3.1. Chemistry
Reagents were purchased from Sigma-Aldrich (3050 Spruce Street St. Louis, MO, 63103, USA), Fluorochem (Unit 14 Graphite Way, Hadfield, Glossop SK13 1QH, UK), Fisher Scientific (168 3rd Ave, Waltham, MA 02451, USA), or TCI chemicals (9211 North Harborgate Street, Portland, OR 97203, USA) and used without further purification. Microwave reactions were performed using monomode reactors: Biotage Initiator® classic (Uppsala, Sweden) in sealed vials with output power from 0 to 400 W. The following adsorbent was used for column chromatography: silica gel 60 (Merck KgaA, Darmstadt, Germany, particle size 0.063–0.200 mm, 70–230 mesh ASTM). Reaction monitoring of intermediary compounds 2, 2′, and 3 was performed either using aluminum TLC plates (5 × 5 cm) with silica gel coated 60F-254 (Merck) in an appropriate eluent and visualized using ultraviolet light in a UV-Lamp VL-6.CL., 254 nm (6 W) and 365 nm (6 W) or using an LC-MS apparatus Thermo Scientific Accela High Speed LC System® coupled to a Thermo MSQ Plus® quadrupole mass spectrometer, with an HPLC column Thermo Hypersil Gold® (168 3rd Ave, Waltham, MA 02451, USA) 50 × 2.1 mm (C18 bounded), with particles of a diameter of 1.9 mm. The volume of sample injected into the column was 1 µL. Chromatographic analysis, total duration of 8 min, was on the gradient of the following solvents: t = 0 min, methanol/water 50:50; 0 < t < 4 min, linear increase in the proportion of methanol to a methanol/water ratio of 95:5; 4 < t < 6 min, methanol/water 95:5; 6 < t < 7 min, linear decrease in the proportion of methanol to return to a methanol/water ratio of 50:50; 6 < t < 7 min, methanol/water 50:50. The water used was buffered with ammonium acetate 5 mM. The flow rate of the mobile phase was 0.3 mL/min.
Low-resolution mass spectra were recorded for products 2, 2′, 3, and A, in an Agilent SQ G6120B mass spectrometer (5301 Stevens Creek Blvd, Santa Clara, CA 95051, USA) in positive and negative electrospray mode using liquid chromatography with Diode-Array Detection at 254 nM, column Agilent Poroshell (5301 Stevens Creek Blvd, Santa Clara, CA 95051, USA) 120 EC-C18 2.7 µm (4.6 × 50 mm), mobile phase (A: H2O + 0.1% Formic acid, B: MeCN + 0.1% Formic acid), method flow rate 0.5 mL/min, time/%B 0/10, 5/100, 9/100, at the Faculté de Pharmacie of Marseille.
The high-resolution mass spectrum of compound A was recorded on an SYNAPT G2 HDMS (Waters, 34 Maple St., Milford, MA 01757, USA) equipped with a pneumatically assisted atmospheric pressure ionization (API) source. The sample was ionized in positive electrospray mode under the following conditions: electrospray voltage: 2.8 kV; orifice voltage: 20 V; nebulizing gas flow rate (nitrogen): 100 L/h. The sample was dissolved in 300µL of dichloromethane and then diluted 1:103 in a solution of methanol with 3 mM ammonium acetate. The extract solution was introduced into the ionization source via infusion at a flow rate of 10 µL/min. Exact mass measurement was performed in triplicate with external calibration. HRMS was performed at the Faculté des Sciences de Saint-Jérôme (Marseille).
NMR spectra were recorded on a Bruker Avance NEO 400 MHz NanoBay spectrometer at the Faculté de Pharmacie of Marseille. Residual 1H and 13C peaks in deuterated solvents (CDCl3 and DMSO-d6) were used for chemical shift calibration without the need for an additional internal standard. 1H NMR: reference CDCl3 δ = 7.26 ppm, reference DMSO-d6 δ = 2.50 ppm and 13C NMR: reference CDCl3 δ = 77.16 ppm, reference DMSO-d6 δ = 39.52 ppm. Data for 1H NMR are reported as follows: chemical shifts (δ) in parts per million (ppm), multiplicity (described as follows: s, singlet; bs, broad singlet; d, doublet; t, triplet; q, quadruplet; dd, doublet of doublet; ddd, doublet of doublet of doublet; m, multiplet), coupling constants (J) in Hertz (Hz) and integration. Data for 13C NMR are reported as follows: chemical shifts (δ) in parts per million (ppm).
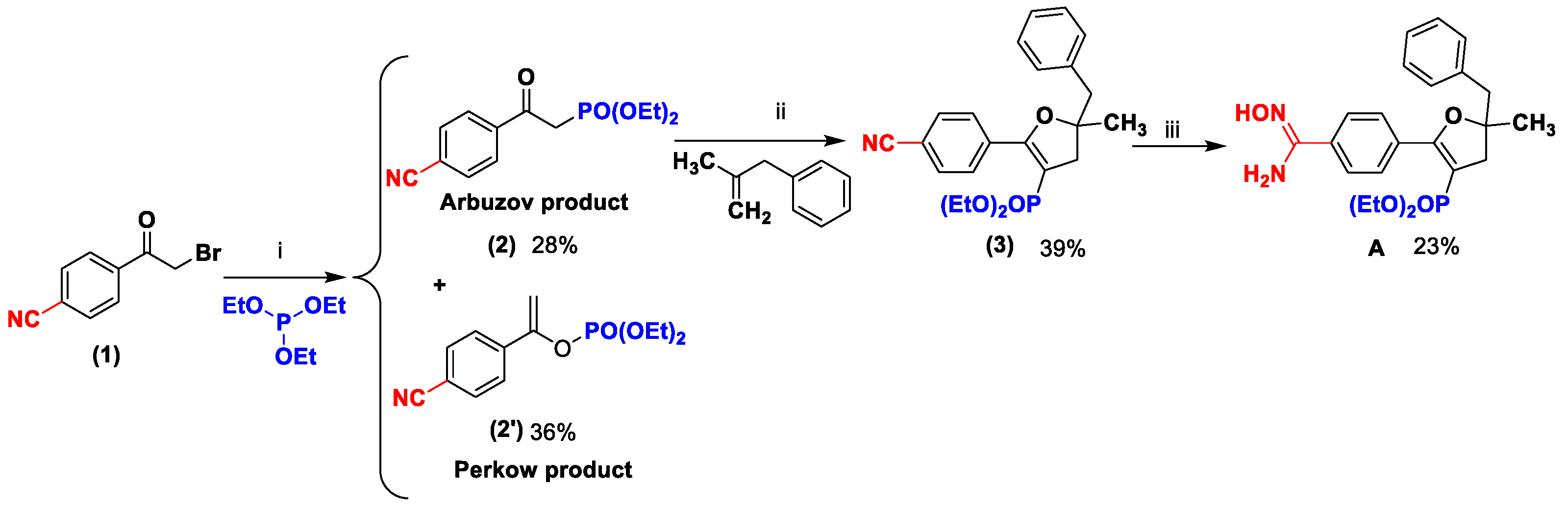
Diethyl (2-(4-cyanophenyl)-2-oxoethyl)phosphonate (2)
The Michaelis–Arbuzov reaction was performed in a round-bottom flask dried, mixing 4-(2-bromoacetyl)benzonitrile (2 g, 8.92 mmol, 1 equiv.) and triethyl phosphite (1 equiv.) and heated at 120 °C for 18 h. The TLC monitoring reaction was performed using DCM-AcOEt (4:1) as eluent and visualized with ultraviolet light in a UV-Lamp VL-6.CL., 254 nm (6 W), with a retardation factor of 0.28, and verified via low-resolution LC-MS. The mixture was allowed to cool down to room temperature and concentrated in vacuo. The crude product was purified via column chromatography (silica gel; eluent: dichloromethane/cyclohexane/AcOEt 80/10/10) affording the title product. Yield 28% (678 mg). The product was obtained in the keto form principally, with traces of enol form as an orange oily solid. The NMR data were in agreement with the literature values [
13,
14] (
Supplementary Materials).
1H NMR (400 MHz, CDCl
3): δ (ppm) 8.12 (d,
3JH-H = 8.8 Hz, 2H, 2CH
Ar), 7.78 (d,
3JH-H = 8.8 Hz, 2H, 2CH
Ar), 4.18–4.09 (m, 4H, 2CH
2), 3.63 (d,
3JH-H = 22.9 Hz, 2H, CH
2), 1.28 (t,
3JH-H = 7.1 Hz, 6H, 2CH
3).
13C NMR (100 MHz, CDCl
3): δ (ppm) 190.9 (d,
JP-C = 6.6 Hz, C), 139.5 (C), 132.6 (2CH
Ar), 129.6 (2CH
Ar), 117.9 (C), 117.0 (C), 63.1 (d,
JP-C = 6.8 Hz, 2CH
2), 39.2 (d,
JP-C = 131.5 Hz, CH
2), 16.3 (d,
JP-C = 6.0 Hz, 2CH
3). C
13H
16NO
4P: LC/MS ESI+ t
R 4.45 min, (
m/
z) [M + H]
+ 281.25/282.70.
1-(4-Cyanophenyl)vinyl diethyl phosphate (2′)
The Perkow product was achieved with the same method to obtain 2. The TLC monitoring reaction was performed using DCM-AcOEt (4:1) as eluent and visualized with ultraviolet light in a UV-Lamp VL-6.CL., 254 nm (6 W), with a retardation factor of 0.63, and verified via low-resolution LC-MS. The crude product was purified via column chromatography (silica gel; eluent: dichloromethane/cyclohexane/AcOEt 40/50/10) affording the title product. Yield 36% (875 mg). The product was obtained as a brown oily solid. 1H NMR (400 MHz, CDCl3): δ (ppm) 7.72–7.62 (m, 4H, 4CHAr), 5.45–5.38 (m, 2H, CH2), 4.29–4.15 (m, 4H, 2CH2), 1.35 (td, JH-H = 1.2 Hz, JH-H = 7.2 Hz, 6H, 2CH3). 13C NMR (100 MHz, CDCl3): δ (ppm) 150.7 (d, J = 7.4 Hz, C), 138.7 (d, J = 6.6 Hz, C), 132.4 (2CHAr), 125.9 (2CHAr). C13H16NO4P: LC/MS ESI+ tR 5.10 min, (m/z) [M + H]+ 281.25/282.60.
Diethyl (5-benzyl-2-(4-(N′-hydroxycarbamimidoyl)phenyl)-5-methyl-4,5-dihydrofuran-3-yl)phosphonate (3)
In a microwave vial of 20 mL equipped with a stirring bar, a solution of manganese (III) acetate dihydrate (2.1 equiv.) and copper (II) acetate (1 equiv.) in 12 mL of glacial acetic acid was heated at 80 °C under microwave irradiation for 15 min. Then, the reaction mixture was cooled and compound 2 (300 mg, 1.07 mmol, 1 equiv.) and 2-methyl-3-phenyl-1-propene (2 equiv.) in 13 mL of acetic acid was added. The reaction mixture was heated for 2.5 h under microwave irradiation under the same conditions. The TLC monitoring reaction was performed using DCM-AcOEt (4:1) as eluent and visualized with ultraviolet light in a UV-Lamp VL-6.CL., 254 nm and 365 nm (6 W), with a retardation factor of 0.58, and verified via low-resolution LC-MS. The resulting product was poured into 50 mL of cold water and extracted with dichloromethane (3 × 40 mL). The organic extracts were collected and washed with saturated aqueous NaHCO3 (3 × 40 mL) and dried over Na2SO4. The solvent was evaporated under reduced pressure, and the crude product was purified via column chromatography (silica gel; eluent: dichloromethane/MeOH 98/2) affording the title product as a yellow oily solid, Yield 39% (171 mg). 1H NMR (400 MHz, CDCl3) δ (ppm) 7.91 (d, 3JH-H = 8.4 Hz, 2H, 2CHAr), 7.65 (d, 3JH-H = 8.4 Hz, 2H, 2CHAr), 7.31–7.19 (m, 5H, CH), 3.97–3.68 (m, 4H, 2CH2), 3.06 (dd, 4JH-H = 3.4 Hz, 2JH-H = 15.3 Hz, 1H, H-(CH2)), 3.00 (dd, 4JH-H = 14.1 Hz, 2JH-H = 40.1 Hz, 2H, H-(CH2)), 2.80 (dd, 4JH-H = 3.3 Hz, 2JH-H = 15.5 Hz, 1H, H-(CH2)), 1.50 (s, 3H, CH3), 1.18 (t, 3JH-H = 7.0 Hz, 3H, CH3), 1.13 (t, 3JH-H = 7.0 Hz, 3H, CH3). 13C NMR (100 MHz, CDCl3) δ (ppm) 161.7 (d, J = 26.2 Hz, C), 136.5 (C), 134.5 (C), 131.7 (2CHAr), 130.6 (2CHAr), 129.6 (2CHAr), 128.4 (2CHAr), 127.0 (CHAr), 118.7 (C), 113.6 (C), 97.3 (d, J = 213.8 Hz, C), 88.1 (d, J = 11.8 Hz, C), 61.7 (t, J = 6.3 Hz, 2CH2), 46.8 (CH2), 44.3 (d, J = 8.6 Hz, CH2), 27.1 (CH3), 16.3 (t, J = 6.4 Hz, 2CH3). 31P NMR (161.9 MHz, CDCl3) δ (ppm) 17.0. C23H26NO4P: LC/MS ESI+ tR 6.23 min, (m/z) [M + H] + 412.51. [M + H]+ 411.16/411.60.
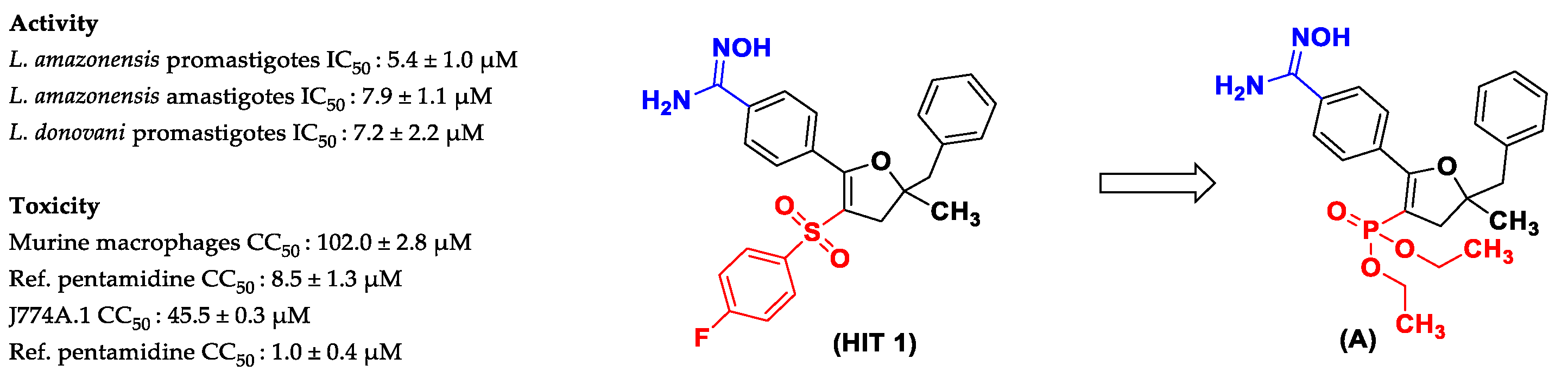
Diethyl (5-benzyl-2-(4-(N′-hydroxycarbamimidoyl)phenyl)-5-methyl-4,5-dihydrofuran-3-yl)phosphonate (A)
A suspension of hydroxylamine hydrochloride (10 equiv.) in DMSO was stirred under inert atmosphere and cooled to 0 °C. Potassium tert-butoxide (10 equiv.) was added gradually, and the reaction mixture was stirred for 30 min. Then, compound 3 was added (160 mg, 0.38 mmol, 1 equiv.), and the reaction mixture was stirred for 18 h at room temperature. The TLC monitoring reaction was performed using DCM-MeOH (95:5) as eluent and visualized with ultraviolet light in a UV-Lamp VL-6.CL., 254 and 365 nm (6 W), with a retardation factor of 0.55, and verified via low-resolution LC-MS. The resulting mixture was poured into cold water. Then, the reaction mixture was extracted with EtOAc (3 × 15 mL), and the organic layers were combined, washed with water (1 × 20 mL), brine (1 × 20 mL), dried over Na2SO4, and concentrated. The crude product was purified via column chromatography (eluent: dichloromethane/MeOH 98/2), Rf 0.32. Yield 23% (40 mg). The product was obtained as a yellow solid and verified via HRMS. Mp 110–111 °C. 1H NMR (400 MHz, DMSO) δ (ppm) 9.82 (s, 1H, OH), 7.77–7.66 (m, 4H, 4CHAr), 7.33–7.17 (m, 5H, 5CHAr), 5.96 (br s, 2H, NH2), 3.83–3.68 (m, 2H, CH2), 3.65–3.54 (m, 2H, CH2), 3.01 (q, JH-H = 16.5 Hz, 2H, CH2), 2.93 (dd, 4JH-H = 2.9 Hz, 2JH-H = 15.2 Hz, 1H, H-(CH2)), 2.70 (dd, 4JH-H = 2.8 Hz, 2JH-H = 15.3 Hz, 1H, H-(CH2)), 1.44 (s, 3H, CH3), 1.08 (t, 3JH-H = 7.0 Hz, 3H, CH3), 1.01 (t, 3JH-H = 7.0 Hz, 3H, CH3). 13C NMR (100 MHz, CDCl3) δ (ppm) 163.5 (d, J = 26.2 Hz, C), 152.5 (C), 136.6 (C), 133.6 (C), 131.8 (C), 130.6 (2CHAr), 129.2 (2CHAr), 128.3 (2CHAr), 126.8 (CHAr), 125.4 (2CHAr), 94.8 ((d, J = 214.7 Hz, C), 87.6 (d, J = 11.8 Hz, C), 61.6 (t, J = 6.3 Hz, 2CH2), 46.8 (CH2), 44.3 (d, J = 8.6 Hz, CH2), 27.1 (CH3), 16.3 (t, J = 6.4 Hz, 2CH3). 31P NMR (161.9 MHz, CDCl3) δ (ppm) 17.0. C23H29N2O5P: LC/MS ESI+ tR 5.14 min, (m/z) [M + H]+ 444.47/445.90; HRMS: m/z [M + H]+ calculated 445.1887; found 445.1884.


 ,
, 
{kind=link}
{kind=link}

