
Synthesis and Characterization of Novel Indazole–Sulfonamide Compounds with Potential MAPK1 Inhibitory Activity for Cancer Treatment


Abstract
:1. Introduction
2. Results
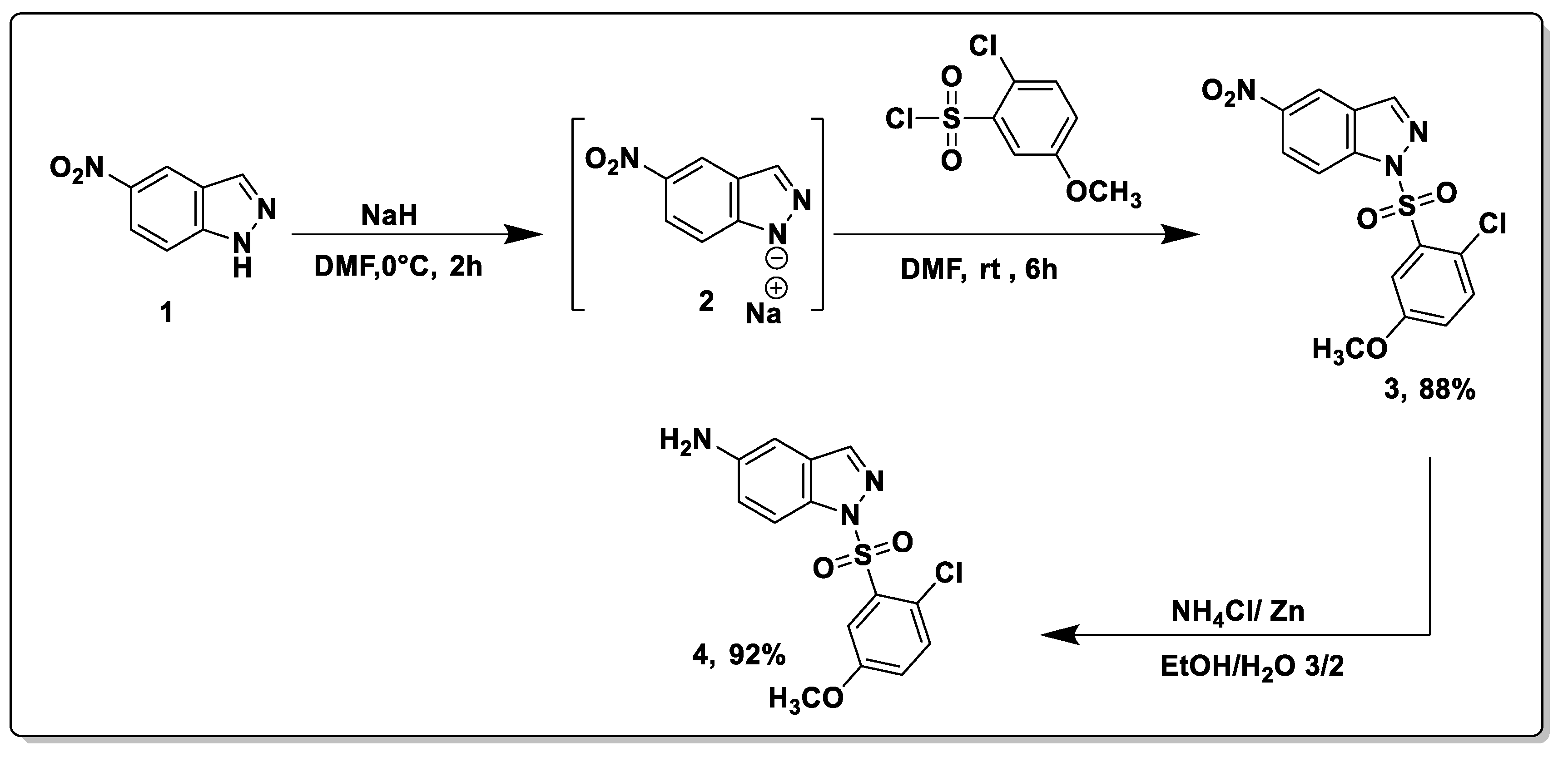
2.1. Synthesis
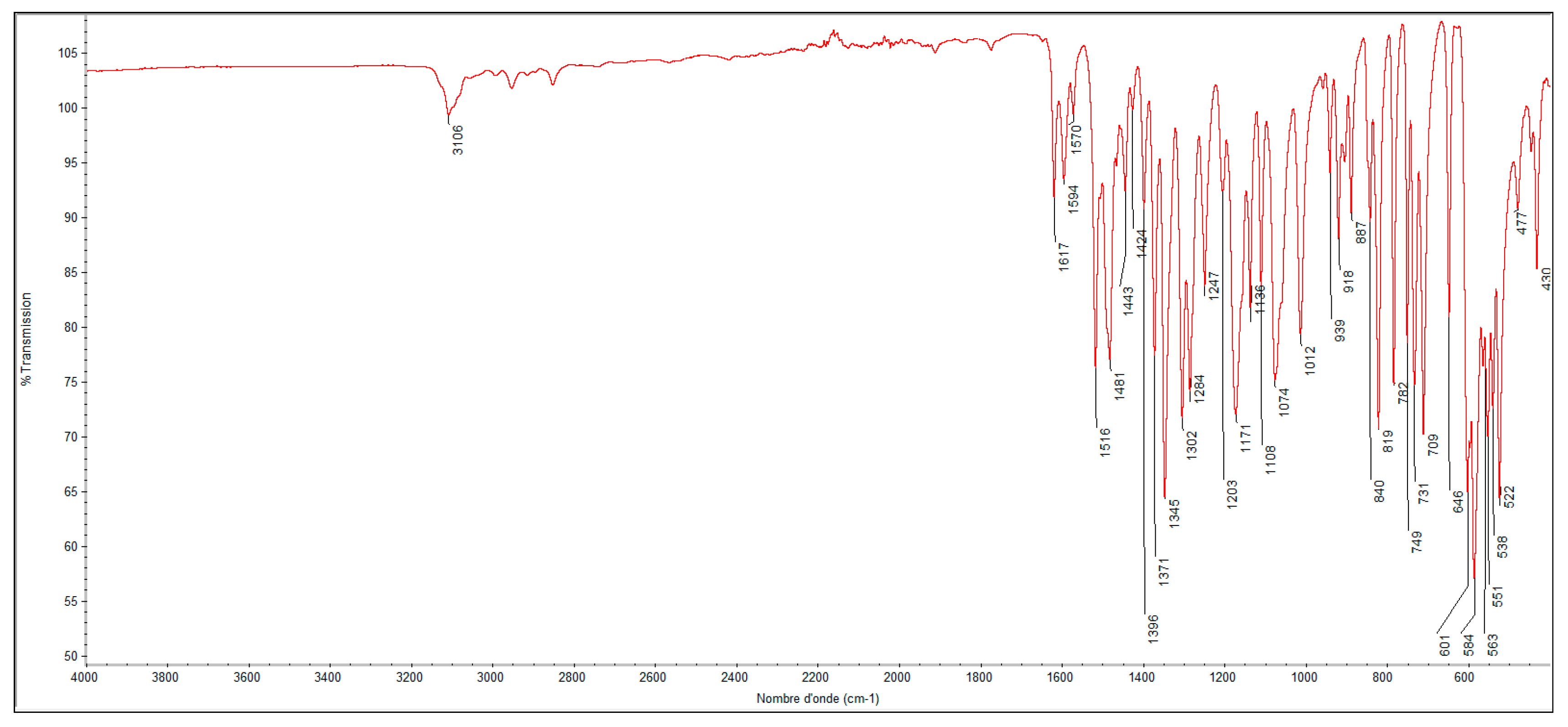
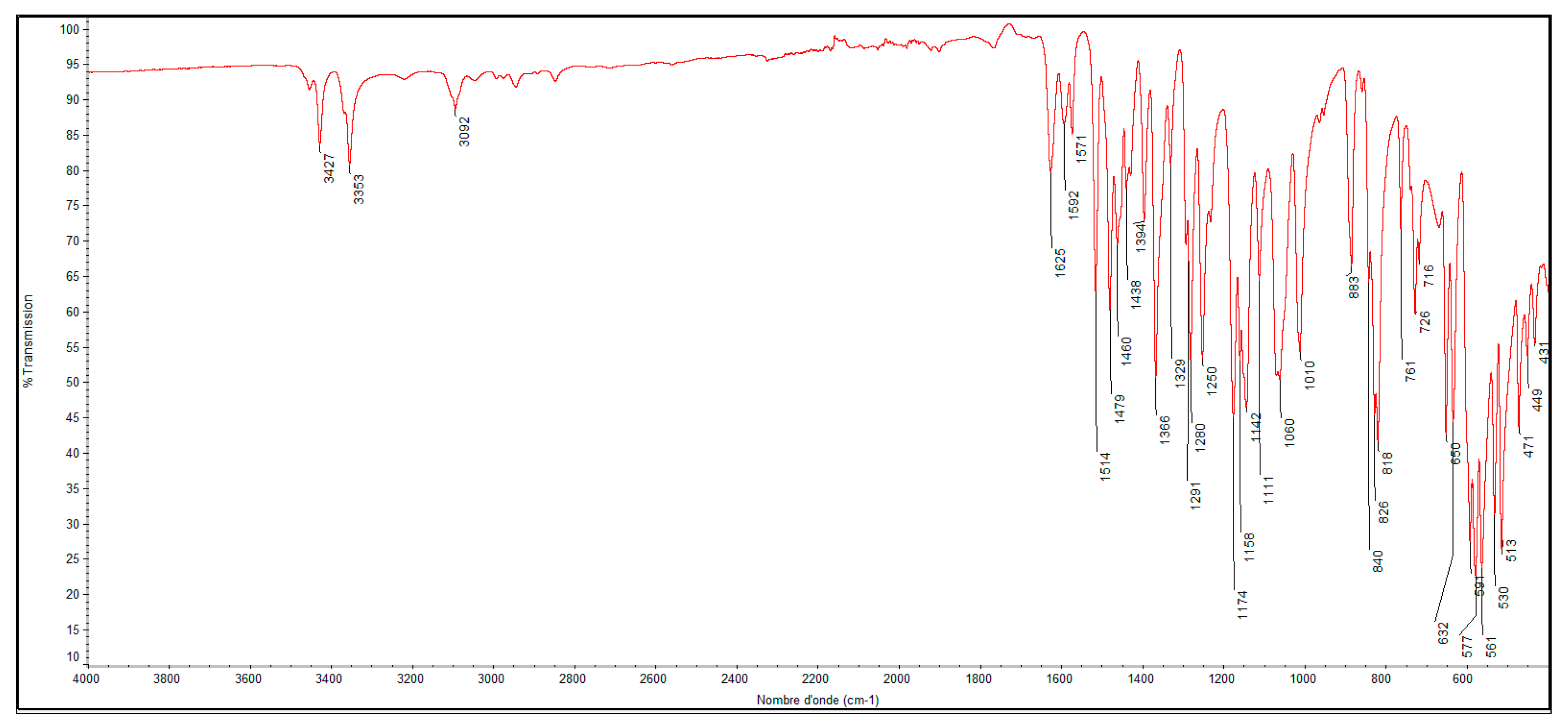
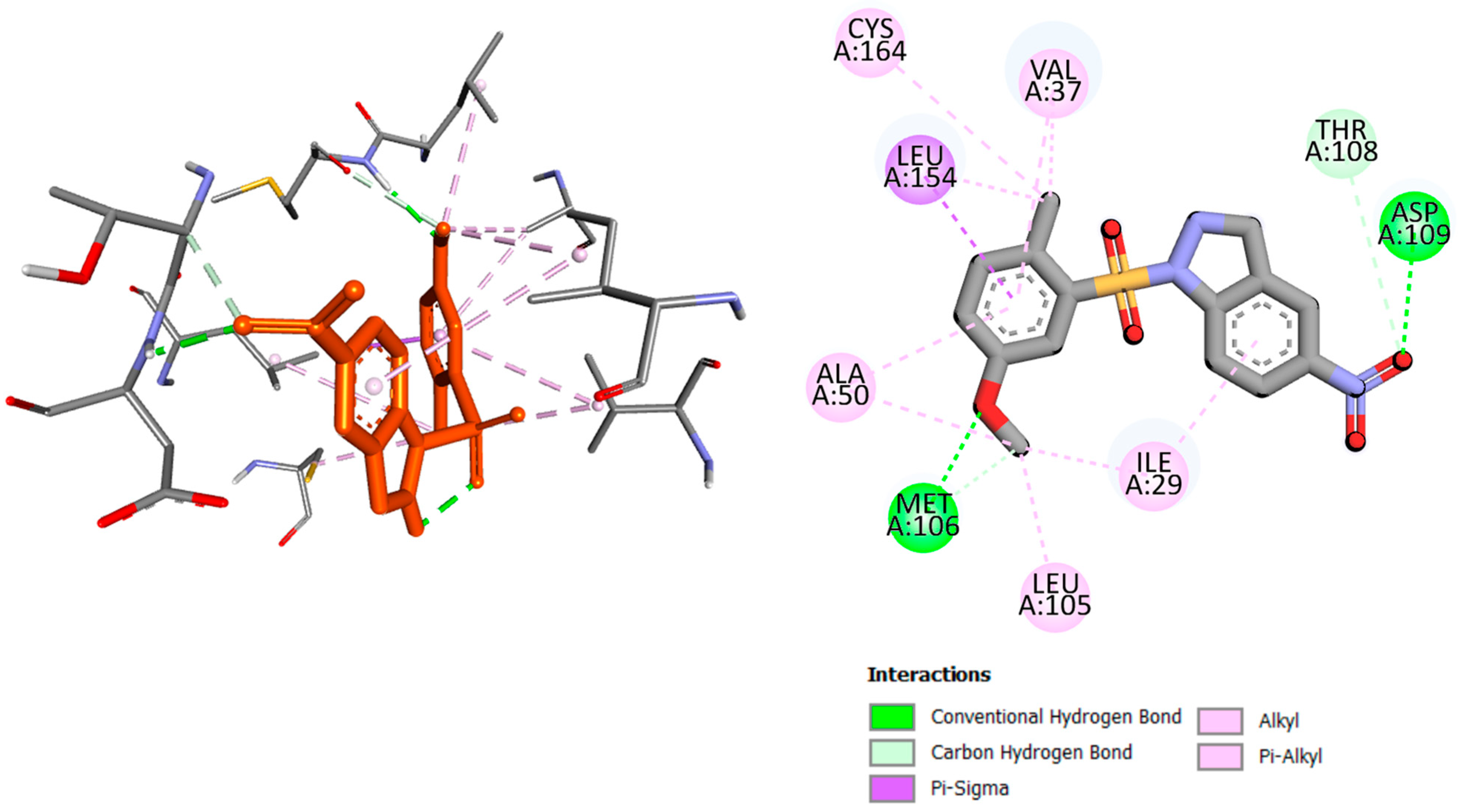
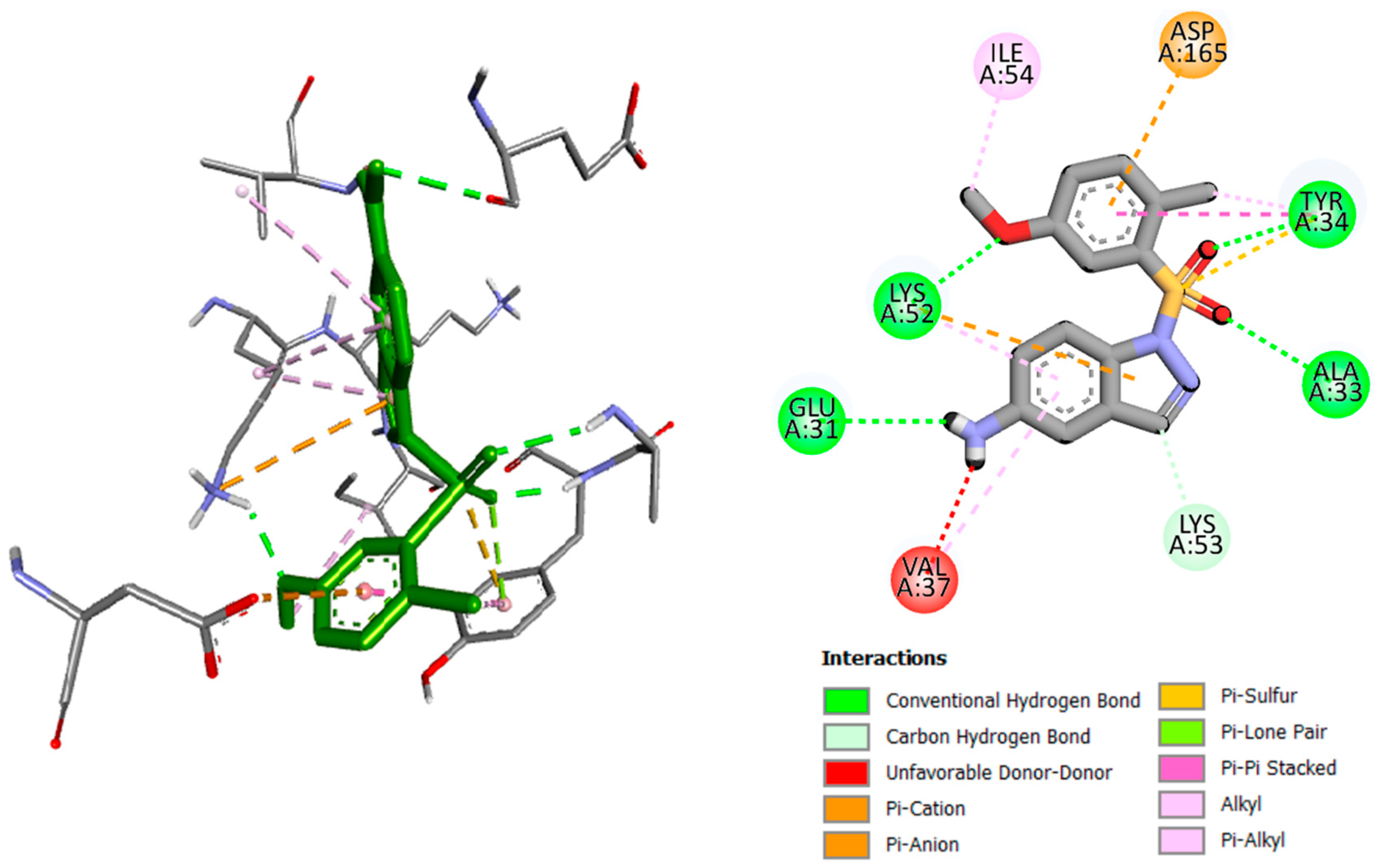
2.2. Molecular Docking
3. Materials and Methods
3.1. General Methods
3.2. General Synthetic Procedures
3.3. Protein Retrieval and Preparation
3.4. Ligands Preparation
3.5. Docking
3.6. Visualization
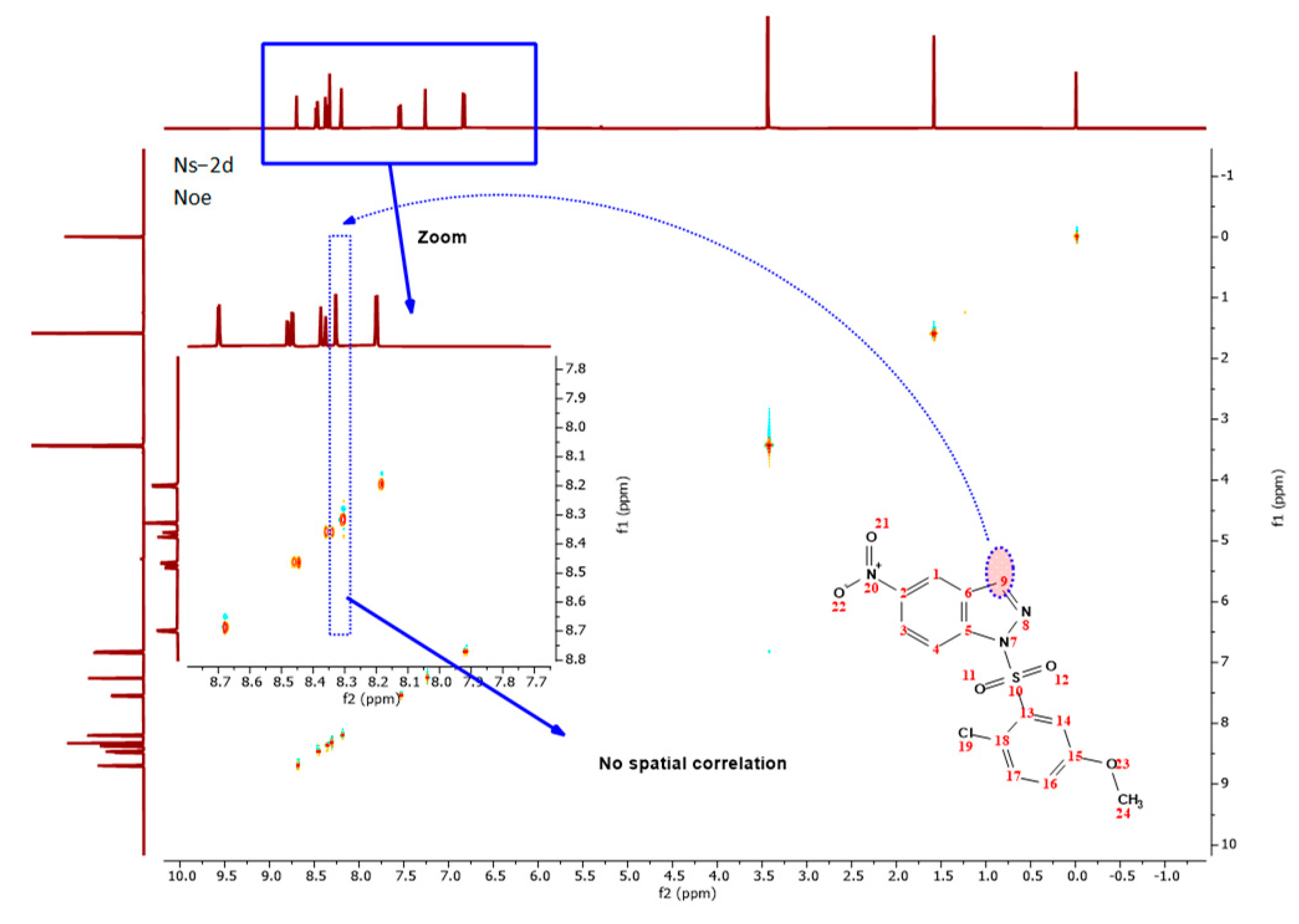
3.7. NOE Experiment
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Amewu, R.K.; Sakyi, P.O.; Osei-Safo, D.; Addae-Mensah, I. Synthetic and Naturally Occurring Heterocyclic Anticancer Compounds with Multiple Biological Targets. Molecules 2021, 26, 7134. [Google Scholar] [CrossRef] [PubMed]
- Vitaku, E.; Smith, D.T.; Njardarson, J.T. Analysis of the Structural Diversity, Substitution Patterns, and Frequency of Nitrogen Heterocycles among U.S. FDA Approved Pharmaceuticals: Miniperspective. J. Med. Chem. 2014, 57, 10257–10274. [Google Scholar] [CrossRef] [PubMed]
- Santos, A.S.; Raydan, D.; Cunha, J.C.; Viduedo, N.; Silva, A.M.S.; Marques, M.M.B. Advances in Green Catalysis for the Synthesis of Medicinally Relevant N-Heterocycles. Catalysts 2021, 11, 1108. [Google Scholar] [CrossRef]
- Bourzikat, O.; El Abbouchi, A.; Ghammaz, H.; El Brahmi, N.; El Fahime, E.; Paris, A.; Daniellou, R.; Suzenet, F.; Guillaumet, G.; El Kazzouli, S. Synthesis, Anticancer Activities and Molecular Docking Studies of a Novel Class of 2-Phenyl-5,6,7,8-Tetrahydroimidazo [1,2-b]Pyridazine Derivatives Bearing Sulfonamides. Molecules 2022, 27, 5238. [Google Scholar] [CrossRef] [PubMed]
- Gambouz, K.; Abbouchi, A.E.; Nassiri, S.; Suzenet, F.; Bousmina, M.; Akssira, M.; Guillaumet, G.; El Kazzouli, S. Palladium-Catalyzed Oxidative Arylation of 1 H-Indazoles with Arenes. Eur. J. Org. Chem. 2020, 2020, 7435–7439. [Google Scholar] [CrossRef]
- Wan, Y.; He, S.; Li, W.; Tang, Z. Indazole Derivatives: Promising Anti-Tumor Agents. ACAMC 2019, 18, 1228–1234. [Google Scholar] [CrossRef]
- Dong, J.; Zhang, Q.; Wang, Z.; Huang, G.; Li, S. Recent Advances in the Development of Indazole-based Anticancer Agents. ChemMedChem 2018, 13, 1490–1507. [Google Scholar] [CrossRef]
- Pérez-Villanueva, J.; Yépez-Mulia, L.; González-Sánchez, I.; Palacios-Espinosa, J.; Soria-Arteche, O.; Sainz-Espuñes, T.; Cerbón, M.; Rodríguez-Villar, K.; Rodríguez-Vicente, A.; Cortés-Gines, M.; et al. Synthesis and Biological Evaluation of 2H-Indazole Derivatives: Towards Antimicrobial and Anti-Inflammatory Dual Agents. Molecules 2017, 22, 1864. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Chu, S.; Feher, V.A.; Khalili, M.; Nie, Z.; Margosiak, S.; Nikulin, V.; Levin, J.; Sprankle, K.G.; Tedder, M.E.; et al. Structure-Based Design, Synthesis, and Antimicrobial Activity of Indazole-Derived SAH/MTA Nucleosidase Inhibitors. J. Med. Chem. 2003, 46, 5663–5673. [Google Scholar] [CrossRef]
- Kim, S.-H.; Markovitz, B.; Trovato, R.; Murphy, B.R.; Austin, H.; Willardsen, A.J.; Baichwal, V.; Morham, S.; Bajji, A. Discovery of a New HIV-1 Inhibitor Scaffold and Synthesis of Potential Prodrugs of Indazoles. Bioorg. Med. Chem. Lett. 2013, 23, 2888–2892. [Google Scholar] [CrossRef]
- Degnan, A.P.; Tora, G.O.; Huang, H.; Conlon, D.A.; Davis, C.D.; Hanumegowda, U.M.; Hou, X.; Hsiao, Y.; Hu, J.; Krause, R.; et al. Discovery of Indazoles as Potent, Orally Active Dual Neurokinin 1 Receptor Antagonists and Serotonin Transporter Inhibitors for the Treatment of Depression. ACS Chem. Neurosci. 2016, 7, 1635–1640. [Google Scholar] [CrossRef] [PubMed]
- Tan, C.; Yang, S.-J.; Zhao, D.-H.; Li, J.; Yin, L.-Q. Antihypertensive Activity of Indole and Indazole Analogues: A Review. Arab. J. Chem. 2022, 15, 103756. [Google Scholar] [CrossRef]
- Shang, C.; Hou, Y.; Meng, T.; Shi, M.; Cui, G. The Anticancer Activity of Indazole Compounds: A Mini Review. Curr. Top. Med. Chem. 2021, 21, 363–376. [Google Scholar] [CrossRef]
- López-Vallejo, F.; Castillo, R.; Yépez-Mulia, L.; Medina-Franco, J.L. Benzotriazoles and Indazoles Are Scaffolds with Biological Activity against Entamoeba Histolytica. SLAS Discov. 2011, 16, 862–868. [Google Scholar] [CrossRef]
- Tzvetkov, N.T.; Hinz, S.; Küppers, P.; Gastreich, M.; Müller, C.E. Indazole- and Indole-5-Carboxamides: Selective and Reversible Monoamine Oxidase B Inhibitors with Subnanomolar Potency. J. Med. Chem. 2014, 57, 6679–6703. [Google Scholar] [CrossRef]
- El Abbouchi, A.; El Brahmi, N.; Hiebel, M.-A.; Bignon, J.; Guillaumet, G.; Suzenet, F.; El Kazzouli, S. Synthesis and Biological Evaluation of Ethacrynic Acid Derivatives Bearing Sulfonamides as Potent Anti-Cancer Agents. Bioorg. Med. Chem. Lett. 2020, 30, 127426. [Google Scholar] [CrossRef] [PubMed]
- Boztaş, M.; Çetinkaya, Y.; Topal, M.; Gülçin, İ.; Menzek, A.; Şahin, E.; Tanc, M.; Supuran, C.T. Synthesis and Carbonic Anhydrase Isoenzymes I, II, IX, and XII Inhibitory Effects of Dimethoxybromophenol Derivatives Incorporating Cyclopropane Moieties. J. Med. Chem. 2015, 58, 640–650. [Google Scholar] [CrossRef] [PubMed]
- Ballatore, C.; Huryn, D.M.; Smith, A.B. Carboxylic Acid (Bio)Isosteres in Drug Design. ChemMedChem 2013, 8, 385–395. [Google Scholar] [CrossRef]
- Cao, X.; Sun, Z.; Cao, Y.; Wang, R.; Cai, T.; Chu, W.; Hu, W.; Yang, Y. Design, Synthesis, and Structure–Activity Relationship Studies of Novel Fused Heterocycles-Linked Triazoles with Good Activity and Water Solubility. J. Med. Chem. 2014, 57, 3687–3706. [Google Scholar] [CrossRef]
- Feng, Z.; Lu, X.; Gan, L.; Zhang, Q.; Lin, L. Xanthones, a Promising Anti-Inflammatory Scaffold: Structure, Activity, and Drug Likeness Analysis. Molecules 2020, 25, 598. [Google Scholar] [CrossRef]
- Gao, F.; Wang, T.; Xiao, J.; Huang, G. Antibacterial Activity Study of 1,2,4-Triazole Derivatives. Eur. J. Med. Chem. 2019, 173, 274–281. [Google Scholar] [CrossRef] [PubMed]
- Scozzafava, A.; Owa, T.; Mastrolorenzo, A.; Supuran, C. Anticancer and Antiviral Sulfonamides. Curr. Med. Chem. 2003, 10, 925–953. [Google Scholar] [CrossRef]
- Rodés, B.; Sheldon, J.; Toro, C.; Jiménez, V.; Álvarez, M.Á.; Soriano, V. Susceptibility to Protease Inhibitors in HIV-2 Primary Isolates from Patients Failing Antiretroviral Therapy. J. Antimicrob. Chemother. 2006, 57, 709–713. [Google Scholar] [CrossRef]
- Gao, F.; Zhang, X.; Wang, T.; Xiao, J. Quinolone Hybrids and Their Anti-Cancer Activities: An Overview. Eur. J. Med. Chem. 2019, 165, 59–79. [Google Scholar] [CrossRef] [PubMed]
- Wan, Y.; Long, J.; Gao, H.; Tang, Z. 2-Aminothiazole: A Privileged Scaffold for the Discovery of Anti-Cancer Agents. Eur. J. Med. Chem. 2021, 210, 112953. [Google Scholar] [CrossRef] [PubMed]
- Hong, L.; Wang, Y.; Chen, W.; Yang, S. MicroRNA-508 Suppresses Epithelial-Mesenchymal Transition, Migration, and Invasion of Ovarian Cancer Cells through the MAPK1/ERK Signaling Pathway. J. Cell. Biochem. 2018, 119, 7431–7440. [Google Scholar] [CrossRef]
- Wang, M.; Liao, Q.; Zou, P. PRKCZ-AS1 Promotes the Tumorigenesis of Lung Adenocarcinoma via Sponging miR-766-5p to Modulate MAPK1. Cancer Biol. Ther. 2020, 21, 364–371. [Google Scholar] [CrossRef]
- Hamadneh, L.; Bahader, M.; Abuarqoub, R.; AlWahsh, M.; Alhusban, A.; Hikmat, S. PI3K/AKT and MAPK1 Molecular Changes Preceding Matrix Metallopeptidases Overexpression during Tamoxifen-Resistance Development Are Correlated to Poor Prognosis in Breast Cancer Patients. Breast Cancer 2021, 28, 1358–1366. [Google Scholar] [CrossRef]
- Guo, Y.J.; Pan, W.W.; Liu, S.B.; Shen, Z.F.; Xu, Y.; Hu, L.L. ERK/MAPK signalling pathway and tumorigenesis. Exp. Ther. Med. 2020, 19, 1997–2007. [Google Scholar] [CrossRef]
- Wang, Y.; Guo, Z.; Tian, Y.; Cong, L.; Zheng, Y.; Wu, Z.; Shan, G.; Xia, Y.; Zhu, Y.; Li, X.; et al. MAPK1 Promotes the Metastasis and Invasion of Gastric Cancer as a Bidirectional Transcription Factor. BMC Cancer 2023, 23, 959. [Google Scholar] [CrossRef]
- Aronov, A.M.; Baker, C.; Bemis, G.W.; Cao, J.; Chen, G.; Ford, P.J.; Germann, U.A.; Green, J.; Hale, M.R.; Jacobs, M.; et al. Flipped out: Structure-Guided Design of Selective Pyrazolylpyrrole ERK Inhibitors. J. Med. Chem. 2007, 50, 1280–1287. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
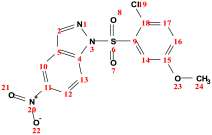 | ||||
|---|---|---|---|---|
| δ (ppm) | Integration | Multiplicity | Coupling Constant (Hz) | Assignment |
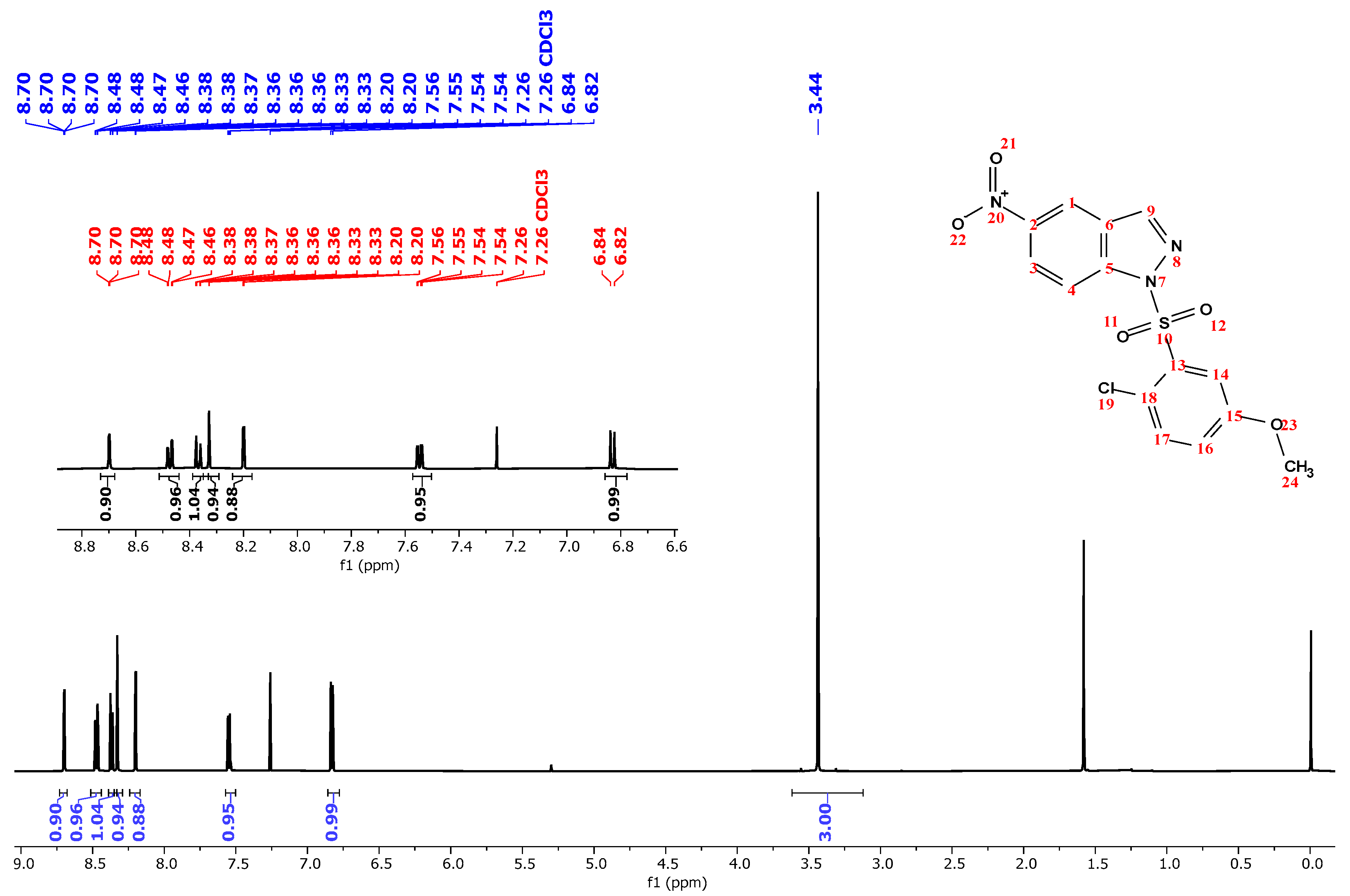
| 3.44 | 3 | s | − | 3H (24) |
| 6.83 | 1 | d | 8.9 | H (17) |
| 7.55 | 1 | dd | 8.9, 2.6 | H (16) |
| 8.20 | 1 | d | 2.2 | H (14) |
| 8.33 | 1 | s | − | H (2) |
| 8.37 | 1 | d | 8.9 | H (13) |
| 8.47 | 1 | dd | 8.9, 2.6 | H (12) |
| 8.69 | 1 | d | 2.6 | H (10) |
| 56.32 | − | − | − | C24 |
| 102.14 | − | − | − | C14 |
| 113.81 | − | − | − | C13 |
| 114.74 | − | − | − | C10 |
| 118.30 | − | − | − | C16 |
| 123.68 | − | − | − | C12 |
| 124.56 | − | − | − | C18 |
| 126.31 | − | − | − | C5 |
| 131.10 | − | − | − | C17 |
| 136.64 | − | − | − | C9 |
| 141.42 | − | − | − | C4 |
| 143.66 | − | − | − | C2 |
| 144.56 | − | − | − | C11 |
| 156.26 | − | − | − | C15 |
 | ||||
|---|---|---|---|---|
| δ (ppm) | Integration | Multiplicity | Coupling Constant (Hz) | Assignment |
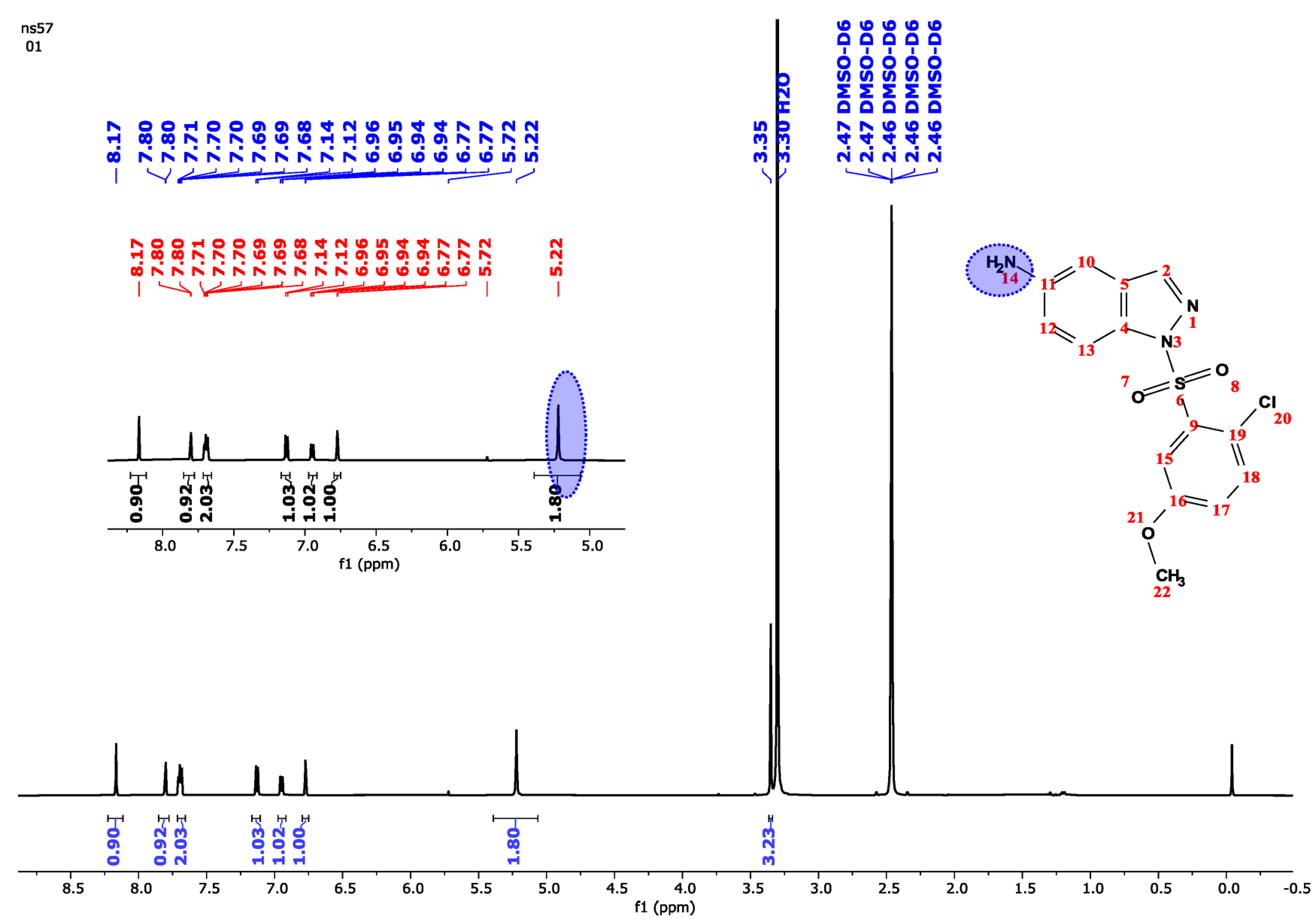
| 3.35 | 3 | s | − | 3H (22) |
| 5.22 | 2 | Bs | − | 2H (14) |
| 6.77 | 1 | d | 9 | H (18) |
| 6.89 | 1 | d | 2.2 | H (15) |
| 7.00 | 1 | dd | 9, 2.2 | H (17) |
| 7.45 | 1 | dd | 9, 2.2 | H (12) |
| 7.96–8.00 | 2 | m | − | 2H (2, 10) |
| 8.13 | 1 | d | 9 | H (13) |
| 57.97 | − | − | − | C22 |
| 102.14 | − | − | − | C10 |
| 114.08 | − | − | − | C15 |
| 116.12 | − | − | − | C13 |
| 119.70 | − | − | − | C12 |
| 124.56 | − | − | − | C17 |
| 126.77 | − | − | − | C19 |
| 126.92 | − | − | − | C5 |
| 129.49 | − | − | − | C4 |
| 134.69 | − | − | − | C18 |
| 136.58 | − | − | − | C9 |
| 141.96 | − | − | − | C2 |
| 146.34 | − | − | − | C11 |
| 156.71 | − | − | − | C16 |
| N° | Name | Molecular Wight (g/mol) | 3D Structure | B.E. (Kcal/mol) |
|---|---|---|---|---|
| 3 | 1-((2-chloro-5-methoxyphenyl) sulfonyl)-5-nitro-1H indazole | 367.76 | 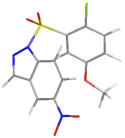 | −7.55 |
| 4 | 1-((2-chloro-5-methoxyphenyl) sulfonyl)-1H-indazol-5-amine | 337.02 | 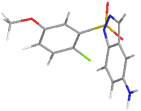 | −8.34 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Saghdani, N.; Chihab, A.; El Brahmi, N.; El Kazzouli, S. Synthesis and Characterization of Novel Indazole–Sulfonamide Compounds with Potential MAPK1 Inhibitory Activity for Cancer Treatment. Molbank 2024, 2024, M1858. https://doi.org/10.3390/M1858
Saghdani N, Chihab A, El Brahmi N, El Kazzouli S. Synthesis and Characterization of Novel Indazole–Sulfonamide Compounds with Potential MAPK1 Inhibitory Activity for Cancer Treatment. Molbank. 2024; 2024(3):M1858. https://doi.org/10.3390/M1858
Chicago/Turabian StyleSaghdani, Nassima, Abdelali Chihab, Nabil El Brahmi, and Saïd El Kazzouli. 2024. "Synthesis and Characterization of Novel Indazole–Sulfonamide Compounds with Potential MAPK1 Inhibitory Activity for Cancer Treatment" Molbank 2024, no. 3: M1858. https://doi.org/10.3390/M1858
APA StyleSaghdani, N., Chihab, A., El Brahmi, N., & El Kazzouli, S. (2024). Synthesis and Characterization of Novel Indazole–Sulfonamide Compounds with Potential MAPK1 Inhibitory Activity for Cancer Treatment. Molbank, 2024(3), M1858. https://doi.org/10.3390/M1858