Genetic Diversity of Northern Wheatgrass (Elymus lanceolatus ssp. lanceolatus) as Revealed by Genotyping-by-Sequencing

 ,
, 

Abstract
:1. Introduction
2. Materials and Methods
2.1. Plant Materials
2.2. Genotyping-by-Sequencing
2.3. Bioinformatics Analysis
2.4. Genetic Diversity Analysis
2.5. Flow Cytometry Analysis
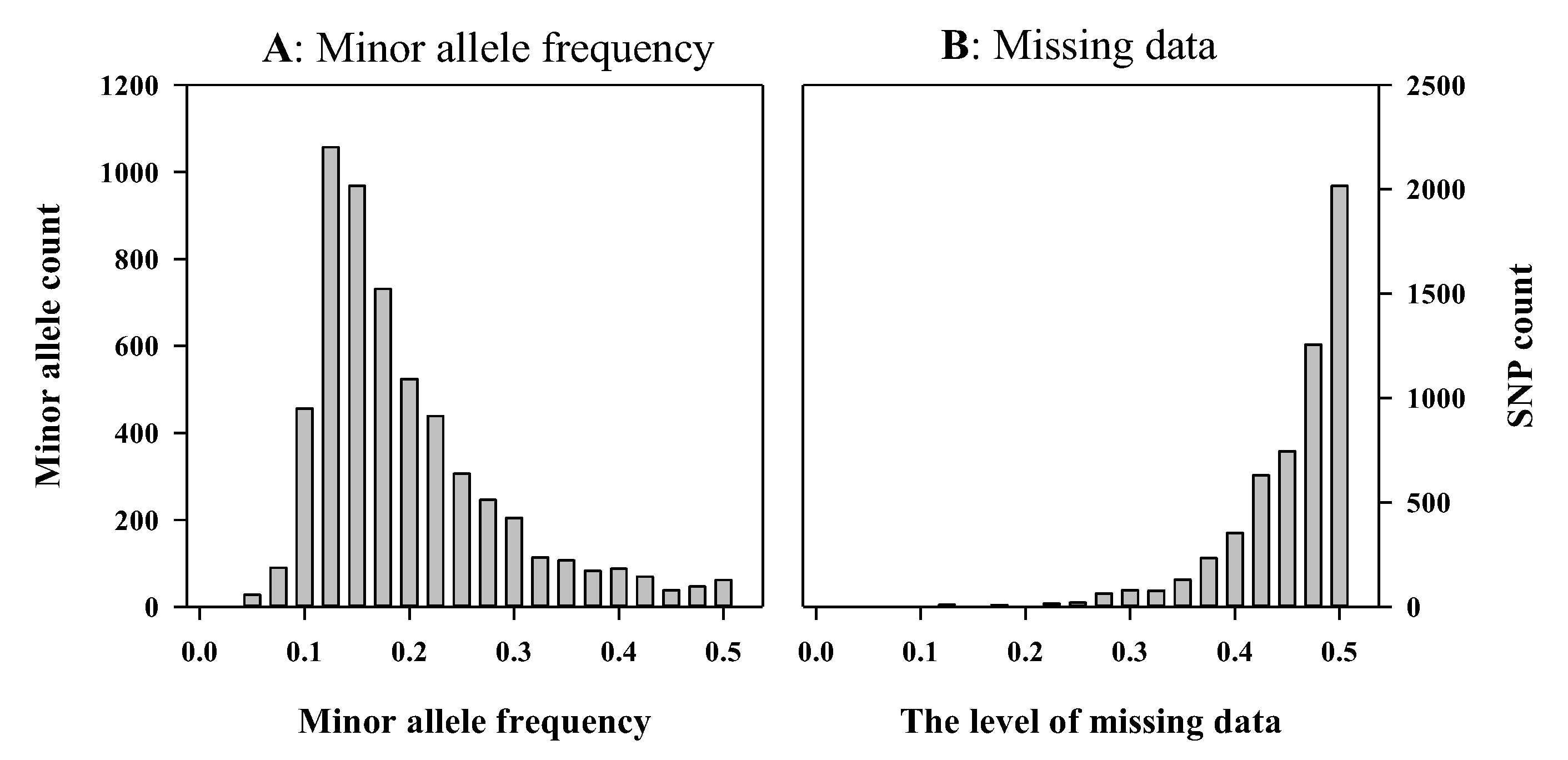
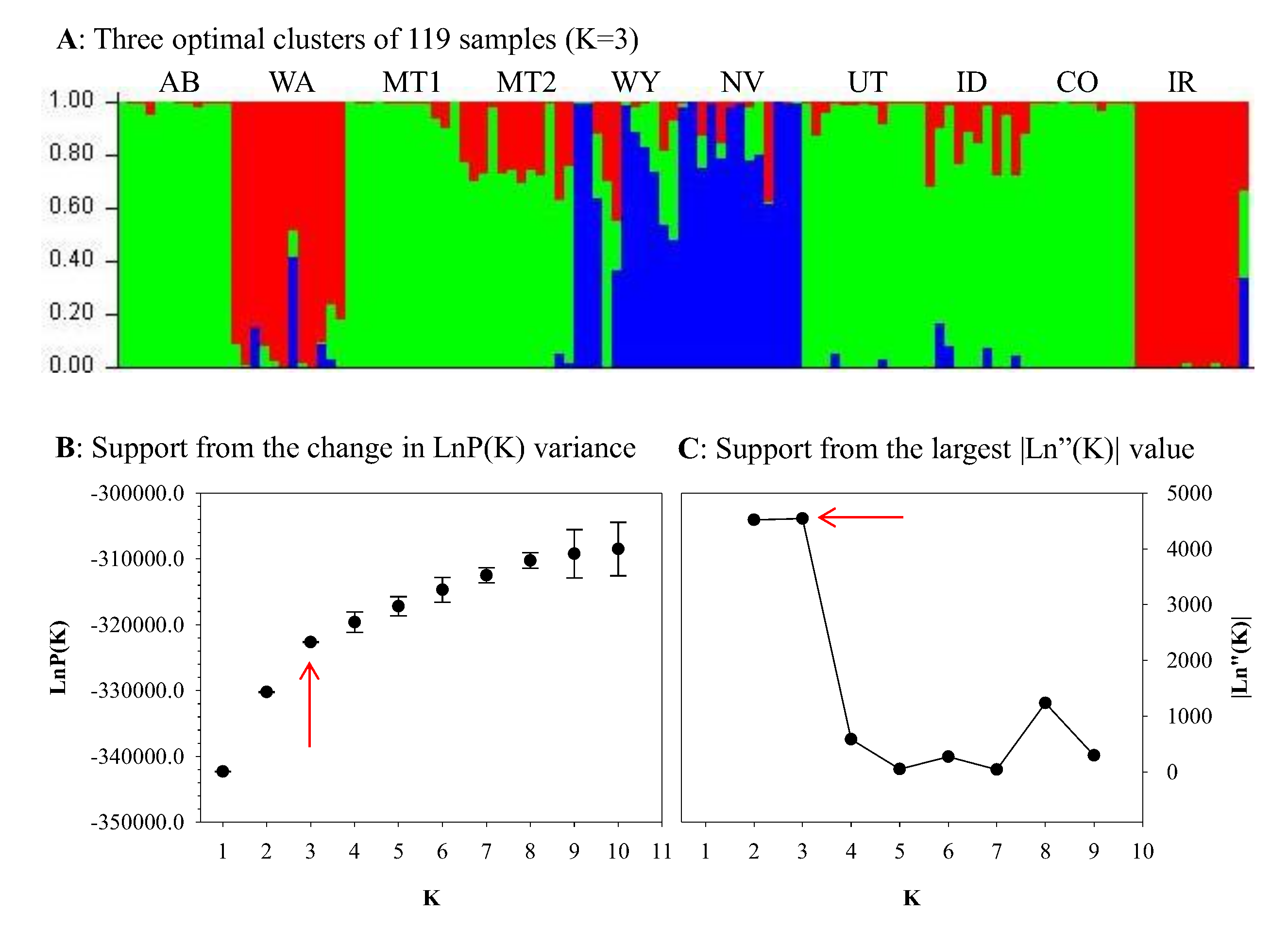
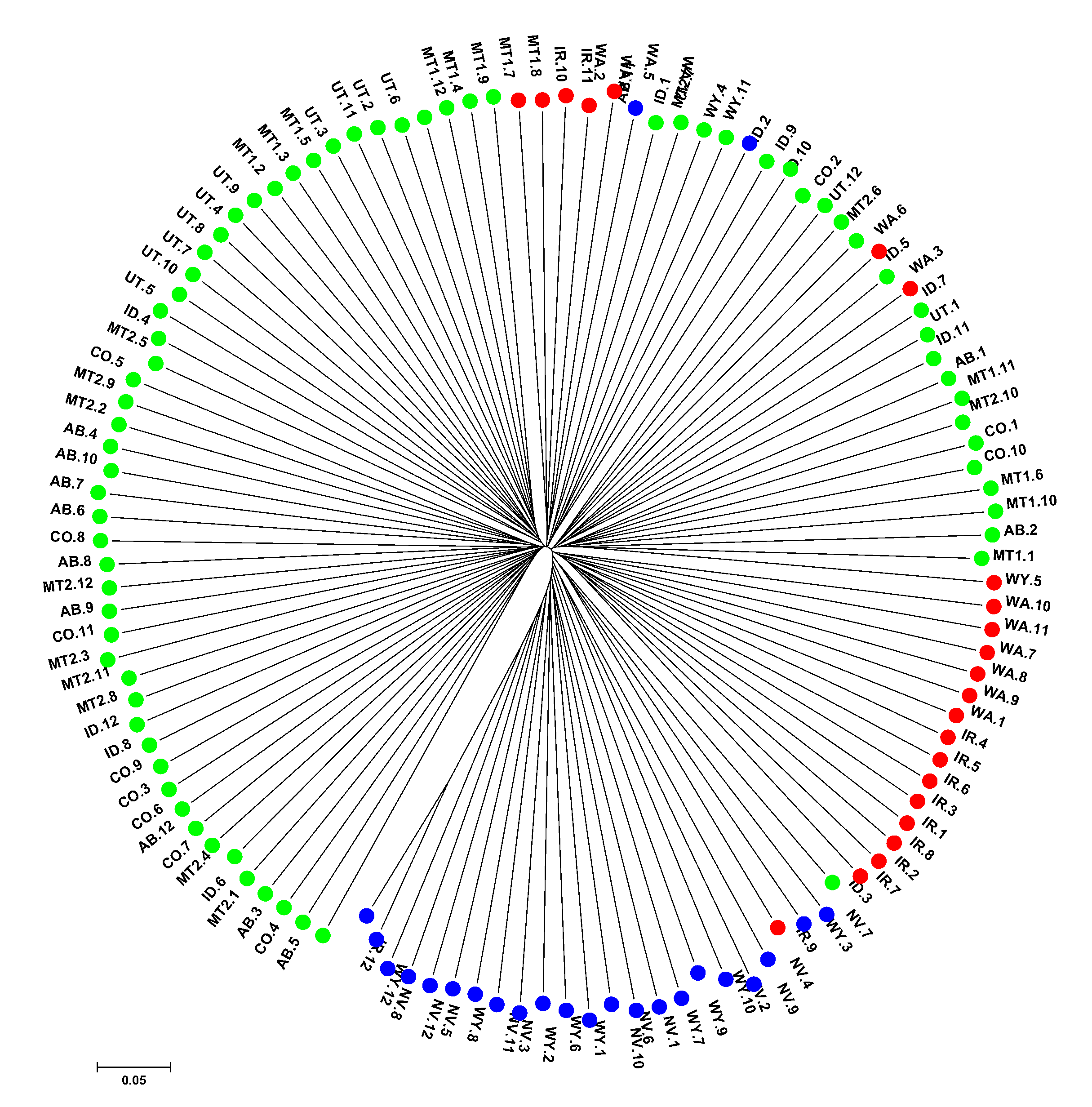
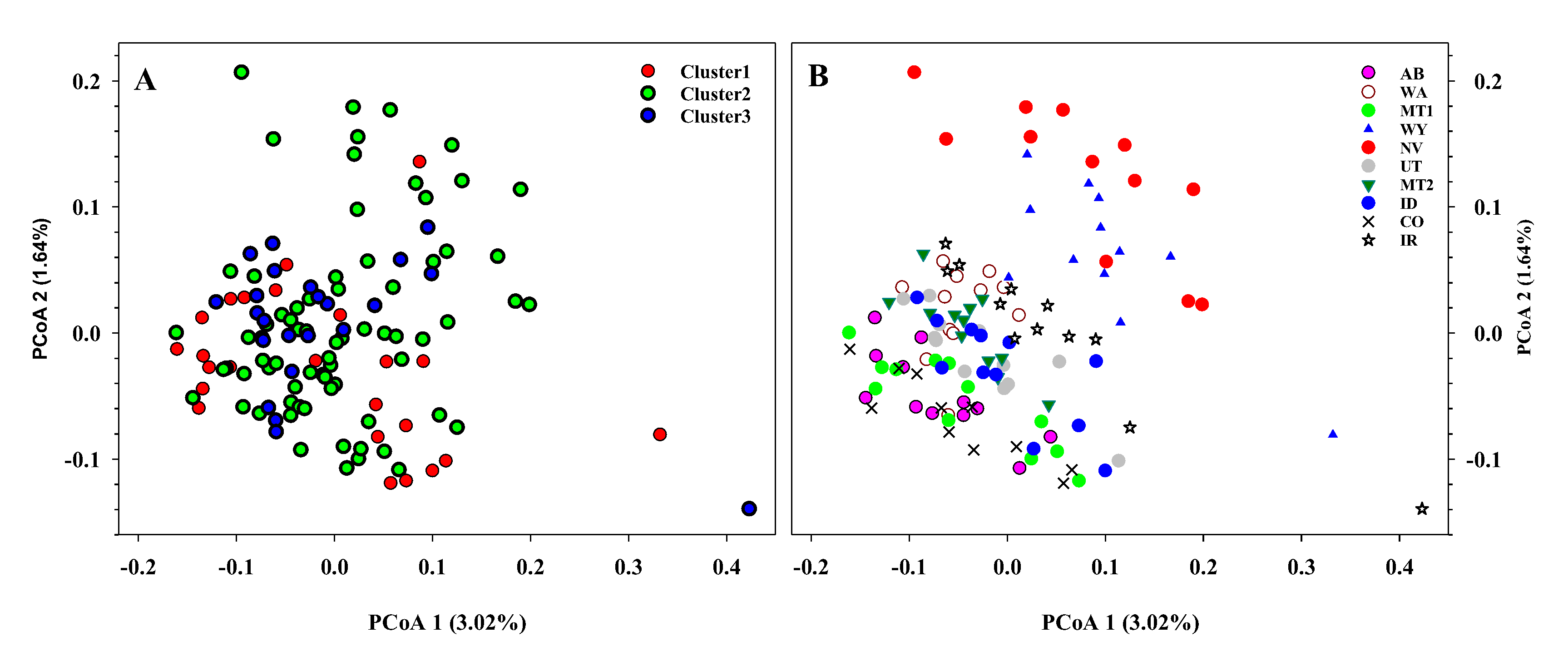
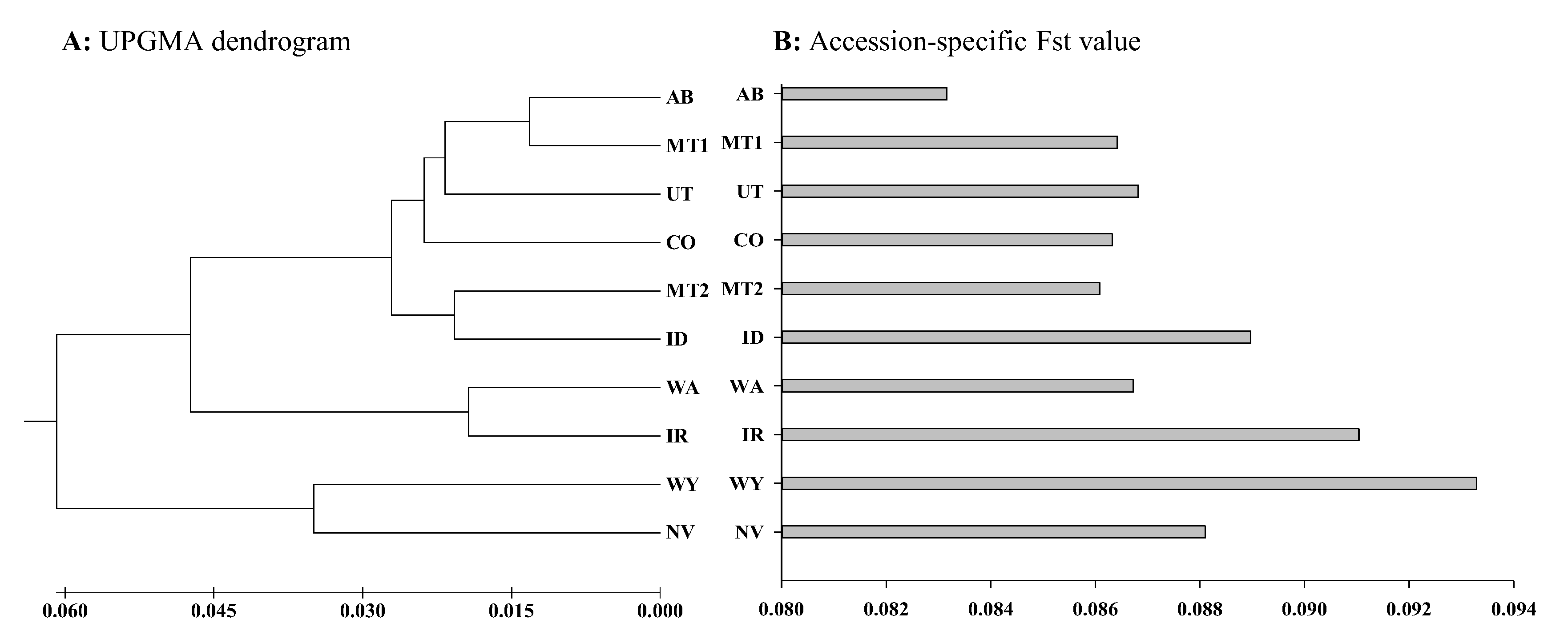
3. Results
4. Discussion
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
Ethical Standards
References
- Huang, X.; Feng, Q.; Qian, Q.; Zhao, Q.; Wang, L.; Wang, A.; Guan, J.; Fan, D.; Weng, Q.; Huang, T.; et al. High throughput genotyping by whole-genome resequencing. Genome Res. 2009, 19, 1068–1076. [Google Scholar] [CrossRef] [PubMed]
- Elshire, R.J.; Glaubitz, J.C.; Sun, Q.; Poland, J.A.; Kawamoto, K.; Buckler, E.S.; Mitchell, S.E. A robust, simple genotyping-by-sequencing (GBS) approach for high diversity species. PLoS ONE 2011, 6, e19379. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.B.; Peterson, G.W. Genetic diversity analysis with 454 pyrosequencing and genomic reduction confirmed the eastern and western division in the cultivated barley gene pool. Plant Genome 2011, 4, 226–237. [Google Scholar] [CrossRef]
- Peterson, B.; Weber, J.N.; Kay, E.H.; Fisher, H.S.; Hoekstra, H.E. Double digest RADseq: An inexpensive method for de novo SNP discovery and genotyping in model and non-model species. PLoS ONE 2012, 7, e37135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peterson, G.W.; Dong, Y.B.; Horbach, C.; Fu, Y.B. Genotyping-by-sequencing for plant genetic diversity analysis: A lab guide for SNP genotyping. Diversity 2014, 6, 665–680. [Google Scholar] [CrossRef]
- Poland, J.A.; Rife, T.W. Genotyping-by-sequencing for plant breeding and genetics. Plant Genome 2012, 5, 92–102. [Google Scholar] [CrossRef]
- Poland, J.A.; Brown, P.J.; Sorrells, M.E.; Jannink, J.L. Development of high-density genetic maps for barley and wheat using a novel two-enzyme genotyping-by-sequencing approach. PLoS ONE 2012, 7, e32253. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.F.; Poland, J.A.; Wight, C.P.; Jackson, E.W.; Tinker, N.A. Using genotyping-by-sequencing (GBS) for genomic discovery in cultivated oat. PLoS ONE 2014, 9, e102448. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.B.; Peterson, G.W.; Dong, Y. Increasing genome sampling and improving SNP genotyping for genotyping-by-sequencing with new combinations of restriction enzymes. G3 2016, 6, 845–856. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.B.; Yang, M.H. Genotyping-by-sequencing and its application to oat genomic research. In Oat—Methods and Protocols; Gasparis, S., Ed.; Springer Science + Business Media: New York, NY, USA, 2017; pp. 169–187. [Google Scholar]
- Tinker, N.A.; Bekele, W.A.; Hattori, J. Haplotag: Software for haplotype-based genotyping-by-sequencing analysis. G3 2016, 6, 857–863. [Google Scholar] [CrossRef] [PubMed]
- Yan, H.; Bekele, W.A.; Wight, C.P.; Peng, Y.; Langdon, T.; Latta, R.G.; Fu, Y.B.; Diederichsen, A.; Howarth, C.J.; Jellen, E.N.; et al. High-density markers profiling confirms ancestral genomes of Avena species and identifies D-genome chromosomes of hexaploid oat. Theor. Appl. Genet. 2016, 129, 2133–2149. [Google Scholar] [CrossRef] [PubMed]
- Bekele, W.A.; Wight, C.P.; Chao, S.; Howarth, C.J.; Tinker, N.A. Haplotype based genotyping-by-sequencing in oat genome research. Plant Biotechnol. J. 2018. [Google Scholar] [CrossRef] [PubMed]
- Dewey, D.R. The genomic system of classification as a guide to intergeneric hybridization with the perennial Triticeae. In Gene Manipulation in Plant Improvement; Gustafson, J.P., Ed.; Columbia University Press: New York, NY, USA, 1984; pp. 209–279. [Google Scholar]
- Love, A. Conspectus of the Triticeae. Feddes Repert. 1984, 95, 425–521. [Google Scholar] [CrossRef]
- Jensen, K.B. Cytology and fertility of advanced populations of Elymus lanceolatus (Scribn. & Smith) Gould × Elymus caninus (L.). hybrids. Crop Sci. 2005, 45, 1211–1215. [Google Scholar]
- Dewey, D.R. Cytogenetics of Agropyron pringlei and its hybrids with A. spicatum, A. seribneri, A. vidaceum, and A. dasystachyum. Bot. Gazette 1976, 137, 179–185. [Google Scholar] [CrossRef]
- Smoliak, S.; Johnston, A. Elbee northern wheatgrass. Can. J. Plant Sci. 1980, 60, 1473–1475. [Google Scholar] [CrossRef]
- Cronquist, A.; Holmgren, A.H.; Holmgren, N.H.; Reveal, J.L.; Holmgren, P.K. Intermountain Flora: Vascular Plants of the Intermountain West, U.S.A. Vol. 6: The Monocotyledons; Columbia University Press: New York, NY, USA, 1977; p. 584. [Google Scholar]
- Kowalenko, B.L.; Romo, J.T. Regrowth and rest requirements of northern wheatgrass following defoliation. J. Range Manag. 1998, 51, 73–78. [Google Scholar] [CrossRef]
- Harniss, R.O.; Murray, R.B. 30 years of vegetal change following burning of sagebrush-grass range. J. Range Manag. 1973, 26, 322–325. [Google Scholar] [CrossRef]
- Wright, H.A.; Bailey, A.W. Fire Ecology: United States and Southern Canada; John Wiley and Sons: New York, NY, USA, 1982; p. 501. [Google Scholar]
- Knapp, E.E.; Rice, K.J. Genetic structure and gene flow in Elymus glaucus (blue wildrye): Implications for native grassland and management. Restor. Ecol. 1996, 4, 40–45. [Google Scholar] [CrossRef]
- Rogers, D.L.; Montalvo, A.M. Genetically Appropriate Choices for Plant Materials to Maintain Biological Diversity; Report to the USDA Forest Service, Rocky Mountain Region, CO, USA; University of California: Davis, CA, USA, 2004; p. 343. [Google Scholar]
- Diaz, O.; Salomon, B.; Bothmer, R.V. Genetic diversity and structure in populations of Elymus caninus (L.) L. (Poaceae). Hereditas 1999, 131, 63–74. [Google Scholar] [CrossRef]
- Sun, G.L.; Diaz, O.; Salomon, B.; Bothmer, R.V. Genetic diversity in Elymus caninus as revealed by isozyme, RAPD and microsatellite markers. Genome 1999, 42, 420–431. [Google Scholar] [CrossRef] [PubMed]
- Sun, G.L.; Diaz, O.; Salomon, B.; Bothmer, R.V. Microsatellite variation and its comparison with isozyme and RAPD variation in Elymus fibrosus. Hereditas 1999, 129, 275–282. [Google Scholar] [CrossRef]
- Diaz, O.; Sun, G.L.; Salomon, B.; Bothmer, R.V. Level and distribution of allozyme and RAPD variation in populations of Elymus fibrosus (Poaceae). Genet. Resour. Crop Evol. 2000, 47, 11–24. [Google Scholar] [CrossRef]
- Wu, D.C.; He, D.M.; Gu, H.L.; Wu, P.P.; Yi, X.; Wang, W.J.; Shi, H.F.; Wu, D.X.; Sun, G. Origin and evolution of allopolyploid wheatgrass Elymus fibrosus (Schrenk) Tzvelev (Poaceae: Triticeae) reveals the effect of its origination on genetic diversity. PLoS ONE 2016, 11, e0167795. [Google Scholar] [CrossRef] [PubMed]
- Diaz, O.; Salomon, B.; Bothmer, R.V. Genetic variation and differentiation in Nordic populations of Elymus alaskanus (Scrib. ex Merr.) LoÈve (Poaceae). Theor. Appl. Genet. 1999, 99, 210–217. [Google Scholar]
- Sun, G.L.; Salomon, B.; Bothmer, R.V. Microsatellite polymorphism and genetic differentiation in three Norwegian populations of Elymus alaskanus (Poaceae). Plant Syst. Evol. 2002, 234, 101–110. [Google Scholar] [CrossRef]
- Zhang, X.Q.; Salomon, B.; Bothmer, R.V. Application of random amplified polymorphic DNA markers to evaluate intraspecific genetic variation in the Elymus alaskanus complex (Poaceae). Genet. Resour. Crop Evol. 2002, 49, 397–407. [Google Scholar] [CrossRef]
- Sun, G.L.; Salomon, B. Microsatellite variability and the heterozygote deficiency in arctic-alpine species Elymus alaskanus complex. Genome 2003, 46, 729–737. [Google Scholar] [CrossRef] [PubMed]
- Gaudett, M.; Salomon, B.; Sun, G.L. Molecular variation and population structure in Elymus trachycaulus and comparison with its morphologically similar E. alaskanus. Plant Syst. Evol. 2005, 250, 81–91. [Google Scholar] [CrossRef]
- Sun, G.L.; Li, W.B. Molecular diversity of Elymus trachycaulus complex species and their relationships to non-North American taxa. Plant Syst. Evol. 2006, 256, 179–191. [Google Scholar] [CrossRef]
- Ma, X.; Zhang, X.Q.; Zhou, Y.H.; Bai, S.Q.; Liu, W. Assessing genetic diversity of Elymus sibiricus (Poaceae:Triticeae) populations from Qinghai-Tibet Plateau by ISSR markers. Biochem. Syst. Ecol. 2008, 36, 514–522. [Google Scholar] [CrossRef]
- Ma, X.; Chen, S.Y.; Bai, S.Q.; Zhang, X.Q.; Li, D.X.; Zhang, C.B.; Yan, J.J. RAPD analysis of genetic diversity and population structure of Elymus sibiricus (Poaceae) native to the southeastern Qinghai-Tibet Plateau, China. Genet. Mol. Res. 2012, 11, 2708–2718. [Google Scholar] [CrossRef] [PubMed]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed]
- Lu, F.; Lipka, A.E.; Glaubitz, J.; Elshire, R.; Cherney, J.H.; Casler, M.D.; Buckler, E.S.; Costich, D.E. Switchgrass genomic diversity, ploidy, and evolution: Novel insights from a network-based SNP discovery protocol. PLoS Genet. 2013, 9, e1003215. [Google Scholar] [CrossRef] [PubMed]
- Pritchard, J.; Stephens, M.; Donnelly, P. Influence of population structure using multilocus genotype data. Genetics 2000, 155, 945–959. [Google Scholar] [PubMed]
- Falush, D.; Stephens, M.; Pritchard, J.K. Inference of population structure using multilocus genotype data: Dominant markers and null alleles. Mol. Ecol. Notes 2007, 7, 574–578. [Google Scholar] [CrossRef] [PubMed]
- Evanno, G.; Regnaut, S.; Goudet, J. Detecting the number of clusters of individuals using the software STRUCTURE: a simulation study. Mol. Ecol. 2005, 14, 2611–2620. [Google Scholar] [CrossRef] [PubMed]
- Swofford, D.L. PAUP*: Phylogenetic Analysis Using Parsimony (*and Other Methods), Version 4; Sinauer Associates: Sunderland, MA, USA, 1998. [Google Scholar]
- Tamura, K.; Peterson, D.; Peterson, N.; Stecher, G.; Nei, M.; Kumar, S. MEGA5: Molecular Evolutionary Genetics Analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol. Biol. Evol. 2011, 28, 2731–2739. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.H.; Fu, Y.B. AveDissR: An R function for assessing genetic distinctness and genetic redundancy. Appl. Plant Sci. 2017, 5, 1700018. [Google Scholar] [CrossRef] [PubMed]
- R Development Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2016; ISBN 3-900051-07-0. Available online: http://www.r-project.org/ (accessed on 10 April 2018).
- Excoffier, L.; Laval, G.; Schneider, S. Arlequin ver. 3.1: An integrated software package for population genetics data analysis. Evol. Bioinform. Online 2005, 1, 47–50. [Google Scholar] [CrossRef]
- Bennett, R.A.; Séguin-Swartz, G.; Rahman, H. Broadening genetic diversity in Canola using the C-Genome species Brassica oleracea L. Crop Sci. 2012, 52, 2030–2039. [Google Scholar] [CrossRef]
- Hamrick, J.L.; Godt, M.J.W. Allozyme diversity in plant species. In Plant Population Genetics, Breeding and Genetic Resources; Brown, A.H.D., Clegg, M.T., Kahler, A.L., Weir, B.S., Eds.; Sinauer Associates: Sunderland, MA, USA, 1989; pp. 43–63. [Google Scholar]
- Fu, Y.B.; Coulman, B.E.; Ferdinandez, Y.S.N.; Cayouette, J.; Peterson, P.M. Genetic diversity of fringed brome (Bromus ciliatus) as determined by amplified fragment length polymorphism. Can. J. Bot. 2005, 83, 1322–1328. [Google Scholar] [CrossRef]
- Biligetu, B.; Schellenberg, M.P.; Fu, Y.B. Detecting genetic diversity of side-oats grama grass populations using AFLP Marker. Can. J. Plant Sci. 2013, 93, 1105–1114. [Google Scholar] [CrossRef]
- Sun, G.L.; Salomon, B.; Bothmer, R.V. Analysis of tetraploid Elymus species using wheat microsatellite markers and RAPD markers. Genome 1997, 40, 806–814. [Google Scholar] [CrossRef] [PubMed]
- Phan, A.T.; Fu, Y.B.; Smith, S.R., Jr. RAPD variations in selected and unselected blue grama populations. Crop Sci. 2003, 43, 1852–1857. [Google Scholar] [CrossRef]
- Glover, N.A.; Redestig, H.; Dessimoz, C. Homeologs: What are the and how do we infer them? Trends Plant Sci. 2016, 21, 609–621. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, R.; Paul, J.S.; Albrechtsen, A.; Song, Y.S. Genotype and SNP calling from next-generation sequencing data. Nat. Rev. Genet. 2011, 12, 443–451. [Google Scholar] [CrossRef] [PubMed]
- Vos, P.; Hogers, R.; Bleeker, M.; Reijans, M.; van De Lee, T.; Hornes, M.; Frijters, A.; Pot, J.; Peleman, J.; Kuiper, M.; et al. AFLP: A new technique for DNA fingerprinting. Nucleic Acids Res. 1995, 23, 4407–4414. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.B.; Phan, A.T.; Coulman, B.; Richards, K.W. Genetic diversity in natural populations and corresponding seed collections of little bluestem as revealed by AFLP markers. Crop Sci. 2004, 44, 2254–2260. [Google Scholar] [CrossRef]
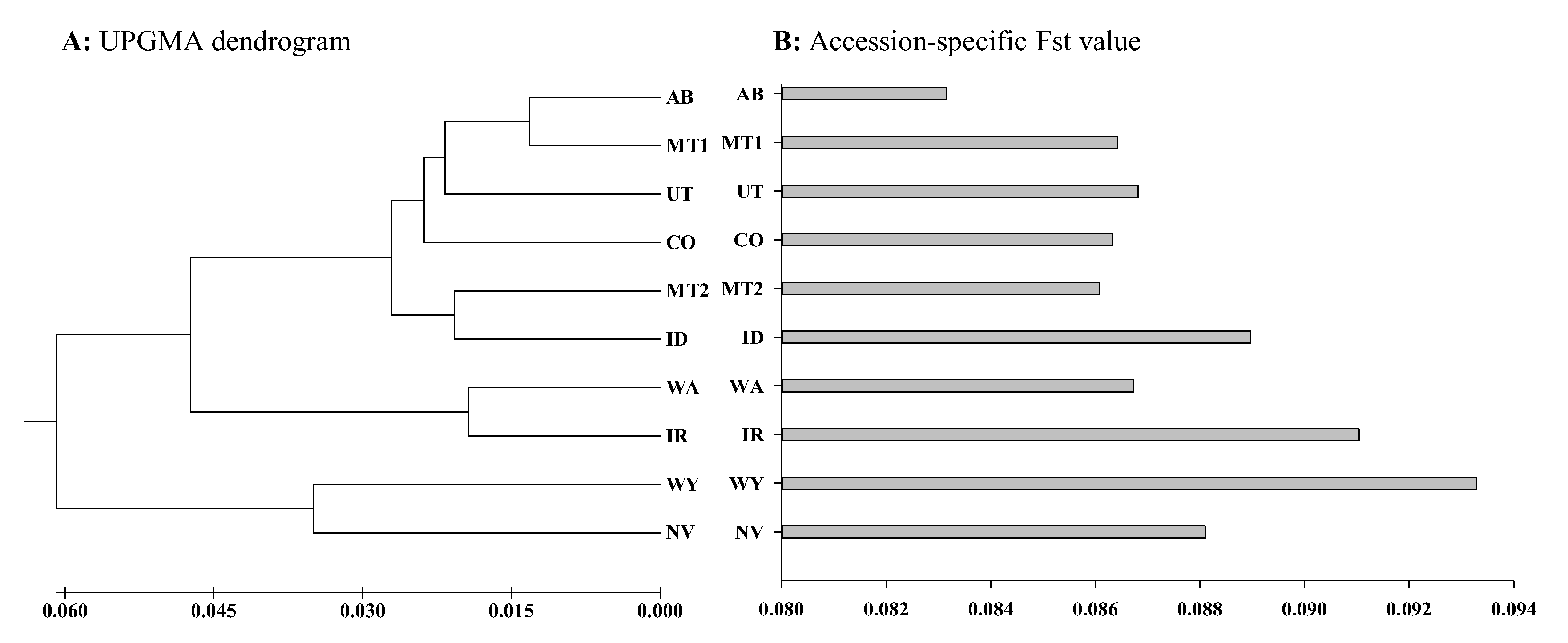

{kind=link}
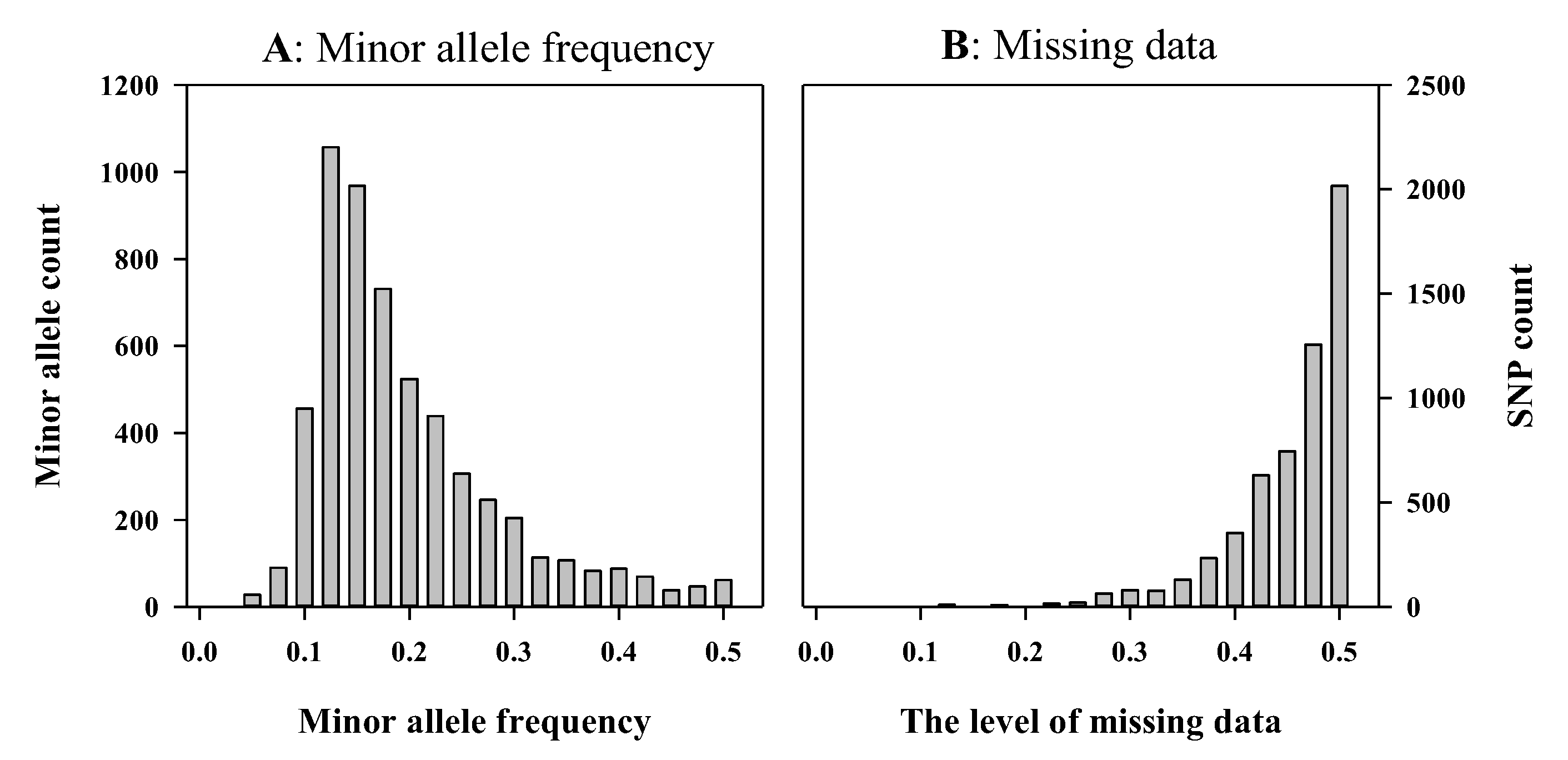
{kind=link}
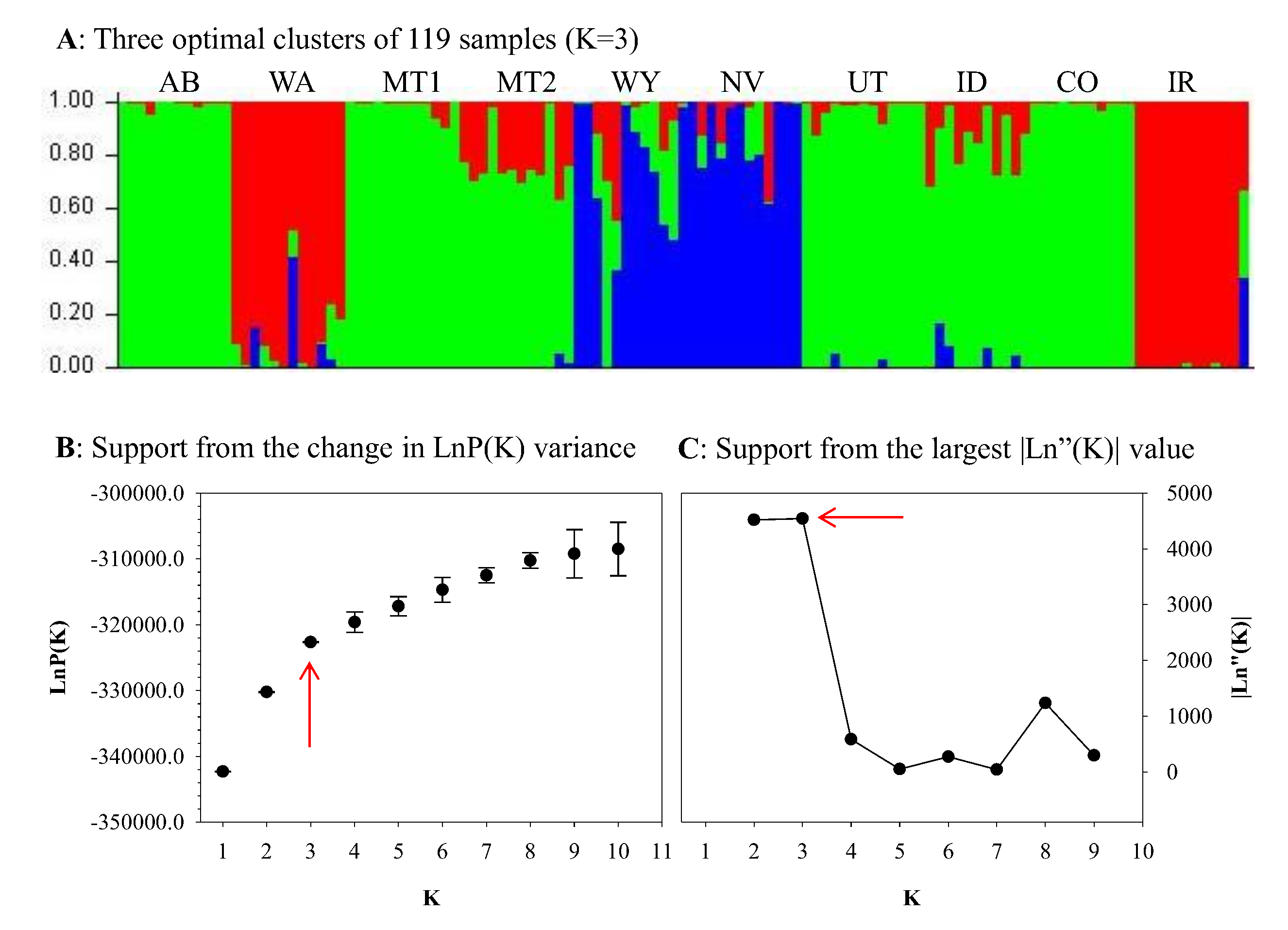
{kind=link}
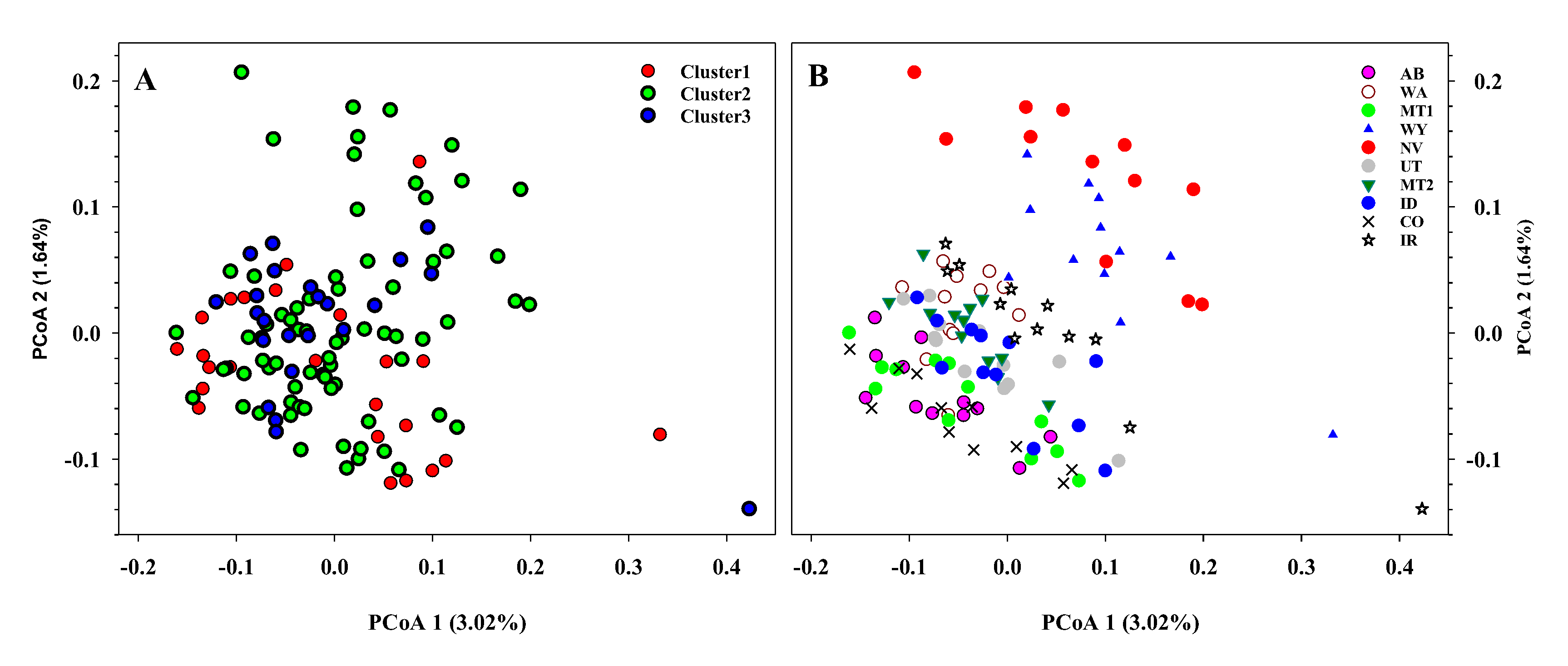{kind=link}
{kind=link}
| Accession a | Alt Acc a | Germplasm Type | Collected/Received a | Label | GsrTa a | GsrTd a |
|---|---|---|---|---|---|---|
| CN37154 | cultivar ‘Elbee’ | AB, Canada (1980) | AB | 9.195 | 8.841 | |
| TMP24006 | PI632756 | cultivar ‘Schwendimar’ | WA, USA (2003) | WA | 9.531 | 9.397 |
| TMP24007 | PI469235 | cultivar ‘Critana’ | MT, USA (1982) | MT1 | 9.574 | 9.478 |
| TMP24013 | PI562038 | Cultivated | MT, USA (1992) | MT2 | ||
| TMP24010 | W614632 | wild accession | WY, USA (1993) | WY | ||
| TMP24011 | W616745 | wild accession | NV, USA (1995) | NV | 9.412 | 9.294 |
| TMP24012 | PI564552 | wild accession | UT, USA (1992) | UT | ||
| TMP24015 | PI562037 | wild accession | ID, USA (1992) | ID | ||
| TMP24016 | PI552794 | wild accession | CO, USA (1991) | CO | ||
| TMP24018 | PI380620 | wild accession | Iran (1972) | IR |
| Model/Source of Variation | df | Sum of Squares | Variance Explained | Variance (%) a |
|---|---|---|---|---|
| 10 accessions | ||||
| Among accessions | 9 | 6854.6 | 22.4 | 8.77 |
| Within accessions | 226 | 52,675.3 | 233.1 | 91.24 |
| Cultivated vs. wild accessions | ||||
| Between groups | 1 | 742.6 | 4.3 | 1.69 |
| Within groups | 234 | 58,787.3 | 251.2 | 98.31 |
| Three clusters from STRUCTURE | ||||
| Among clusters | 2 | 3176.5 | 20.7 | 7.88 |
| Within clusters | 233 | 56,353.5 | 241.1 | 92.12 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, P.; Bhattarai, S.; Peterson, G.W.; Coulman, B.; Schellenberg, M.P.; Biligetu, B.; Fu, Y.-B. Genetic Diversity of Northern Wheatgrass (Elymus lanceolatus ssp. lanceolatus) as Revealed by Genotyping-by-Sequencing. Diversity 2018, 10, 23. https://doi.org/10.3390/d10020023
Li P, Bhattarai S, Peterson GW, Coulman B, Schellenberg MP, Biligetu B, Fu Y-B. Genetic Diversity of Northern Wheatgrass (Elymus lanceolatus ssp. lanceolatus) as Revealed by Genotyping-by-Sequencing. Diversity. 2018; 10(2):23. https://doi.org/10.3390/d10020023
Chicago/Turabian StyleLi, Pingchuan, Surendra Bhattarai, Gregory W. Peterson, Bruce Coulman, Michael P. Schellenberg, Bill Biligetu, and Yong-Bi Fu. 2018. "Genetic Diversity of Northern Wheatgrass (Elymus lanceolatus ssp. lanceolatus) as Revealed by Genotyping-by-Sequencing" Diversity 10, no. 2: 23. https://doi.org/10.3390/d10020023
APA StyleLi, P., Bhattarai, S., Peterson, G. W., Coulman, B., Schellenberg, M. P., Biligetu, B., & Fu, Y.-B. (2018). Genetic Diversity of Northern Wheatgrass (Elymus lanceolatus ssp. lanceolatus) as Revealed by Genotyping-by-Sequencing. Diversity, 10(2), 23. https://doi.org/10.3390/d10020023