


1-{(1S,2S,4R)-7,7-Dimethyl-1-[(pyrrolidin-1-yl)methyl]bicyclo [2.2.1]heptan-2-yl}-1H-benzo[d]imidazole




{kind=link}
{kind=link}
Abstract
1. Introduction
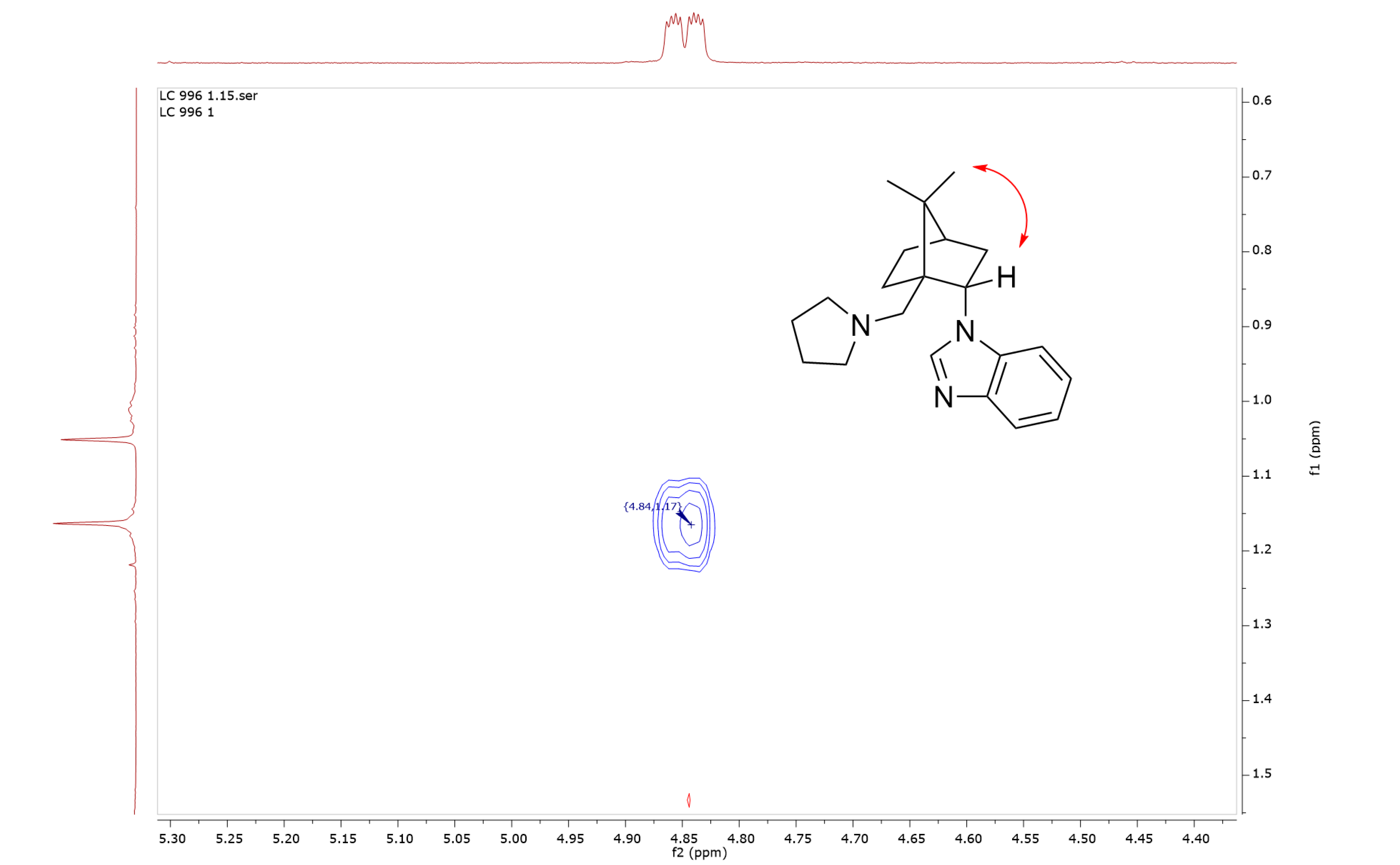
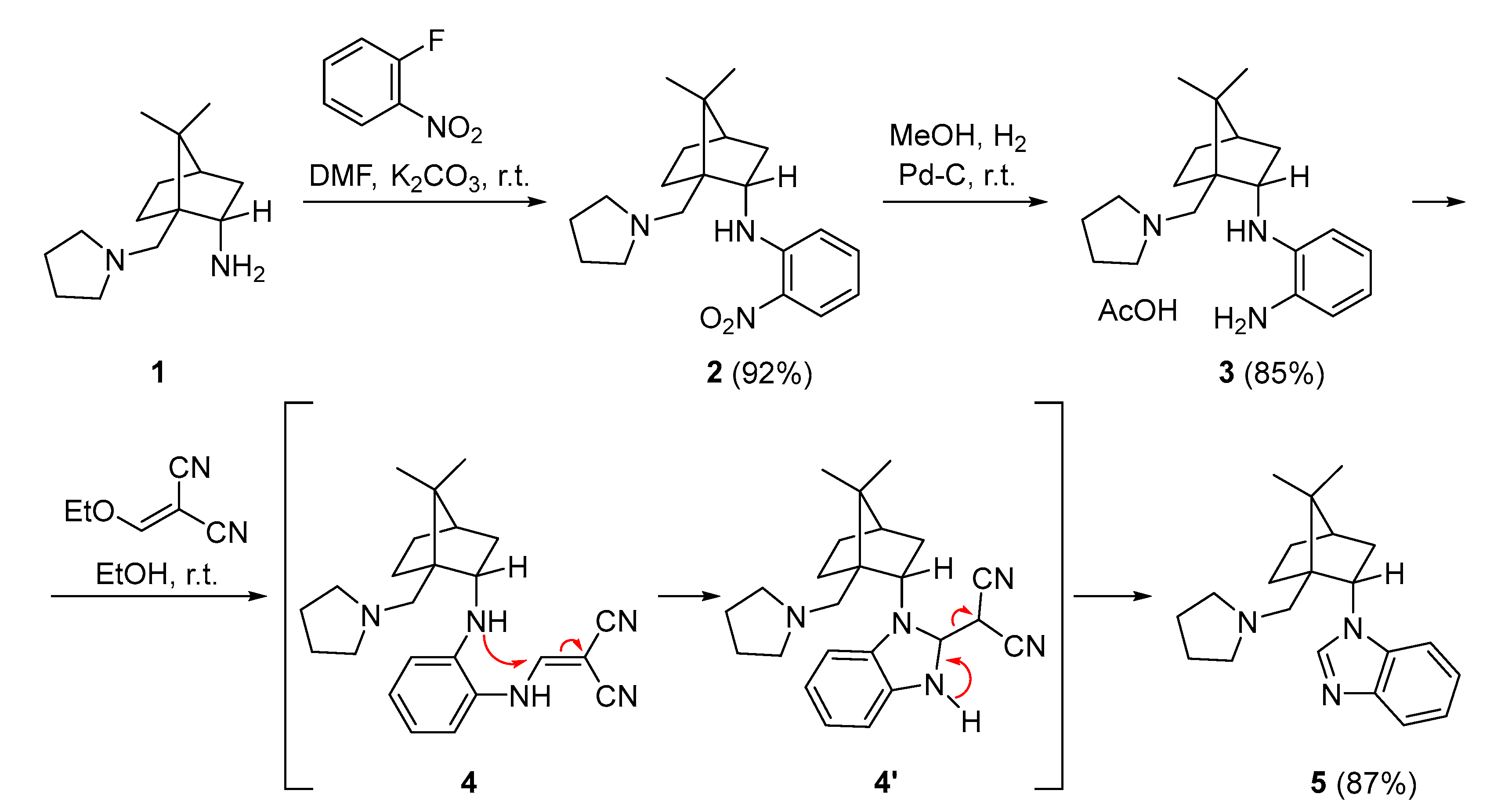
2. Results and Discussion
3. Materials and Methods
3.1. Synthesis of (1S,2S,4R)-7,7-Dimethyl-N-(2-nitrophenyl)-1-[(pyrrolidin-1-yl)methyl]bicyclo[2.2.1]heptan-2-amine (2)
3.2. Synthesis of N1-{(1S,2S,4R)-7,7-Dimethyl-1-[(pyrrolidin-1-yl)methyl]bicyclo[2.2.1]heptan-2-yl}benzene-1,2-diamine (3)
3.3. Synthesis of 1-{(1S,2S,4R)-7,7-Dimethyl-1-[(pyrrolidin-1-yl)methyl]bicyclo[2.2.1]heptan-2-yl}-1H-benzo[d]imidazole (5)
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Torres, R.R. (Ed.) Stereoselective Organocatalysis: Bond Formation Methodologies and Activation Modes, 1st ed.; JohnWiley & Sons: Hoboken, NJ, USA, 2013. [Google Scholar]
- List, B. Asymmetric Organocatalysis 1. Lewis Base and Acid Catalysis. In Science of Synthesis; Georg Thieme Verlag KG: Stuttgart, Germany, 2012. [Google Scholar]
- Dalko, P.I. Comprehensive Enantioselective Organocatalysis: Catalysts, Reactions, and Applications; Wiley-VCH: Weinheim, Germany, 2013. [Google Scholar]
- Pellissier, H. Asymmetric organocatalysis. Tetrahedron 2007, 63, 9267–9331. [Google Scholar] [CrossRef]
- List, B.; MacMillan, D.W.C. For the Development of Asymmetric Organocatalysis. Available online: https://www.nobelprize.org/prizes/chemistry/2021/advanced-information/ (accessed on 30 November 2022).
- De Figueiredo, R.M.; Christmann, M. Organocatalytic Synthesis of Drugs and Bioactive Natural Products. Eur. J. Org. Chem. 2007, 2007, 2575–2600. [Google Scholar] [CrossRef]
- Sun, B.-F. Total synthesis of natural and pharmaceutical products powered by organocatalytic reactions. Tetrahedron Lett. 2015, 56, 2133–2140. [Google Scholar] [CrossRef]
- Okino, T.; Hoashi, Y.; Takemoto, Y. Enantioselective Michael Reaction of Malonates to Nitroolefins Catalyzed by Bifunctional Organocatalysts. J. Am. Chem. Soc. 2003, 125, 12672–12673. [Google Scholar] [CrossRef] [PubMed]
- Malerich, J.P.; Hagihara, K.; Rawal, V.H. Chiral Squaramide Derivatives are Excellent Hydrogen Bond Donor Catalysts. J. Am. Chem. Soc. 2008, 130, 14416–14417. [Google Scholar] [CrossRef] [PubMed]
- Rombola, M.; Sumaria, C.S.; Montgomery, T.D.; Rawal, V.H. Development of Chiral, Bifunctional Thiosquaramides: Enantioselective Michael Additions of Barbituric Acids to Nitroalkenes. J. Am. Chem. Soc. 2017, 139, 5297–5300. [Google Scholar] [CrossRef]
- Alemán, J.; Parra, A.; Jiang, H.; Jørgensen, K.A. Squaramides: Bridging from Molecular Recognition to Bifunctional Organocatalysis. Chem. Eur. J. 2011, 17, 6890–6899. [Google Scholar] [CrossRef] [PubMed]
- Ričko, S.; Svete, J.; Štefane, B.; Perdih, A.; Golobič, A.; Meden, A.; Grošelj, U. 1,3-Diamine-Derived Bifunctional Organocatalyst Prepared from Camphor. Adv. Synth. Catal. 2016, 358, 3786–3796. [Google Scholar] [CrossRef]
- Ričko, S.; Meden, A.; Ivančič, A.; Perdih, S.; Štefane, B.; Svete, J.; Grošelj, U. Organocatalyzed Deracemization of Δ2-Pyrrolin-4-ones. Adv. Synth. Catal. 2017, 359, 2288–2296. [Google Scholar] [CrossRef]
- Ciber, L.; Požgan, F.; Brodnik, H.; Štefane, B.; Svete, J.; Grošelj, U. Synthesis and Catalytic Activity of Organocatalysts Based on Enaminone and Benzenediamine Hydrogen Bond Donors. Catalysts 2022, 12, 1132. [Google Scholar] [CrossRef]
- Grošelj, U. Camphor-Derivatives in Asymmetric Organocatalysis—Synthesis and Application. Curr. Org. Chem. 2015, 19, 2048–2074. [Google Scholar] [CrossRef]
- Singer, R.A.; Ginsburg, P.H. Process for The Preparation Of 1,3-Substituted Indenes and Aryl-Fusedazapolycyclic Componunds. U.S. Patent 2003/0060624 A1, 27 March 2003. [Google Scholar]
- Faheem, M.; Rathaur, A.; Pandey, A.; Kumar Singh, V.; Tiwari, A.K. A Review on the Modern Synthetic Approach of Benzimidazole Candidate. ChemistrySelect 2020, 5, 3981–3994. [Google Scholar] [CrossRef]
- Zhu, X.; Zhang, F.; Kuang, D.; Deng, G.; Yang, Y.; Yu, J.; Liang, Y. K2S as Sulfur Source and DMSO as Carbon Source for the Synthesis of 2-Unsubstituted Benzothiazoles. Org. Lett. 2020, 22, 3789–3793. [Google Scholar] [CrossRef] [PubMed]
- Ryabukhin, S.V.; Plaskon, A.S.; Volochnyuk, D.M.; Tolmachev, A.A. Synthesis of Fused Imidazoles and Benzothiazoles from (Hetero)Aromatic ortho-Diamines or ortho-Aminothiophenol and Aldehydes Promoted by Chlorotrimethylsilane. Synthesis 2006, 21, 3715–3726. [Google Scholar] [CrossRef]
- Bahrami, K.; Khodaei, M.M.; Naali, F. Mild and Highly Efficient Method for the Synthesis of 2-Arylbenzimidazoles and 2-Arylbenzothiazoles. J. Org. Chem. 2008, 73, 6835–6837. [Google Scholar] [CrossRef] [PubMed]
- Bastug, G.; Eviolitte, C.; Markó, I.E. Functionalized Orthoesters as Powerful Building Blocks for the Efficient Preparation of Heteroaromatic Bicycles. Org. Lett. 2012, 14, 3502–3505. [Google Scholar] [CrossRef] [PubMed]
- Nale, D.B.; Bhanage, B.M. N-Substituted Formamides as C1-Sources for the Synthesis of Benzimidazole and Benzothiazole Derivatives by Using Zinc Catalysts. Synlett 2015, 26, 2835–2842. [Google Scholar] [CrossRef]
- Wang, Y.; Sarris, K.; Sauer, D.R.; Djuric, S.W. A simple and efficient one step synthesis of benzoxazoles and benzimidazoles from carboxylic acids. Tetrahedron Lett. 2006, 47, 4823–4826. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ciber, L.; Požgan, F.; Svete, J.; Štefane, B.; Grošelj, U. 1-{(1S,2S,4R)-7,7-Dimethyl-1-[(pyrrolidin-1-yl)methyl]bicyclo [2.2.1]heptan-2-yl}-1H-benzo[d]imidazole. Molbank 2023, 2023, M1538. https://doi.org/10.3390/M1538
Ciber L, Požgan F, Svete J, Štefane B, Grošelj U. 1-{(1S,2S,4R)-7,7-Dimethyl-1-[(pyrrolidin-1-yl)methyl]bicyclo [2.2.1]heptan-2-yl}-1H-benzo[d]imidazole. Molbank. 2023; 2023(1):M1538. https://doi.org/10.3390/M1538
Chicago/Turabian StyleCiber, Luka, Franc Požgan, Jurij Svete, Bogdan Štefane, and Uroš Grošelj. 2023. "1-{(1S,2S,4R)-7,7-Dimethyl-1-[(pyrrolidin-1-yl)methyl]bicyclo [2.2.1]heptan-2-yl}-1H-benzo[d]imidazole" Molbank 2023, no. 1: M1538. https://doi.org/10.3390/M1538
APA StyleCiber, L., Požgan, F., Svete, J., Štefane, B., & Grošelj, U. (2023). 1-{(1S,2S,4R)-7,7-Dimethyl-1-[(pyrrolidin-1-yl)methyl]bicyclo [2.2.1]heptan-2-yl}-1H-benzo[d]imidazole. Molbank, 2023(1), M1538. https://doi.org/10.3390/M1538