9 (CH2Ph), 69.1 (C-6), 52.8 (CH3), 52.6 (CH3); HR-ESI-TOF calcd. for C39H42O8Na [M + Na]+: 661.2772 Found: 661.2777; Anal. calcd. for C39H42O8: C, 73.33; H, 6.63. Found: C, 73.04; H, 6.65.
3.6. Benzyl 3,4,6-Tri-O-benzyl-2-C-((1,3-dioxan-2-yl)ethynyl)-β-d-mannopyranoside (3c)
To a solution of dibenzyl acetal 3a (465 mg; 0.59 mmol) in anhydrous toluene (15 mL) was added 1,3-propanediol (426 µL; 5.88 mmol) and a catalytic amount of p-toluenesulfonic acid hydrate (14 mg) and the solution was stirred at 100 °C for 22 h until TLC (toluene/ethyl acetate, 4:1) indicated the complete transformation of the starting material. The reaction mixture was diluted with toluene (20 mL), washed with satd. aqueous NaHCO3 solution (20 mL) and water (20 mL). After drying with Na2SO4, filtration and concentration in vacuo chromatographic purification of the residue (SiO2, toluene/ethyl acetate, 7:1) afforded 3c (320 mg; 84%) as a colorless amorphous solid. Rf = 0.26 (toluene/ethyl acetate, 4:1); [α] −2.4 (c 1, CHCl3); 1H-NMR (400 MHz, CDCl3) δ: 7.48–7.42 (m, 4H, HAr), 7.40–7.26 (m, 14H, HAr), 7.21–7.16 (m, 2H, HAr), 5.46 (s, 1H, H-9), 5.10 (d, 1H, Jgem = 10.9 Hz, CH2Ph), 5.01 (d, 1H, Jgem = 12.4 Hz, CH2Ph), 4.91 (d, 1H, Jgem = 10.9 Hz, CH2Ph), 4.85–4.76 (m, 2H, CH2Ph), 4.69–4.52 (m, 4H, CH2Ph, H-1), 4.22–4.15 (m, 2H, -CH2-CH2-CH2-), 3.86–3.69 (m, 6H, -CH2-CH2-CH2-, H-6a, H-6b, H-4, H-3, visible coupling constants: J3,4 = 8.8 Hz), 3.48 (ddd, 1H, J4,5 = 9.8, J5,6 = 5.1, 2.0 Hz, H-5), 3.07 (bs, 1H, OH), 1.81–1.67 (m, 2H, -CH2-CH2-CH2-); 13C-NMR (100 MHz, CDCl3) δ: 138.3, 138.1, 137.9, 136.9 (4 × CAr,quart), 128.5, 128.4, 128.4, 128.3, 128.0, 127.9, 127.9, 127.8, 127.7 (20 × CAr), 99.9 (C-1), 90.2 (C-9), 85.4 (C-3), 85.1 (C-7), 80.7 (C-8), 76.1 (CH2Ph), 75.4 (C-5), 75.2 (CH2Ph), 75.0 (C-4), 73.6 (CH2Ph), 71.7 (C-2), 70.9 (CH2Ph), 69.1 (C-6), 64.6 (-CH2-CH2-CH2-), 64.6 (-CH2-CH2-CH2-), 25.7 (-CH2-CH2-CH2-); HR-ESI-TOF calcd. for C40H42O8Na [M + Na]+: 673.2772 Found: 673.2777; Anal. calcd. for C40H42O8: C, 73.83; H, 6.51. Found: C, 73.67; H, 6.66.
3.7. Benzyl 2-O-Acetyl-3,4,6-tri-O-benzyl-2-C-(3,3-dibenzyloxypropyn-1-yl)-β-d-mannopyranoside (3d)
A solution 3a (394 mg; 0.50 mmol), acetic anhydride (4 mL), triethylamine (76 µL; 0.548 mmol) and DMAP (122 mg; 1.0 mmol) in chloroform (4 mL) was heated to 65 °C for 4 d until TLC (toluene/ethyl acetate, 4:1) indicated complete conversion of the starting material. The reaction mixture was diluted with chloroform (50 mL) and poured onto an ice-cold satd. aqueous NaHCO3 solution (50 mL). The organic layer was washed with sat. aqueous NaHCO3 solution and water (20 mL each), dried with Na2SO4, filtered, concentrated in vacuo and coevaporated with toluene (2 × 10 mL). Chromatography of the residue (SiO2, toluene/acetone, 50:1) gave 3d (335 mg; 0.40 mmol; 81%) as a colorless oil. Rf = 0.70 (toluene/ethyl acetate, 4:1); [α] −11.7 (c 0.85, CHCl3); 1H-NMR (400 MHz, CDCl3) δ: 7.43–7.18 (m, 28H, HAr), 7.15–7.10 (m, 2H, HAr), 5.53 (s, 1H, H-9), 5.30 (d, 1H, Jgem = 10.9 Hz, CH2Ph), 4.97 (d, 1H, Jgem = 12.4 Hz, CH2Ph), 4.87–4.62 (m, 8H, CH2Ph), 4.56 (d, 1H, Jgem = 12.1 Hz, CH2Ph), 4.51–4.44 (m, 2H, H-1, CH2Ph), 3.79–3.67 (m, 4H, H-6a, H-6b, H-3), 3.49 (ddd, 1H, J4,5 = 9.6, J5,6 = 4.5, 2.5 Hz, H-5), 2.18 (s, 3H, CH3); 13C-NMR (100 MHz, CDCl3) δ: 168.3 (C=O acetyl), 138.3, 138.2, 138.0, 137.6, 137.6, 136.7 (6 × CAr,quart), 128.7, 128.5, 128.5, 128.5, 128.4, 128.4, 128.3, 128.2, 128.0, 127.9, 127.8, 127.8, 127.7 (30 × CAr), 100.6 (C-1), 91.0 (C-9), 86.1 (C-3), 84.4 (C-7), 82.2 (C.8), 75.7 (C-5), 75.4 (CH2Ph), 2 × 75.2 (CH2Ph, C-2), 74.7 (C-4), 73.6 (CH2Ph), 71.2 (CH2Ph), 69.2 (C-6), 67.6 (CH2Ph), 67.6 (CH2Ph), 22.2 (CH3); HR-ESI-TOF calcd. for C53H52O9Na [M + Na]+: 855.3504 Found: 855.3505; Anal. calcd. for C53H52O9: C, 76.42; H, 6.29. Found: C, 76.36; H, 6.32.
3.8. Lindlar Hydrogenations: General Procedure for the Synthesis of 4a, 4b, 4c and 4d
Compound 3 was dissolved in anhydrous DMF and Lindlar catalyst was added. The suspension was stirred under an atmosphere of hydrogen (at a pressure slightly above atmospheric pressure evoked by a column of water) at r.t. until TLC (toluene/ethyl acetate, 4:1) indicated complete conversion of the starting material. The mixture was diluted with CH2Cl2. Filtration through a layer of Celite provided, after evaporation of the solvent and coevaporation with n-heptane, the cis-alkene 4, typically in high purity. If necessary, chromatographic purification/recrystallization provided the further purified products.
3.9. Benzyl 3,4,6-Tri-O-benzyl-2-C-((Z)-3,3-dibenzyloxyprop-1-en-1-yl)-β-d-mannopyranoside (4a)
A solution of 3a (129 mg; 0.163 mmol) in anhydrous DMF (2 mL) was treated with Lindlar catalyst (64 mg; 50 wt %) for 48 h as described in the general procedure above. Chromatography (SiO2, toluene/acetone, 20:1) gave 4a (120 mg; 0.151 mmol; 93%) as a colorless solid. An analytical sample was recrystallized from n-hexane/ethyl acetate. Crystals suitable for X-ray structural analysis were obtained from n-pentane/ethyl acetate. Rf = 0.76 (toluene/ethyl acetate, 4:1); [α] −34.0 (c 0.5, CHCl3); m.p. 95 °C (n-hexane/ethyl acetate); 1H-NMR (400 MHz, DMSO-d6) δ: 7.40–7.15 (m, 30H, HAr), 6.19 (d, 1H, J8,9 = 6.8 Hz, H-9), 5.74 (dd, 1H, J7,8 = 12.4, J8,9 = 6.8 Hz, H-8), 5.67 (d, J7,8 = 12.4 Hz, H-7), 4.85–4.40 (m, 13H, CH2Ph, H-1), 4.34 (bs, 1H, OH), 3.78–3.63 (m, 4H, H-3, H-4, H-6a, H-6b), 3.58–3.52 (m, 1H, H-5); 13C-NMR (100 MHz, DMSO-d6) δ: 138.5, 138.5, 138.4, 138.4, 138.3, 137.7 (6 × CAr,quart), 133.2 (C-7), 129.9 (C-8), 128.2, 128.2, 128.1, 128.1, 128.0, 127.7, 127.6, 127.6, 127.5, 127.4, 127.4, 127.3, 127.2, 127.2 (30 × CAr), 100.8 (C-1), 97.5 (C-9), 84.3 (C-3), 78.7 (C-2), 75.6 (C-4), 74.9 (CH2Ph), 74.5 (C-5), 74.1 (CH2Ph), 72.3 (CH2Ph), 70.2 (CH2Ph), 69.1 (C-6), 67.5 (CH2Ph), 66.3 (CH2Ph); HR-ESI-TOF calcd. for C51H52O8Na [M + Na]+: 815.3554 Found: 815.3544; Anal. calcd. for C51H52O8: C, 77.25; H, 6.61. Found: C, 77.31; H, 6.56.
3.10. Benzyl 3,4,6-Tri-O-benzyl-2-C-((Z)-3,3-dimethoxyprop-1-en-1-yl)-β-d-mannopyranoside (4b)
Alkyne 3b (95 mg; 0.149 mmol) in anhydrous DMF (3 mL) was treated with Lindlar catalyst (71 mg; 75 wt %) for 5 d as described in the general procedure above. After chromatographic purification (SiO2, toluene + 2% Et3N), alkene 4b (87 mg; 0.136 mmol; 91%) was obtained as a colorless, amorphous solid. Rf = 0.23 (toluene + 2% Et3N); [α] −57.0 (c 1, CHCl3); 1H-NMR (400 MHz, DMSO-d6) δ: rotameric isomerism, significant signals: 5.69 (d, 1H, J8,9 = 6.3 Hz, H-9), 5.59 (d, 1H, J7,8 = 12.4 Hz, H-7), 5.54 (dd, 1H, J7,8 = 12.4, J8,9 = 6.3 Hz, H-8), 3.14 (s, 6H, 2 × CH3); 13C-NMR (100 MHz, DMSO-d6) δ: rotameric isomerism, significant signals: 133.2 (C-7), 129.8 (C-8), 100.7 (C-1), 99.0 (C-9), 74.5 (C-5), 52.8, 51.6 (2 × CH3); HR-ESI-TOF calcd. for C39H44O8Na [M + Na]+: 663.2928 Found: 663.2933; Anal. calcd. for C39H44O8: C, 73.10; H, 6.92. Found: C, 72.97; H, 7.01.
3.11. Benzyl 3,4,6-Tri-O-benzyl-2-C-((Z)-2-(1,3-dioxan-2-yl)vinyl)-β-d-mannopyranoside (4c)
Alkyne 3c (239 mg; 0.367 mmol) in anhydrous DMF (5 mL) was treated with Lindlar catalyst (144 mg; 60 wt %) for 3 d as described in the general procedure above. Alkene 4c (194 mg; 0.297 mmol; 81%) was obtained as a colorless, amorphous solid. Rf = 0.65 (toluene/ethyl acetate, 2:1); [α] –61.2 (c 1, CHCl3); 1H-NMR (400 MHz, CDCl3) δ: 7.41–7.25 (m, 18H, HAr), 7.23–7.17 (m, 2H, HAr), 5.80 (d, 1H, J8,9 = 5.1 Hz, H-9), 5.70 (dd, 1H, J7,8 = 12.1, J8,9 = 5.2 Hz, H-8), 5.33 (d, 1H, J7,8 = 12.1 Hz, H-7), 4.93 (d, 1H, Jgem = 12.1 Hz, CH2Ph), 4.86 (d, 1H, Jgem = 10.9 Hz, CH2Ph), 4.76–4.52 (m, 6H, CH2Ph), 4.26 (s, 1H, H-1), 4.10–4.01 (m, 2H, -CH2-CH2-CH2-), 3.92–3.72 (m, 6H, H-4, OH, -CH2-CH2-CH2-, H-6a, H-6b), 3.49–3.43 (m, 1H, H-5), 3.40 (d, 1H, J3,4 = 9.1 Hz, H-3), 2.17–2.03 (m, 1H, -CH2-CHaHb-CH2-), 1.31 (d, 1H, Jvic = 13.4 Hz, -CH2-CHaHb-CH2-); 13C-NMR (100 MHz, CDCl3) δ: 138.5, 138.4, 138.0, 137.2 (4 × CAr,quart), 133.2 (C-7), 132.2 (C-8), 128.8, 128.5, 128.5, 128.4, 128.4, 128.3, 127.9, 127.9, 127.9, 127.8, 127.7 (20 × CAr), 99.8 (C-1), 98.5 (C-9), 84.6 (C-3), 78.7 (C-2), 2 × 76.1 (C-4, CH2Ph), 75.5 (C-5), 75.2 (CH2Ph), 73.7 (CH2Ph), 70.4 (CH2Ph), 69.5 (C-6), 66.7 (CH2-CH2-CH2-), 66.6 (CH2-CH2-CH2-), 25.6 (CH2-CH2-CH2-); HR-ESI-TOF calcd. for C40H44O8Na [M + Na]+: 675.2932 Found: 675.2928; Anal. calcd. for C40H44O8: C, 73.60; H, 6.79. Found: C, 73.65; H, 6.88.
3.12. Benzyl 2-O-Acetyl-3,4,6-tri-O-benzyl-2-C-((Z)-3,3-dibenzyloxyprop-1-en-1-yl)-β-d-mannopyranoside (4d)
Alkyne 3d (249 mg; 0.299 mmol) in anhydrous DMF (3 mL) was treated with Lindlar’s catalyst (113 mg; 45 wt %) for 3 d as described in the general procedure above. Alkene 4d (220 mg; 0.263 mmol; 88%) was obtained as a colorless oil. Rf = 0.78 (toluene/ethyl acetate, 2:1); [α] –18.6 (c 1, CHCl3); 1H-NMR (400 MHz, CDCl3) δ: 7.35–7.16 (m, 30H, HAr), 6.21 (d, 1H, J7,8 = 12.4 Hz, H-7), 5.76 (dd, 1H, J7,8 = 12.4, J8,9 = 7.8 Hz, H-8), 5.67 (d, 1H, J8,9 = 7.8 Hz, H-9), 5.27 (s, 1H, H-1), 4.87 (d, 1H, Jgem = 11.6 Hz, CH2Ph), 4.65–4.55 (m, 3H, CH2Ph), 4.51–4.46 (m, 3H, CH2Ph), 4.44–4.37 (m, 5H, CH2Ph), 4.95–4.82 (m, 4H, H-5, H-3, H-6a, H-4), 3.77 (dd, 1H, J6a,6b = 9.6, J5,6b = 5.8 Hz, H-6b), 1.83 (s, 3H, CH3); 13C-NMR (100 MHz, CDCl3) δ: 169.4 (C=O acetyl), 138.5, 138.4, 138.2, 138.1, 138.0, 137.8 (6 × CAr,quart), 133.1 (C-7), 128.6, 128.5, 128.5, 128.4, 128.4, 128.3, 128.2, 127.9, 127.8, 127.8, 127.7, 127.6, 127.6, 127.5 (30 × CAr), 127.1 (C-8), 99.9 (C-1), 95.8 (C-9), 80.2 (C-2), 79.7 (C-3), 75.1 (*C-4), 74.9 (*C-5; assignments may be interchanged), 74.8 (CH2Ph), 73.3 (CH2Ph), 72.3 (CH2Ph), 71.1 (CH2Ph), 70.6 (C-6), 66.9 (CH2Ph), 65.2 (CH2Ph), 22.2 (CH3); HR-ESI-TOF calcd. for C53H54O9Na [M + Na]+: 857.3660 Found: 857.3663; Anal. calcd. for C53H54O9: C, 76.24; H, 6.52. Found: C, 76.14; H, 6.48.




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}