Abstract
In toluene solution, the reaction of [Co2(CO)8] with an equimolar amount of [P(OPh)3] yields first [Co2(CO)7{P(OPh)3}] 1. Following heating of 1, an alternative synthetic access to the tetranuclear cluster [{Co4(μ2-CO)3(CO)8{P(OPh)3}] 2 is provided in a condensation reaction. Compound 2 has been characterized by IR and 31P NMR spectroscopy. The tetranuclear cluster framework has been ascertained by a single-crystal X-ray diffraction study performed at 100 K.
1. Introduction
The tetrahedral carbonyl cluster [Co4(μ2-CO)3(CO)9] was first described by Hieber in 1932 and is commonly prepared by decarbonylation of dicobaltoctacarbonyl [1,2]. This 60-electron cluster, which contains both terminal and bridging carbonyls, has been the subject of several crystallographic investigations [3,4] and has served as the starting material for a number of organic (for example, the formation of arene clusters [Co4(CO)9(arene)]) and inorganic transformations [2,5,6]. Among the latter reaction, several papers have been devoted to the kinetics and mechanistic aspects of substitution reactions with various phosphine PR3, diphosphine (such as bis(diphenylphosphino)amine dppa and bis(diphenylphosphino)methane dppm), and phosphite P(OR)3 ligands have been published [7,8,9,10,11,12,13]. Some examples of structurally characterized mono- and di-substituted derivatives [Co4(μ2-CO)3(CO)9-n(PR3)n] (n = 1, 2) are depicted in Scheme 1. There is also a report on the crystal structure of [Co4(CO)10(PMe2Pr)2] with one PR3 ligand at the apical and a second one at the axial position (see Figure 4) [14].
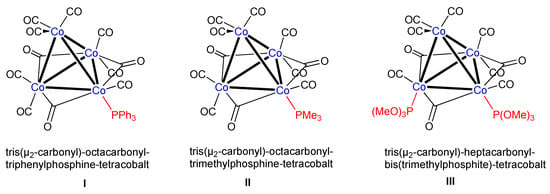
Scheme 1.
Examples of some phosphine- and phosphite-substituted tetranuclear cobalt clusters [11,12].
It is worth noting that P(OMe)3 and P(OPh)3 can even form tetra-substituted clusters [Co4(CO)8{P(OR)3}4] [8]. In the context of our research on P(OPh)3-substituted Co-Co carbonyl complexes towards alkynes producing dicobaltatetrahedranes [15,16], we attempted to synthetize the monosubstituted dinuclear complex [Co2(CO)7{P(OPh)3}] 1 by adding a stoichiometric amount of P(OPh)3 to a solution of [Co2(CO)8]. The existence of this substitution product has been mentioned in the literature, but apart from its IR spectrum, no further characterization data have been communicated [17]. We repeated this reaction under similar conditions with the goal of isolating this species, but upon heating, we isolated the title compound [Co4(μ2-CO)3(CO)8{P(OPh)3}] 2 by serendipity as the major component.
2. Results
Marko et al. first mentioned the title compound 2 in 1975, and it was obtained by treating [Co4(CO)9(arene)] with triphenylphosphite at 20 °C under CO atmosphere, along with [Co4(CO)10{P(OPh)3}2] and [Co4(CO)9{P(OPh)3}3] [5]. There was no additional characterization data presented aside from a detailed IR analysis in solution. To prepare the compound [Co2(CO)7{P(OPh)3}], we first treated a solution of [Co2(CO)8] in toluene at ambient temperature in a 1:1 ratio (Scheme 2). The formation of 1 (2086, 2034, 1999, and 1978 cm−1) was revealed by IR monitoring as well as traces of [Co2(CO)6{P(OPh)3}2] (1978 cm−1, very strong). Formation of this dinuclear bisphosphite complex is also corroborated by an NMR 31P{1H} analysis performed on a sample of the reaction mixture, which shows a singlet at 167.3 ppm [18]. To complete the reaction, the mixture was then heated for 5 h at 60 °C. Surprisingly, the IR bands attributed to 1 had disappeared and replaced by novel ones at 2089, 2050, 2042, 2032, 2012 and 1882, 1850, and 1839 cm−1, the latter being in the characteristic region of bridging carbonyl ligand. The formation of minor amounts of [Co4(CO)10{P(OPh)3}2] is suggested by a CO stretching vibration at 2073 cm−1 in the IR spectrum, in accordance with value reported by Marko et al. [5].
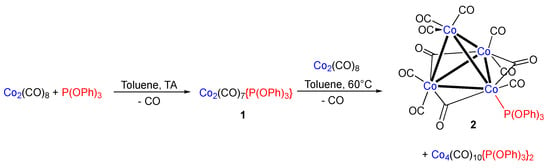
Scheme 2.
Synthesis of the title compound 2.
After workup, a product in the form of dark crystals was isolated, and elemental analysis revealed a composition of [Co4(CO)11{P(OPh)3}]. The IR spectrum of this moderate air-stable product in cyclohexane, shown in Figure 1, reveals that in addition to the ν(CO) vibrations at 2088, 2049, 2043, 2032, and 2011 cm−1, three further absorptions at 1885, 1856, and 1842 cm−1, are attributed to bridging carbonyls. These values fit well with those reported in heptane by Marko et al. [5]. The infrared band pattern is similar to that reported for [Co4(μ2-CO)3(CO)8(PPh3)] in heptane, the CO vibrational frequencies being slightly shifted to higher wavenumbers due to the weaker electron-donating propensity exerted by P(OPh)3 with respect to PPh3.
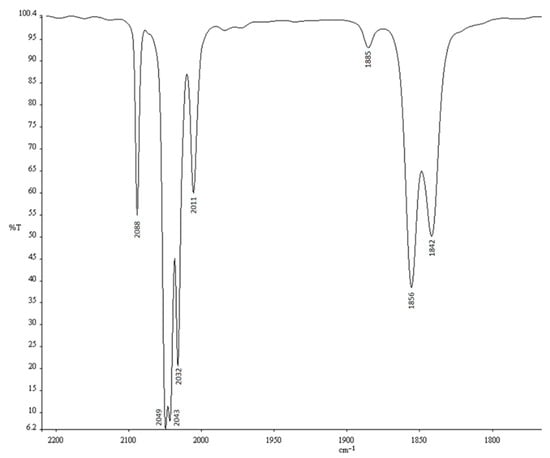
Figure 1.
IR spectrum of compound 2 recorded in cyclohexane.
The proton-decoupled 31P-NMR recorded in CDCl3 reveals a strongly broadened singlet at δ 130.4 due to the coordinated triphenylphosphite ligand, suggesting a fluxional behavior in solution (Figure 2). In line with this hypothesis, we were unable to identify at ambient temperature distinct carbonyl resonances in the proton-decoupled 13C NMR spectrum despite long data acquisition overnight. Only a broad hump centered at about δ 196 could be observed for the 11 carbonyl groups (Figure 3). It should be noted that no 31P or 13C NMR data recorded at ambient temperature on related PR3 and P(OR)3 clusters are available in the literature. There is only one report on [Co4(CO)11{P(OMe)3}] at low temperature using 13C-enriched CO, which allows for the differentiation between bridging and terminal carbonyls [19].
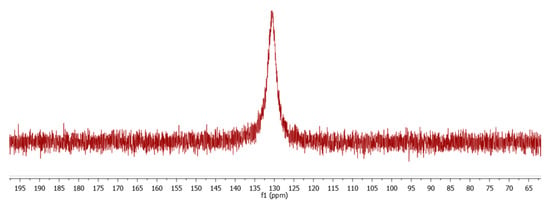
Figure 2.
31P{1H} NMR spectrum (161.99 MHz, CDCl3) of compound 2 at 25 °C.
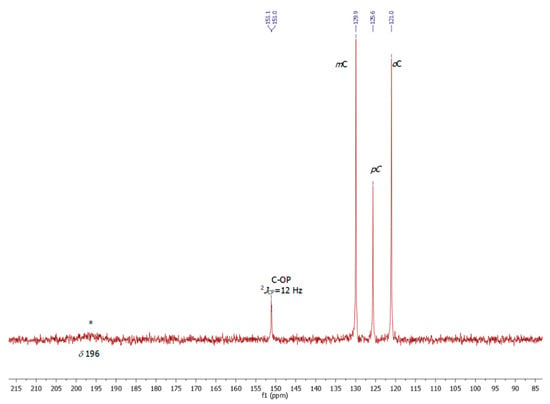
Figure 3.
13C{1H} NMR spectrum (100.62 MHz, CDCl3) of compound 2 at 25 °C.
In order to check whether [{Co4(μ2-CO)3(CO)8{P(OPh)3}] 2 is isostructural to [{Co4(μ2-CO)3(CO)8(PPh3)], we examined the product by an X-ray diffraction study performed at 100 K. Indeed, cluster 2 crystallizes like its PPh3 analogue in the monoclinic crystal system but has been refined with space group P21/c instead of the P21/n employed for the latter. Darensbourg and Incorva proposed that a monosubstituted [Co4(μ2-CO)3(CO)8L] skeleton can have a priori three isomeric motifs (Figure 4) [11].
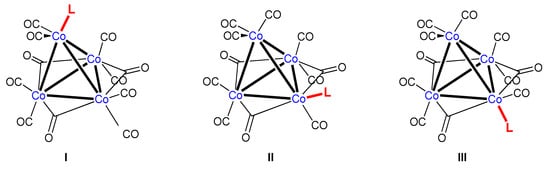
Figure 4.
Presentation of the three conceivable isomers of [{Co4(μ2-CO)3(CO)8L] with L at the apical (I), equatorial (II), or axial site (III).
As shown in Figure 5, the tetrahedral nido-shaped cluster (according to the Wade–Mingos rules) [20,21] exhibits an isomer III-type structure since the phosphite ligand occupies an axial site with respect to the basal triangular plane formed by Co1, Co2, and Co3. The Co1 center, which bears the P(OPh)3 ligand is also ligated by one terminal CO ligand and shares two symmetrically bridging carbonyls with the adjacent Co2 and Co3 centers. Co2 and Co3 in turn bear each of the two terminal Cos and have one shared edge bridged by a μ2-CO ligand. The fourth vertex of the tetrahedral core is composed of the Co4 fragment, bearing exclusively three terminal COs. The Co1–P bond is quite colinear with the Co1–Co4 vector, the angle Co4–Co1–P being 166.108(11)°. Overall, the molecular structures of 2 and [{Co4(μ2-CO)3(CO)8(PPh3)] are very similar, the corresponding Co–Co–PPh3 angle of 174.98(4)° being slightly more linear.
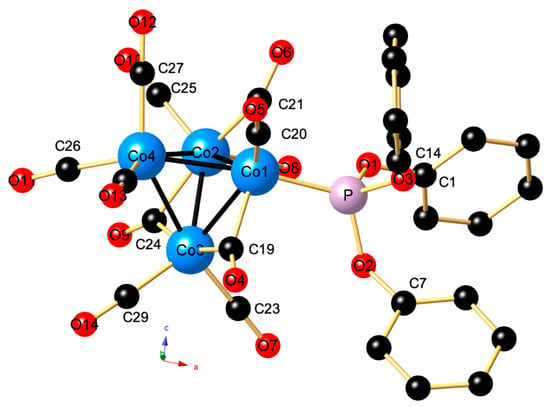
Figure 5.
Molecular structure of 2. Selected angles (deg): For clarity’s sake, all H atoms are omitted. Co1–Co2–Co3 60.008(8), Co1–Co2–Co4 60.369(9), Co1–Co3–Co2 60.284(8), Co1–Co4–Co3 58.646(7), Co1–Co4–Co2 58.796(7), Co3–Co4–Co2 58.305(7), Co4–Co1–P 166.108(11), Co3–Co1–P 107.602(11), and Co2–Co1–P 107.385(11).
Table 1 compares the most relevant bond lengths of 2 to those of [Co4(μ2-CO)3(CO)8(PR3)]. As a result, the mean Co–Co bond length of 2 is slightly shorter than that of its PPh3 and PMe3, analogues, as well as that reported for its parent compound [Co4(μ2-CO)3(CO)9] (2.4837 vs. 2.492 Å). When compared to the reported Co-P distances for [Co4(μ2-CO)3(CO)8(PR3)], that of 2 is shortened and matches the average Co–P distances of [Co4(CO)10{P(OMe)3}2] shown in Figure 1 (2.1546(4) vs. 2.158(2) Å). The mean length of the Co–C bond decreases in the following order: P(OPh)3 > PPh3 > PMe3. Notable is the observation that the Co4–C distances are elongated compared to the other ones.

Table 1.
Relevant bond lengths (Å) in 2 and crystallographically characterized monosubstituted [Co4(μ2-CO)3(CO)8(PR3)] clusters.
Inspection of the crystal structure reveals the existence of various other weak intermolecular contacts. A partial view of the crystal packing is shown in Figure 6. The shortest contact implies two carbonyl ligands, one axial and one bridging [d(C23···O9′) = 3.029 Å; symmetry code (‘) 1 − x, −½ + y, ½ − z]. An apical carbonyl also has two weak interactions with phenyl ring atoms [d(O11···C4″) = 3.073 Å; symmetry code (‘’) −1+x, y, z] and [d(H11···O11′) = 2.611 Å; symmetry code (‘) 1 − x, −½ + y, ½ − z]. There are two intermolecular C-H·π interactions observed, but because all hydrogen atoms were not freely re-fined, a discussion is not appropriate.
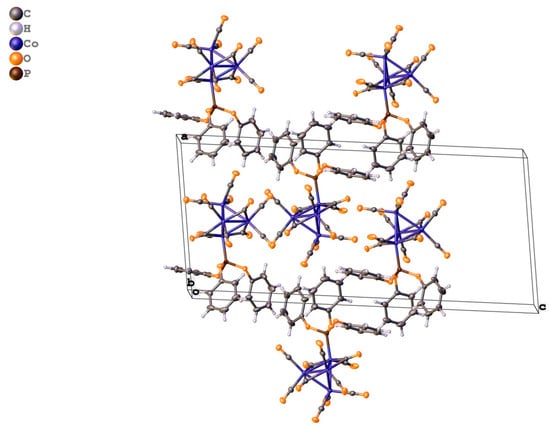
Figure 6.
OLEX-generated view of the packing of 2 along the a axis.
3. Discussion
Several methods have been described in the past to synthesize phosphine and phosphite-substituted tetranuclear clusters [Co4(μ2-CO)3(CO)9-n(PR3)n]. [{Co4(μ2-CO)3(CO)8(PMe3)] has been obtained in low yield by reaction of Me2PCl3 with Na[Co(CO)4] (12). More common is the use of a preformed tetranuclear scaffold, such as [Co4(CO)12] or [Co4(CO)9(arene)], followed by substitution with P(OR)3 or PR3 [5,8,9,10,11]. Since the elucidation of the mechanism of the formation of 2 was not the objective of this short note, we did not investigate this in detail. However, we can rule out the initial formation of [Co4(μ2-CO)3(CO)9] followed by CO/P(OPh)3 exchange, since the formation of [Co2(CO)7{P(OPh)3}] 1 is detectable by IR monitoring. We believe that cluster 2 is formed by the initial formation of [Co2(CO)7{P(OPh)3}] 1, which reacts in a thermal cluster condensation reaction with [Co2(CO)8] yielding 2. There are also IR vibrations in the reaction mixture that can be attributed to the disubstituted cluster [{Co4(μ2-CO)3(CO)3{P(OPh)3}2] [5], but we were unable to isolate the compound in pure form.
In addition, we attempted to condensate [Co2(CO)6{P(OPh)3}2] with [Co2(CO)8] in hot toluene. The formation of cluster 2 as well as [Co4(CO)10{P(OPh)3}2] and minor amounts of unreacted [Co2(CO)6{P(OPh)3}2] was revealed by spectroscopic examination of the reaction mixture. No attempts were undertaken to separate the mixture.
4. Experimental Section
P(OPh)3 (0.26 mL, 1.0 mmol) was added to a stirred solution of Co2(CO)8 (342 mg, 1.0 mmol) in toluene (5 mL). An immediate evolution of gas was observed. The reaction mixture was heated to 60 °C for 5 h. The solution was cooled to room temperature prior to lowering its temperature to 4 °C. The product 2 crystallized as dark plates collected by filtration. Yield: 39%. Anal. Calc. for C29H15Co4O14P (M.W = 854.14 g.mol−1): C, 40.78; H, 1.77%. Found: C, 40.93; H, 1.84%. 13C{1H} NMR (CDCl3) at 298 K: δ 121.0 (Co), 125.6 (Cp), 129.9 (Cm), 151.2 (Cipso-O, d, 2JPC = 12 Hz), 196 (br, CO) ppm.
For the refinement of the crystallographic data, a disorder model was applied for the heavy atoms Co2, Co3, and Co4 using an occupation ratio of 96:4.
Crystal data for C29H15Co4O14P, M = 854.10 g.mol−1, black crystals, crystal size 0.377 × 0.194 × 0.15 mm3, monoclinic, space group P21/c; a = 12.5987(10) Å, b = 8.9773(9) Å, c = 27.953(2) Å, α = 90°, β = 96.829(3)°, γ = 90°; V = 3139.2(5) Å3, Z = 4, Dcalc = 1.807 g/cm3, T = 100 K, GOF = 1.034; R1 = 0.0236, wR2 = 0.0546 for 11,958 reflections with I > = 2σ (I) and 11,958 independent reflections. Largest difference in peak/hole = Å−3 0.58/−0.41. Data were collected using graphite monochromated MoKα radiation with l = 0.71073 Å and deposited at the Cambridge Crystallographic Data Centre as CCDC 2174841. (Supplementary Materials). The data can be obtained free of charge from the Cambridge Crystallographic Data Centre via http://www.ccdc.cam.ac.uk/getstructures. The structure was solved by direct methods and refined by full-matrix least-squares against F2 (SHELXL, 2015 [22,23,24]).
5. Conclusions
We have demonstrated that direct addition of P(OR)3 to [Co2(CO)8] provides an alternative route to [{Co4(CO)11{P(OAr)3}] species, avoiding the use of quite expensive [Co4(CO)12] as starting material. We have crystallographic evidence that cluster 2 adopts a structure quite reminiscent to that reported for [{Co4(μ2-CO)3(CO)8(PPh3)] and bears the P(OPh)3 ligand at the axial site of a Co vertex.
Supplementary Materials
The following supporting information can be downloaded at online. Figure S1: IR ATR spectrum of compound 2; CIF file and Check-CIF report.
Author Contributions
Compound preparation, I.J.; X-ray data collection and data analysis, C.S. and R.S.; conceptualization, data analysis, and manuscript—writing, I.J. and M.K. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Data Availability Statement
The X-ray data are available at CCDC as stated in the paper.
Acknowledgments
We thank Stéphanie Boullanger for recording the IR and NMR spectra.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Hieber, W.; Mühlbauer, F.; Ehmann, E.A. Derivate des Kobalt-und Nickelcarbonyls (XVI. Mitteil. über Metallcarbonyle). Ber. Der Dtsch. Chem. Ges. A B Ser. 1932, 65, 1090–1101. [Google Scholar] [CrossRef]
- Chini, P. The Closed Metal Carbonyl Clusters. Inorg. Chim. Acta 1968, 2, 31–51. [Google Scholar] [CrossRef]
- Wei, C.H. Structural Analyses of tetracobalt Dodecarbonyl and tetrarhodium Dodecarbonyl. Crystallographic Treatments of a disordered Structure and a Twinned Composite. Inorg. Chem. 1969, 8, 2384–2397. [Google Scholar] [CrossRef]
- Farrugia, L.J.; Braga, D.; Grepioni, F. A structural redetermination of Co4(CO)12: Evidence for dynamic disorder and the pathway of metal atom migration in the crystalline phase. J. Organomet. Chem. 1999, 573, 60–66. [Google Scholar] [CrossRef]
- Sisak, A.; Sisak, C.; Ungvary, F.; Palyi, G.; Marko, L. Some chemical properties of (arene)nonacarbonylteracobalt complexes and the preparation of (arene)octacarbonyltetracobalt trialkylphosphites. J. Organomet. Chem. 1975, 90, 77–83. [Google Scholar] [CrossRef]
- Bor, G.; Sbrignadello, G.; Marcati, F. Infrared Spectroscopic Studies on Metal Carbonyl Compounds: XV. Assignments of the C-O stretching Infrared Spectra and Force Constants of some (arene)Co4(CO)9 Complexes. J. Organomet. Chem. 1972, 46, 357–368. [Google Scholar] [CrossRef]
- Fliedel, C.; Pattacini, R.; Braunstein, P. Mono-, Di- and Tetranuclear Complexes and Clusters with Bromine-functionalized Bis(diphenylphosphino)amine Ligands. J. Clust. Sci. 2010, 21, 397–415. [Google Scholar] [CrossRef]
- Labroue, D.; Poilblanc, R. Trivalent Phosphorus Derivatives of Cobalt Carbonyls. I. Infrared and NMR Studies of new Substituted Tetracobaltdecacarbonyl Complexes. Inorg. Chim. Acta 1972, 6, 387–390. [Google Scholar] [CrossRef]
- Darensbourg, D.J.; Incorvia, M.J. Ligand Substitution Processes in Tetranuclear Carbonyl Clusters. I. Co4(CO)9(μ2-CO)3 derivatives. J. Organomet. Chem. 1979, 171, 89–96. [Google Scholar] [CrossRef]
- Darensbourg, D.J.; Incorvia, M.J. Ligand Substitution Processes in Tetranuclear Carbonyl Clusters. 2. Co4(CO)9(μ2-CO)3 derivatives. Inorg. Chem. 1980, 19, 2585–2590. [Google Scholar] [CrossRef]
- Darensbourg, D.J.; Incorvia, M.J. Ligand Substitution Processes in Tetranuclear Carbonyl Clusters. 3. Molecular Structures of compound Co4(CO)8(μ-CO)3[P(C6H5)3] and Co4(CO)7(μ-CO)3[P(OCH3)3]2. Inorg. Chem. 1981, 20, 1911–1918. [Google Scholar] [CrossRef]
- Bartl, K.; Boese, R.; Schmid, G. Heteronucleare Clustersysteme XIX. Strukturuntersuchungen an tetraedrischen Carbonylcobalt-clustern; ein Beitrag zur Frage der Bildung von Carbonylbrücken. J. Organomet. Chem. 1981, 206, 331–345. [Google Scholar] [CrossRef]
- Macchi, P.; Garlaschelli, L.; Martinengo, S.; Sironi, A. Charge Density in Transition Metal Clusters: Supported vs Unsupported Metal-Metal Interactions. J. Am. Chem. Soc. 1999, 121, 10428–10429. [Google Scholar] [CrossRef]
- Keller, E.; Vahrenkamp, H. Eisen- und Cobalt-Cluster nach der Propen-Eliminierungs-Methode. Die Cluster Co4(CO)10[P(CH3)2(CH2CHCH2)]2 und Fe2Co(CO)8[μ-SCH3]2[μ-P(CH3)2]. Chem. Ber. 1981, 114, 1111–1123. [Google Scholar] [CrossRef]
- Clément, S.; Guyard, L.; Khatyr, A.; Knorr, M.; Rousselin, Y.; Kubicki, M.M.; Mugnier, Y.; Richeter, S.; Gerbier, P.; Strohmann, C. Synthesis, crystallographic and electrochemical study of ethynyl [2.2] paracyclophane-derived cobalt metallatetrahedranes. J. Organomet. Chem. 2012, 699, 56–66. [Google Scholar] [CrossRef]
- Jourdain, I.; Knorr, M.; Brieger, L.; Strohmann, C. Synthesis of Tris(arylphosphite)-ligated Cobalt(0) Complexes [Co2(CO)6{P(OAr)3}2], and their Reactivity towards Phenylacetylene and Diphenylacetylene. Adv. Chem. Res. 2020, 2. [Google Scholar] [CrossRef]
- Szabo, P.; Fekete, L.; Nagy-Magos, Z.; Marko, L. Phosphorus-containing cobalt carbonyls. III. Monosubstituted derivatives of dicobaltoctacarbonyl with phosphine and phosphites. J. Organomet. Chem. 1968, 12, 245–248. [Google Scholar] [CrossRef]
- Haumann, M.; Meijboom, R.; Moss, J.R.; Roodt, A. Synthesis, crystal structure and hydroformylation activity of triphenylphosphite modified cobalt catalysts. Dalton Trans. 2004, 4, 1679–1686. [Google Scholar] [CrossRef]
- Cohen, M.A.; Kidd, D.R.; Brown, T.L. The structures of Co4(CO)12 and Co4(CO)11[P(OCH3)3] in solution. J. Am. Chem. Soc. 1975, 97, 4408–4409. [Google Scholar] [CrossRef]
- Wade, K. Structural and bonding patterns in cluster chemistry. Adv. Inorg. Chem. Radiochem. 1976, 18, 1–66. [Google Scholar] [CrossRef]
- Mingos, D.M.P. Polyhedral skeletal electron pair approach. Acc. Chem. Res. 1984, 17, 311–319. [Google Scholar] [CrossRef]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.; Puschmann, H. OLEX2: A complete structure solution, refinement and analysis program. J. Appl. Cryst. 2009, 42, 339–341. [Google Scholar] [CrossRef]
- Sheldrick, G. SHELXT-Integrated space-group and crystal-structure determination. Acta Crystallogr. A 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Sheldrick, G. Crystal structure refinement with SHELXL. Acta Crystallogr. C 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).