Abstract
Mobile genetic elements (MGEs) play an essential role in bacterial adaptation and evolution. These elements are enriched within bacterial communities from extreme environments. However, very little is known if specific genes co-occur with MGEs in extreme environments and, if so, what their function is. We used shotgun-sequencing to analyse the metagenomes of 12 soil samples and characterized the composition of MGEs and the genes co-occurring with them. The samples ranged from less arid coastal sites to the inland hyperarid core of the Atacama Desert, as well as from sediments below boulders, protected from UV-irradiation. MGEs were enriched at the hyperarid sites compared with sediments from below boulders and less arid sites. MGEs were mostly co-occurring with genes belonging to the Cluster Orthologous Group (COG) categories “replication, recombination and repair,” “transcription” and “signal transduction mechanisms.” In general, genes coding for transcriptional regulators and histidine kinases were the most abundant genes proximal to MGEs. Genes involved in energy production were significantly enriched close to MGEs at the hyperarid sites. For example, dehydrogenases, reductases, hydrolases and chlorite dismutase and other enzymes linked to nitrogen metabolism such as nitrite- and nitro-reductase. Stress response genes, including genes involved in antimicrobial and heavy metal resistance genes, were rarely found near MGEs. The present study suggests that MGEs could play an essential role in the adaptation of the soil microbiome in hyperarid desert soils by the modulation of housekeeping genes such as those involved in energy production.
1. Introduction
The old theory “Everything is everywhere, but, the environment selects” has been critically discussed through time [1]. It can be understood as an argument for environmental conditions being essential not only to the structuring of microbial communities but bacterial evolution itself. It has been postulated that microbial populations undergo a strong selective pressure in extreme environments as in hot and cold deserts, deep-sea sediments, hot springs, geysers and permafrost soils [2]. However, if the traces of life detected in some extreme environments are indicative for active microbial communities [3] or whether they reflect dormant and dead cells, remains debated. A recent polyphasic study gave evidence for metabolically active microbial communities in the Atacama Desert, even in the hyperarid region where the water activity is often below the metabolic needs [3]. Likewise, transcriptional data showed active microbial communities in desert soil from the Namib Desert [4]. Some studies have indicated that Mobile Genetic Elements (MGEs) are enriched in the microbiomes of extreme environments, which could facilitate an enhanced evolution and adaptation of the microbiome to environmental stressors [5]. MGEs are fragments of DNA with the ability to mobilize themselves within genomes and between bacterial cells, a process referred to as horizontal gene transfer (HGT). HGT includes three mechanisms, transformation, conjugation and transduction [6]. Even though MGEs have been considered “selfish elements” [7], their high abundance [8] and evolutionary success could indicate their importance for bacterial adaptation [9]. MGEs have been vastly studied in environments with high degrees of anthropogenic pollution, including sites with high loads of antibiotics where the mobilization of antibiotic resistance genes has been documented within the respective microbiome [10,11,12].
MGEs associated with different functional traits enhance genome plasticity, stress response and other environmental adaptations. For instance, insertion sequences can modulate various metabolic pathways, including the utilization of different carbon sources and electron donors, as well as the expression of metabolites increasing antimicrobial/xenobiotic and virulence resistance [13]. Also, restriction-modification systems (RMs) are highly associated with MGEs [14] and can act as independent mobile elements carrying accessory genes [15]. Finally, SOS response and HGT events stimulate each other and could be a mechanism for increasing bacterial genome plasticity [16]. However, data on the role of MGEs and genes co-occurring with MGEs in extreme natural environments is still missing.
In the present study, we analyzed the diversity of MGEs and the genes co-occurring with them in the soil microbiome of the Atacama Desert. The investigated samples represent a precipitation gradient, decreasing from the arid coast to the hyperarid inland region of the desert. Besides, samples below boulders from the hyperarid inland region were included, which are shielded from UV-irradiation. We anticipated that MGEs would be enriched in the hyperarid inland sites due to the higher desiccation and UV-irradiation stress. Furthermore, assuming that the genes co-occurring with the MGEs could represent an adaptive advantage to the bacterial community under hyperarid conditions.
2. Methods
2.1. Soil Sampling
Between 2015 and 2018, surface and near-surface soil samples (0–5 cm depth) were collected from the Atacama Desert in Chile. Shortly before the first sampling campaign, in 2015, the highest rain event since 1978 was recorded in Antofagasta and affected all studied sites. Samples from four sites were selected in the present study, Coastal (481 m.a.s.l., S 23.8247, W 70.4888), Yungay (1004 m.a.s.l., S 24.0883, W 69.9946), Maria Elena (1313 m.a.s.l., S 22.2631, W 69.7243) and Lomas Bayas (1528 m.a.s.l., S 23.3933, W 69.6039). Soils from all sites have been classified as Aridisols. Additionally, sediments below boulders, which have been less exposed to UV-irradiation, were collected from Yungay, Maria Elena and Lomas Bayas. For sample collection, the triangle method was used. Over each sampling location, a triangle with a side length of ~5 m was laid and at each end-point, a sample was taken. Besides, a boulder was selected and available material below the boulder was collected. Sampling equipment, metallic spoons, was sterilized with ethanol previously to the sampling. The coastal site is situated 21 km from Antofagasta, the presence of vegetation and hypolithic communities indicate occasional rain events. In the sampling spot, the top 20 cm was composed of silty to sandy material. The Maria Elena site is placed 50 km inland in the hyperarid core of the desert. Cobbles and boulders can be found on the surface. In the sampling pit, the uppermost 20 cm consisted of weakly cemented silt to coarse sand. The Yungay site is located on the distal part of an alluvial fan, 50 km inland from the coastline. Vegetation and hypoliths were not visible. On the surface near the sampling place, cobbles and boulders rested on a layer of mostly lithic particles. The Lomas Bayas site is situated at the hyperarid core of the desert, approximately 80 km inland from the shoreline. The surface is broadly covered by angular clasts. The sampling site surface was predominantly covered by sandy to gravel material. Samples were grouped into three categories: all the samples from the arid soils near the coast (n = 4), from now on referred to as Arid; the hyperarid inland sites Yungay and Maria Elena (n = 5), from now on referred to as Hyperarid; and unconsolidated sediment samples collected below boulders (Yungay, Maria Elena and Lomas Bayas, n = 3), from now on referred to as Hyperarid-BBoulder. Even though more soil samples were available, not all of them yielded enough DNA for library preparation. For example, surface soils from Lomas Bayas and a third sample from Maria Elena. While the coastal sites are occasionally exposed to fog and rain, the rest of the samples, including the below boulder sites, are located in the hyperarid core of the Atacama Desert. In general, soils of the Atacama Desert contain only little amounts of total organic carbon (~0.1%) and are exposed to relative humidity levels below 30% and a UV-irradiation dose of 30 J m−2. Surface soil pH ranged from 7.57 to 8.08 and it was similar in the Hyperarid and Arid sites (Supplementary file S1: Table S1). Soil conductivity was approximately three times higher in two of the Hyperarid sites compared with the Arid sites, which could indicate differences in moisture levels and salinity. Total carbon, nitrogen and sulphur were low in soil samples from all sites and did not differ significantly. Further data on abiotic soil properties can be found in Reference [3]. Soil samples were kept at −80 °C until use.
2.2. DNA Extraction and Metagenomic Library Preparation
For the extraction of DNA, 10 g of soil material was mixed with 40 mL of cell extraction buffer (1% PEG-8000, 1 M NaCl, pH 9.2) and incubated for 30 min as described before [17]. The suspension was centrifuged at 44,000× g for 2 h at 4 °C. Subsequently, DNA was extracted from the pellet using bead-beating and a phenol-chloroform-isoamyl alcohol (PCI) extraction protocol [18]. As DNA negative extraction control, 10 mL of cell extraction buffer was used. After precipitation, DNA was eluted in 40 µL of sterile water. DNA from three extractions was combined to obtain sufficient DNA for sequence library construction. DNA concentrations were measured using Quant-It™ PicoGreen® dsDNA Assay Kit (Thermo Fisher Scientific, MA, USA) and a spectrofluorometer (SpectraMax Gemini EM microplate reader Molecular Devices, LLC, USA). In total, ~100 ng of DNA per sample was sheared using an E220 Focused-ultrasonicator (Covaris® Inc., MA, USA) targeting 500 bp fragments following Covaris’ instructions. Metagenomic libraries were constructed using the NEBNext® Ultra™ DNA Library Prep Kit for Illumina®. Dual indexing was done employing the NEBNext® Multiplex Oligos kit for Illumina® (Dual index primers set 1, New England BioLabs, UK). Purification and size selection was performed based on Agencourt® AMPure® XP (Beckman-Coulter, MA, USA). Library inserts ranging from 500–700 bp were further evaluated using a Fragment Analyzer™ (Advanced Analytical, IA, USA). Libraries were quantified using the Quant-It™ PicoGreen® dsDNA Assay Kit. Libraries were pooled equimolarly and 15 pM was spiked with 1% PhiX (Illumina, CA, USA). Sequencing was performed on an Illumina MiSeq (Illumina, CA, USA) using the paired-end mode (2 × 300 bp, Reagent v3 for 600 cycles). Sequences have been deposited in GenBank (BioProject ID PRJNA395196).
2.3. Bioinformatic Analysis
Adapters and primers were removed from raw reads using Adapterremoval v.2.1 [19]. Nucleotides with quality values less than 15 were trimmed and sequences shorter than 50 bp discarded. PhiX internal Illumina control was filtered using Deconseq v.0.4 [20]. Clean reads were taxonomically classified by Kaiju v1.4.5 [21] in “greedy” mode allowing 5 substitutions. Nonpareil v2.4 [22] was employed to estimate the metagenomes’ coverage and to calculate the Nonpareil diversity index, which is a way of describing the complexity of the bacterial community. Total cleaned reads were assembled using metaSPADES v 3.10 [23] with a maximum k-mer size of 127; for downstream analysis, only contigs larger than 500 bp were retained. Protein-coding genes were predicted using prodigal v2.6.3 with default parameters using the “meta” mode for metagenomic data. Contigs with two or more Open Reading Frames (ORFs) were used for further analysis. MGEs homologs were searched using the PFAM 31 [24] and TnpPred [25] databases through HMMER v3.1b2 [26]. Hits with a maximum 1 × 10−5 e-value were retained and the best hit per read was used for further analysis. MGEs were grouped into six groups based on identified MGEs’ genes: phage integrases, transposons (transposases related to a specific transposon), transposases, recombinases, resolvases, integrases and other (DDE and DUF elements related to transposases). The position and co-occurrence of genes and MGEs were analyzed using in-home scripts. co-occurrence was considered positive if a gene was found within five ORFs from upstream or downstream of a MGE. The implemented script can be found in Supplementary file S2. Contigs harboring MGEs were taxonomically annotated using Kaiju and the relative abundance was calculated based on the total of contigs harbouring MGEs.
2.4. MGEs and Co-Occurring Open Reading Frames
ORFs co-occurring with MGEs were evaluated between a maximum distance of five ORFs around all the MGEs predicted with PFAM and Tnpred. Cluster Orthologous Groups (COGs) were annotated using the eggNOG database [27] and Diamond v.0.8 [28] with the “more-sensitive” mode and a maximum e value of 10−3. As different COGs have the same protein name/description, those with identical description were merged to evaluate the most abundant protein name/description co-occurring with MGEs. Putative transposases, annotated with eggNOG, were highly abundant among the genes co-occurring with MGEs. Those transposases were excluded for MGE co-occurrence analysis. Additionally, genes co-occurring with MGEs were screened using GhostKOALA v2.0 [29], which allows an automatic KEGG Orthology (KO) assignment against a non-redundant set of KEGG genes.
Normalization of the genes annotated with eggNOG was performed in three different ways: (1) the number of genes in a given COG category relative to the total number of annotated genes (Figure 1A and Figure S1), (2) the number of genes co-occurring with MGEs in a given COG category relative to the total number of annotated genes co-occurring with MGEs (Figure 1B, and Figures 3–5 and (3) the number of genes co-occurring with MGEs in a given COG category relative to the total number of genes annotated to the same COG category (Figure 1C, Figures S4 and S5). The same methods were also used to normalize the data at a lower category level (COG numbers or COG description).
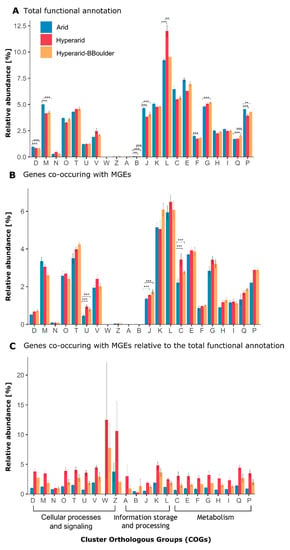
Figure 1.
Abundance of Cluster orthologous group (COG) categories found at Arid, Hyperarid and Hyperarid-BBoulder sites. (A) the total functional potential, (B) only the genes co-occurring with MGEs and (C) proportion of genes co-occurring with MGEs relative to the total amount of genes per category. D: Cell cycle control, cell division, chromosome partitioning, M: Cell wall/membrane/envelope biogenesis, N: Cell motility, O: Post-translational modification, protein turnover and chaperones, T: Signal transduction mechanisms, U: Intracellular trafficking, secretion and vesicular transport, V: Defence mechanisms, W: Extracellular structures, Z: Cytoskeleton, A: RNA processing and modification, B: Chromatin structure and dynamics, J: Translation, ribosomal structure and biogenesis, K: Transcription, L: Replication, recombination and repair, C: Energy production and conversion, E: Amino acid transport and metabolism, F Nucleotide transport and metabolism, G: Carbohydrate transport and metabolism, H: Coenzyme transport and metabolism, I: Lipid transport and metabolism, Q: Secondary metabolites biosynthesis, transport and catabolism, P: Inorganic ion transport and metabolism. The standard error is shown, n = 4, 5 and 3 at Arid, Hyperarid and Hyperarid-BBoulder, respectively. Robust ANOVA, * < 0.05, ** < 0.005 and *** < 0.0005.
Antimicrobial resistance genes (AMRs) were detected by employing the Resistance Gene Identifier v3.2.1 and the “Comprehensive Antibiotic Resistance Database” (CARD) v1.1.9 [30]. Hits classified as “loose” were not used for further analysis. Also, metal resistance genes were predicted using Diamond and the Antibacterial Biocides & Metal Resistance Genes Database (BacMet v2) [31]. Matches with at least 80% of identity, 70% of query coverage and an e value of maximum 10−5 were kept. Stress response genes were predicted using Diamond and a stress response protein (SRPs) database [32]. Matches with at least 65% of identity, 65% of query coverage and an e value of maximum 10−5 were kept.
2.5. Statistical Analysis and Visualization
Statistical analysis and visualization were done using R v3.3.1 [33]. The Likelihood ratio test (LRT, DESeq2) [34] was performed to analyze for differences in terms of the abundance of genes co-occurring with MGEs between the samples using raw counts. Differences between the COG numbers obtained from the total functional annotation were not statistically compared. LRT compares a full model versus a reduced model. All significantly different COGs between soils were used for preparing ternary plots (LRT, p < 0.5) implemented in the R package ‘ggtern.’ Differences in the relative abundance of MGEs and COG categories were evaluated using robust one-way ANOVA and robust post hoc Rand Wilcox’s based on trimmed means and percentile bootstrap [35]. For that, the t1waybt (α = 0.05 and trimmed mean = 10%, bootstrap = 1000) and mcppb20 (trimmed mean = 20% and bootstrap = 2000) functions implemented by Wilcox were utilized for the analysis. Log2 fold changes were calculated by comparing the fraction of genes co-occurring with MGEs per each category to the average fraction of genes co-occurring with MGEs within all the COG categories per site. Differences between the log-fold changes were evaluated using a one-sample T-test. Moreover, the correlation of the log10 transformed relative abundance of MGEs and the Nonpareil diversity index was evaluated by a robust Spearman’s correlation implemented by Wilcox as the function bootTau() (bootstrap = 2000) [36]. Finally, the abundance of stress response genes was compared using the two-sided White’s non-parametric t-test implemented in STAMP v2.1.3 [37].
3. Results
3.1. Reads and Contigs Quality
High-throughput sequencing produced between 1.2 and 12.9 million paired-end reads per sample. The initial quality filtering removed approximately 2.2% of the total paired-end reads. After, clean non-assembled reads were annotated taxonomically and revealed 50.24–77.84% bacterial, 3.40–12.35% eukaryote, 0.37–3.56% archaeal and 0.10–0.21% viral sequences in all the samples (Supplementary file S1: Table S2). Based on the Nonpareil approach, it was established that the obtained depth sequencing of the non-assembled reads covered between 24 and 92% of the expected metagenomes, being higher at the Hyperarid site samples compared with the Arid ones (Supplementary file S1: Table S3). This also indicated a lower diversity at the Hyperarid sites. Regarding the contig quality, assembled reads produced between 72,209 and 602,537 contigs larger than 500 bp with a mean length (N50) between 634 and 1437 bp. The maximum length of the total contigs fluctuated between 14 and 222 kb (sequencing data provided in Supplementary file S1: Table S4).
3.2. General Functional Potential in the Atacama Desert Soils and Taxonomic Diversity
On the basis of the general functional annotation, putative members of the COG categories “replication, recombination and repair (L),” “amino acid transport and metabolism (E)” and “energy production and conversion (C)” were the most abundant in samples from the investigated sites (Figure 1A). Hyperarid soils were significantly enriched with members of the COG category “replication, recombination and repair.” This enrichment was caused by a higher abundance of transposases and integrases. Further, the categories “cell cycle control (D),” “cell wall/membrane/envelope biogenesis (M),” “translation, ribosomal structure and biogenesis (J),” “nucleotide transport and metabolism (F)” and “inorganic ion transport and metabolism (P),” were significantly more abundant at the Arid sites compared with the other two sites (Figure 1A). In more detail, putative transposases were the most abundant genes at all sites (3.57 ± 0.55%), followed by (ABC) transporter (2.04 ± 0.19%), histidine kinase (1.47 ± 0.18%) and transcriptional regulator (1.41 ± 0.08%) associated genes (Supplementary file S1: Figure S1). Dehydrogenase and oxidoreductases were the most abundant gene/function associated with the “energy production (C)” COG category.
Based on a general taxonomic annotation of the metagenomic reads, Actinobacteria was the most abundant phylum detected in the samples with 57.97% at Hyperarid, 44.14% at Arid and 36.80% at Hyperarid-BBoulder sites. Proteobacteria were also highly abundant in samples from the Hyperarid-BBoulder and Arid sites in comparison to the Hyperarid site, 20.15, 18.80 and 13.56%, respectively. Other less abundant phyla found at the sites were Chloroflexi and Cyanobacteria, more abundant at the Hyperarid-BBoulder sites and Firmicutes, Bacteroidetes, Acidobacteria and Deinococcus-Thermus more abundant at the Arid sites (Supplementary file S1: Table S5).
3.3. Diversity of MGEs
Between the three sites, the relative abundance of MGEs was significantly different (robust ANOVA, p = 0.046), being more abundant at the Hyperarid sites compared with the Arid sites (Supplementary file S1: Table S6). Among the sampled sites, transposases were the most dominant MGE, follow by phage integrases, other elements, resolvases, transposons, integrases and recombinases (Figure 2). The relative number of phage integrases, transposons, resolvases and recombinases was significantly increased at the Hyperarid sites compared with the Arid sites. Screening of transposase families in the metagenomic contigs led to identifying several groups, IS630 contributing the most to the total abundance (0.23%) followed by IS5/IS427 (0.15%), IS10 (0.14%) and IS3/IS3 (0.13%) (Supplementary file S1: Table S7). The latter three families were enriched at the Hyperarid sites compared with the Arid sites. In addition, transposase families corresponding to two transposons Tn3 (0.026 ± 0.007%) and Tn7 (0.0003 ± 0.0001%) were identified in all the sites, being again more abundant at the Hyperarid sites (0.036 and 0.0005% respectively). Only for the transposases, the influence of the UV stress was visible with lower relative abundance at the Hyperarid-BBoulder sites (reduced UV light).
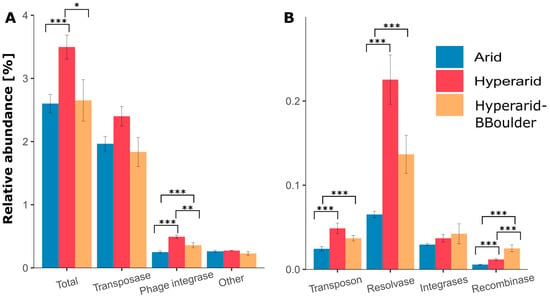
Figure 2.
The relative abundance and diversity of (A) high abundant and (B) low abundant mobile genetic elements (MGEs) in different locations of the Atacama Desert. The standard error is shown, n = 4, 5 and 3 at Arid, Hyperarid and Hyperarid-BBoulder sites, respectively. Robust ANOVA, * < 0.05, ** < 0.005 and *** < 0.0005. Other: DDE (super family endonuclease) and DUF (domain of unknown function) elements related to transposases.
Microbial diversity of the different samples was calculated using the Nonpareil diversity index, which is a measurement of the complexity of the community based on the “sequence space.” Arid sites showed the highest diversity levels (21.41 ± 0.24), contrasted with the Hyperarid-BBoulder (20.42 ± 0.20) and Hyperarid sites (19.04 ± 0.58). Overall, we found a negative correlation between the Nonpareil diversity index and the log10 transformed relative abundance of total MGEs (Spearman’s rank correlation rho −0.78, s = 392, p-value = 0.007, Supplementary file S1: Figure S2). Kaiju was used for the taxonomic prediction of the contigs harboring genes identified as MGEs. Of the total MGEs annotated, Actinobacteria (38.10 ± 6.65%) contributed the most to the MGE population in the Atacama Desert, followed by Proteobacteria (23.67 ± 5.33%), Chloroflexi (9.40 ± 1.31%) and Cyanobacteria (4.71 ± 0.55%) (Supplementary file S1: Figure S3). Actinobacterial MGEs were enriched at the Hyperarid sites compared with the Arid and Hyperarid-BBoulder sites. In contrast, Proteobacterial MGEs were enriched at the Hyperarid-BBoulder sites compared with the Hyperarid and Arid sites. MGEs abundance among the phyla Cyanobacteria and Deinococcus-Thermus was not different between the sites. Between 0.054 and 0.094% of the contigs were identified as MGEs of viral origin.
3.4. Co-Occurrence of MGEs and COG Categories
In total, 121,352 ORFs were found co-occurring with MGEs and between 37.60 and 52.01% were functionally annotated. The most abundant ORFs associated with MGEs belonged to the COG category “replication, recombination and repair (L),” followed by “transcription (K)” and “signal transduction mechanisms (T)” (Figure 1B). However, the genes found co-occurring with MGEs for those categories did not significantly differ between the sites. Besides, the relative abundance of genes associated to the COG categories “energy production and conversion (C),” “translation, ribosomal structure and biogenesis (J),” as well as “intracellular trafficking, secretion and vesicular transport (U)” co-occurring with MGEs was significantly different between the sites. They were more abundant at the Hyperarid and Hyperarid-BBoulder sites compared with the Arid sites. Between 38.3 and 41.6% of the ORFs co-occurring with MGEs were assigned to the unspecific category “function unknown (S).” Furthermore, different trends were observed in comparison with the general functional annotation. For example, “replication, recombination and repair (L)” accompanied by “amino acid transport and metabolism (E),” as well as “energy production and conversion (C)” were the most abundant categories when the total number of annotated genes were compared between the samples (Figure 1A).
3.5. Genes Coding for Enzymes Involved in Energy Production and Housekeeping Co-Occurring with MGEs
Several ORFs annotated as dehydrogenases, reductases and hydrolases were found co-occurring with MGEs (Figure 3). Genes coding for dihydrolipoyl dehydrogenase, monooxygenase and aldo-keto reductase were the most abundant COGs of the category C co-occurring with MGEs. All of them were more abundant at the Hyperarid and Hyperarid-BBoulder sites compared with the Arid sites. In addition, genes coding for enzymes involved in nitrogen metabolism as nitroreductase (COG0778), formamidase (COG2421) and nitrite reductase (COG2421) were more often co-occurring with MGEs at the Hyperarid sites (Figure 3). Furthermore, COGs linked to the sulphur cycle were close to MGEs. For example, (COG3119) was more abundantly co-occurring at the Arid sites compared with Hyperarid-BBoulder and Hyperarid sites. Nevertheless, (COG2897) co-occurrence was higher at the Hyperarid sites compared with Hyperarid-BBoulder and Arid sites. Further analysis showed that COGs associated with the ribosomal structure (category J) as amidotransferase (COG0154), acetyltransferase (COG1670) and Glycyl-tRNA synthetase (COG0423) were the most commonly co-occurring with MGEs from this category. Besides, genes coding for enzymes driving conjugation (COG4962) and maintenance of the outer membrane integrity (tolB-COG0823 and yidC-COG0706) were the most abundant linked to secretion and transport (category U), all co-occurring with MGEs at the Hyperarid sites in comparison with the other sites. The most abundantly co-occurring COGs with MGEs from categories C, J, U and L are listed in the Supplementary file S1: Table S8.
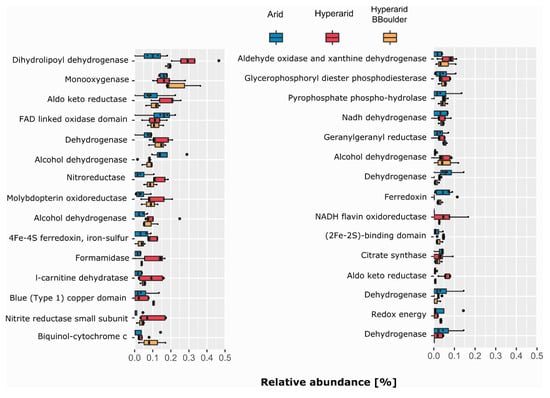
Figure 3.
COGs related to energy production co-occurring with MGEs. The 30 most abundant COGs co-occurring with MGEs were selected. n = 4, 5 and 3 at the Arid, Hyperarid and Hyperarid-BBoulder sites, respectively. COG numbers can be found in the supplementary material.
From the total ORFs functionally annotated co-occurring with MGEs, only 28 COGs were significantly different between the sites (DESeq2, LRT p < 0.05), several of them more abundantly co-occurring with MGEs at the Hyperarid sites (Figure 4A). Histidine kinase (ENOG410XNMH) involved in environmental sensing was the most abundant co-occurring with MGEs and enriched at the Hyperarid and Hyperarid-BBoulder sites. Further, cobyrinic acid a,c-diamide synthase (COG1192), dihydrolipoyl dehydrogenase (COG1249), helicase (COG0553), conjugation (COG4962), chlorite dismutase (ENOG4111JF8), dehydrogenase (COG2133), thioesterase (COG2050) and binding-DNA proteins were COGs co-occurring significantly more at the Hyperarid sites (Supplementary file S1: Table S9).
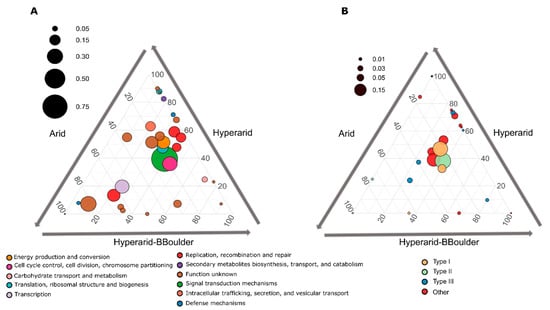
Figure 4.
(A) Significantly enriched Cluster Orthologous groups (COGs) between the Hyperarid, Hyperarid-BBoulder and Arid sites co-occurring with MGEs in the microbial community of the Atacama Desert and (B) COGs associated with DNA restriction. n = 4, 5 and 3 at Arid, Hyperarid and Hyperarid-BBoulder sites, respectively. COGs with a p < 0.05 using DESeq2. The diameter of the circle represents the relative abundance of every COG on average co-occurring with MGEs. Number and lines indicate the contribution of every category to the dot position.
As different COG numbers have the same protein name/description, we decided to merge them and further evaluate which protein name/description was associated with the ORFs co-occurring with MGEs (Figure 5). Transcriptional regulator was the most abundant protein description, representing 1.60, 1.79 and 2.22% (relative abundance of annotated ORF) at the Arid, Hyperarid and Hyperarid-BBoulder sites respectively. Histidine kinase and (ABC) transporter were the following most dominant. We observed that histidine kinase was more abundant at the Hyperarid and Hyperarid-BBoulder sites compared with the Arid sites. (ABC) transporter followed a similar trend being more abundant at the Hyperarid-BBoulder and Hyperarid sites compared with the Arid sites. Besides, dehydrogenase, helicase, DNA methylases and hydrolases were associated in higher frequencies with MGEs at the Hyperarid sites compared with the other two sites. Similar trends were observed when the name/descriptions obtained by the total functional annotation were compared (Supplementary file S1: Figure S1). In addition, between 15.27 and 17.46% of the annotated ORFs could not be linked to a protein description.
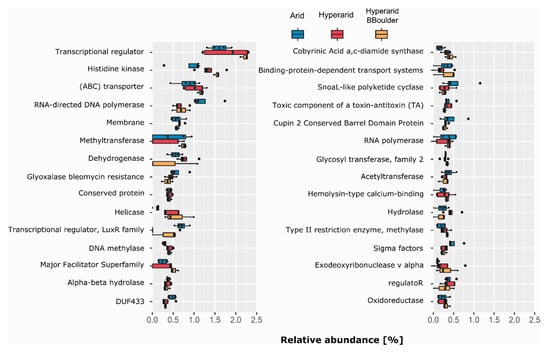
Figure 5.
The most abundant COG protein name/description co-occurring with MGEs. COGs with the same protein name/description were merged. n = 4, 5 and 3 at the Arid, Hyperarid and Hyperarid-BBoulder sites, respectively. The 30 most abundant protein names/descriptions are shown.
3.6. Genes Co-Occurring with MGEs Relative to the Total Functional Potential per COG Category
The fraction of genes co-occurring with MGEs within each COG category was analysed. For most of the COG categories, this fraction was the highest in the Hyperarid sites and the lowest in the Arid sites (Figure 1C). Only the fraction of genes co-occurring with MGEs within the category “chromatin structure and dynamics (B)” was the highest in the Hyperarid-BBoulder sites and the lowest in the Hyperarid sites.
When calculating the average fraction of genes co-occurring with MGEs within each COG category among all sites, the lowest number of genes co-occurred with MGEs within the “chromatin structure and dynamics (B)” category (0.61%) and the highest number co-occurred within the “extracellular structures (W)” category (7.1%). However, high variability between samples was observed for those two categories. Following the category “extracellular structures (W),” the highest fractions of genes co-occurring with MGEs were found within the categories “cytoskeleton (Z)” (6.19%), “transcription (K)” (3.54%) and “defence mechanisms (V)” (3.22%).
Within all COG categories per site, the average highest fraction of genes co-occurring with MGEs was found in the Hyperarid sites (3.91%), followed by Hyperarid-BBoulder (2.23%) and Arid sites (1.12%). Further, the fraction of genes co-occurring with MGEs per each category was compared to the average fraction of genes co-occurring with MGEs within all the COG categories per site (Supplementary file S1: Figure S4). Only in three categories, the fraction of genes co-occurring with MGEs was significantly higher compared to the average, while it was lower in almost half of the categories. The enriched categories were “defence mechanisms (V)” and “transcription (T)” in the Arid sites and “unknown function (S)” in the Arid and Hyperarid sites. Besides, the lowest co-occurrence of genes with MGEs was found for “chromatin structure and dynamics (B)” and “cell motility (N),” both in the Hyperarid sites.
At a lower category level, the 30 most abundant protein functions (genes merged by COG name/description) co-occurring with MGEs were normalized to the total genes predicted by each function. In general, 1.24 to 20.89% of the genes annotated per function were found to co-occur with MGEs. The highest association was observed for RNA-directed DNA polymerase (20.89%), followed by uncharacterized protein (DUF33, 10.77%) and exodeoxyribonuclease (7.84%). Moreover, half of the evaluated protein functions co-occurring with MGEs were significantly different between the sites (Supplementary file S1: Figure S5). The percentage of genes co-occurring with MGEs associated with DNA replication and repair, transcription, environmental sensing, energy production and defence mechanism was consistently higher in samples from the Hyperarid sites compared to the other two sites.
3.7. Genes Related to Restriction-Modification and Co-Occurrence with MGEs
KO assignment using GhostKOALA showed several restriction-modification systems (0.25–1.67% of the annotated ORFs) among the most abundant genes co-occurring with MGEs. For example, Type I restriction enzyme (hsdM, hsdR, hsdS), restriction system protein (mrr) and Type III restriction (res) (Supplementary file S1: Figure S6). As we observed that MGEs in the Atacama Desert soils were co-occurring with genes coding for enzymes involved in restriction-modification and the COG category “Defence mechanism” was enriched at the Hyperarid sites, we manually picked all the COGs associated with them. We found 40 different COGs, which represented 1.47, 1.13 and 0.84% of the ORFs co-occurring with MGEs at the Hyperarid, Hyperarid-BBoulder and Arid sites, respectively. Genes linked to Type II restriction enzyme-methylase (COG1002) and Type I restriction-modification system (COG4096 and COG0286) were the most abundant at the Hyperarid, Hyperarid-BBoulder and Arid sites. Genes coding for restriction enzymes were consistently more abundant at the Hyperarid sites (Figure 4B, Supplementary file S1: Figure S7). Our data indicated that only ORFs annotated as Type I restriction-modification systems were significantly more abundant at the Hyperarid sites (Robust ANOVA, p = 0.02), whereas the Type III restriction enzyme (res subunit) was significantly more abundant at the Hyperarid-BBoulder sites (Robust ANOVA, p = 0.002).
3.8. Stress Response genes and Co-Occurrence with MGE
In total, we found 1017 ORF coding for stress response, representing between 0.024–0.035% of the total genes predicted in all the sites. Stress response genes were more abundant at the Arid and Hyperarid-BBoulder sites compare with the Hyperarid sites (Figure 6A). The most abundant genes of this group coded for the cold-shock protein cspA (0.0086%), chaperone groEL (0.0082%), cold-shock protein (0.0034%), ATP-dependent protease clpC (0.0031%), chaperone danK (0.0021%) and RNA polymerase sigma factor rpoD (0.0013%). Significant differences were found comparing the abundance of groEl and cspB between Arid and Hyperarid sites; a lower abundance of these genes was observed for the Hyperarid sites (Figure 7). In addition, danK, groEl and cspB were significantly enriched at the Hyperarid-BBoulder sites compared with the Hyperarid sites. Vice versa, genes belonging to cold-shock proteins were more abundant in samples from the Hyperarid sites compared with the other sites. Only a small fraction of the stress response genes were co-occurring with MGEs (Figure 6B). KO assignments also showed stress response genes co-occurring with MGEs. For example, DNA replication and repair protein (recF) and glutamine synthetase (glnA, linked to nitrogen stress response), enriched at the Hyperarid sites. The RNA polymerase sigma-70 factor (rpoE), involved in envelope stress response, was the most abundant gene co-occurring with MGE and enriched at the Arid sites (Supplementary file S1: Figure S6).
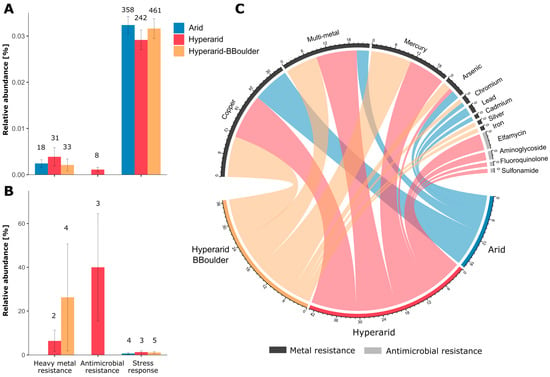
Figure 6.
Bacterial genes coding for antimicrobial resistance, heavy metal resistance and stress response. (A) The relative abundance of heavy metal and antimicrobial resistance and stress response genes, (B) relative abundance of the heavy metal and antimicrobial resistance and stress response genes co-occurring with MGEs, (C) resistance towards antimicrobials and heavy metals conferred by the genes detected. For (A,B) the number on top of the bars indicate the ORFs predicted coding for that function and the standard error is shown. n = 4, 5 and 3 at Arid, Hyperarid and Hyperarid-BBoulder sites, respectively.
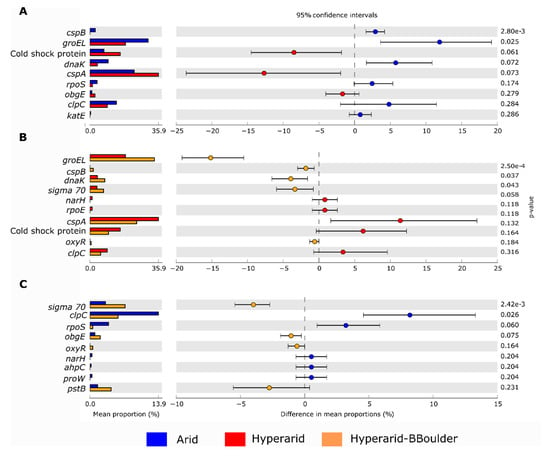
Figure 7.
Comparison of bacterial genes coding for stress response in the soil microbiome of the Atacama Desert between (A) Arid and Hyperarid sites, (B), Hyperarid and Hyperarid-BBoulder sites and (C) Arid and Hyperarid-BBoulder sites. Relative abundance was compared using two-side White’s non-parametric t-test. n = 4, 5 and 3 at the Arid, Hyperarid and Hyperarid-BBoulder sites, respectively.
3.9. Antimicrobial and Metal Resistance Genes
We detected a total of 82 ORFs coding for heavy metal resistance and 8 driving antimicrobial resistance (Figure 6A). Genes coding for heavy metal resistance were more abundant at the Hyperarid sites than both Arid and Hyperarid-BBoulder sites. However, these differences were not significant. On the other hand, genes coding for antimicrobial resistance were only detected at the Hyperarid sites. Predicted metal resistance determinants were mainly related to copper, multi-metal and mercury resistance followed by arsenic, chromium, silver and iron resistance (Figure 6C). Genes driving multi-metal and mercury resistance were principally found in samples from Hyperarid and Hyperarid-BBoulder sites, whereas genes responsible for chromium, lead and cadmium resistance was detected at the Arid sites. Predicted genes driving resistance towards antimicrobial compounds were associated with elfamycin, aminoglycoside, fluoroquinolone and sulfonamide resistance. Only 6.4 (2 genes) and 12.1% (4 genes) of the total heavy metal resistance genes were found co-occurring with MGEs, at the Hyperarid and Hyperarid-BBoulder sites, respectively, whereas 37% (3 genes) of the antimicrobial resistance genes detected were close to MGEs (Figure 6B).
4. Discussion
4.1. MGEs as a Driver for Adaptation of the Soil Microbiome to Extreme Conditions
Mobile genetic elements (MGEs) play an essential role in bacterial evolution and adaptation. Even though it has been postulated that MGEs are selfish elements, their evolutionary success could indicate an advantage for their host under stressful conditions. Several studies have reported an increased abundance and mobility of MGEs in microbiomes under the pressure of antibiotics [38,39,40]. Furthermore, it has been observed that some of the microorganisms most resistant to extreme conditions encode a high amount of MGEs, for example, Deinococcus radiodurans showing remarkable resistance to ionizing radiation, desiccation, UV-irradiation, oxidizing agents and electrophilic mutagens [41]. We postulated that environmental conditions could determine the enrichment of MGEs and which genes co-occur with them. Indeed, we observed that MGEs, mainly transposases, were enriched at the Hyperarid sites, where the most extreme conditions of the desert are found. These findings are supported by a meta-analysis, which has shown that the number of transposases and the evolution rate of microbial communities from extreme environments are higher compared with benign environments [5]. Similar observations like ours were made for other extreme environments, including hydrothermal chimney biofilms [42], hot springs, saline lakes [5] and the Baltic Sea [43]. In this way, microbial communities living in Atacama Desert soils should be adapted to the harsh surroundings and the enrichment of MGEs can indicate an evolutionary adaptation to those conditions.
MGEs associated with Actinobacteria were the most abundant, followed by Proteobacteria. Besides, we also found Actinobacteria and Proteobacteria as the most dominant phyla at the investigated sites in our study. This distribution, to some extent, represents the microbial taxonomy in the Atacama Desert. Actinobacteria have been found as the most dominant phylum on the surface of the most arid regions of the desert, whereas Proteobacteria and Firmicutes were found in subsurface soils [3]. A similar pattern was found in the Baltic Sea, where most of the transposases were harbored by the most dominant taxa. The authors of this study argued that transposases might be positively selected due to the variable and stressful environmental conditions [43]. The dominance of Actinobacteria carrying MGEs at the Hyperarid sites can also be a consequence of the adaptation of members of this phylum to desiccation and UV-irradiation [44,45]. In contrast, MGEs harboured by Proteobacteria increased at the Hyperarid-BBoulder sites compared with the Arid- and Hyperarid sites. However, we could not observe a clear difference in the total abundance of Proteobacteria between the Hyperarid-BBoulder and Arid sites. Below the boulders, lower UV-irradiation and higher humidity could be found in comparison with the surface soils, as it has been reported for porous and translucent rocks [46]. These conditions could be favourable for the exchange of genetic material between the members of Proteobacteria. As described before, the surface below boulders can confer shelter to some bacterial groups. For example, it is known that Cyanobacteria are enriched in hypoliths and are highly adapted to desert-like conditions [32,47]. They also harbour different mechanisms for DNA repair and UV protection, which is an adaptive advantage in desert soils. However, we did not find any difference between the total abundance of this phylum and the MGEs associated with them comparing the Hyperarid-BBoulder with the other two sites. Cyanobacteria populate semi-translucent rocks, where they can receive different amounts of light [48], which is limited below non-translucent boulders in the desert. Interestingly, bacterial communities from the Hyperarid regions were less diverse in contrast to the Arid sites. Likewise, the abundance of MGEs was negatively correlated with this factor. Similarly, in hydrothermal chimney biofilms, where the diversity is shallow, the abundance of transposases is high [42]. It seems that bacterial communities with low diversity need an extensive gene exchange to increase genomic diversity and environmental adaptation [9].
4.2. MGEs and Co-Occurring Genes
In our results, MGEs co-occurred mainly with genes involved in DNA replication and repair, transcription and environmental sensing. DNA replication and repair genes were also found as the most abundant genes in the total functional description of the different sites, which was not observed for transcription and environmental sensing. Thus, transcription- and environmental sensing-like genes seem to be preferentially found close to MGEs. Interestingly, low abundant genes as those coding for extracellular structures and cytoskeleton functions are also highly associated with MGEs. As the most abundant COG detected co-occurring with MGEs largely represents housekeeping functions, we cannot imply that the primary role of the MGEs is the mobilization of those genes, mainly because by definition, the host contains a functioning copy of each housekeeping gene. Besides, some housekeeping genes related to translation and transcription are less likely to be transferred [49]. However, any co-occurrence that results in an advantage would have an increased chance of being maintained. Instead, MGEs can be involved in modulating the expression of these housekeeping genes [13]. In the present study, one of the most enriched genes close to MGEs was related to transcriptional factors or genes from the category “transcription (K).” The function and structure of those genes can be affected by neighboring MGEs. For example, insertion sequences can provide complete or hybrid promoters and can alter the expression of adjacent genes in non-coding regions. In addition, it has been reported that transposases and transposons interact with different essential factors for DNA replication and repair, such as the β-clamps [50,51]. The ability of some MGEs to interact with the β-clamps could result in a transposition event. This might explain the preference of MGEs to co-occurred with this type of genes. In addition, studies using mobilomic approaches in soil samples have found that replication, conjugation, mobilization and stabilization are standard functions among MGEs like plasmids [52]. This is due to the presence of the genetic machinery for their replication and mobilization. This was observed in our data due to the high number of RNA-directed DNA polymerases and conjugation elements close to the MGEs. Additionally, COGs associated with conjugation were more abundant at the Hyperarid sites suggesting that the potential for DNA exchange could be higher. The interaction of MGEs with DNA replication and repair factors could also suggest a potential for gene duplication, which could under harsh environmental conditions represent an advantage. Duplication of DNA repair genes can be induced by the presence of transposons [53]. Besides, the accumulation of environmental sensing factors, such as histidine kinases, can be seen as an asset due to their crucial role in niche adaptation [54]. Bacteria can acquire and accumulate histidine kinases through HGT mechanisms, which usually conserves the operon structure and their response regulators [55]. We found a high co-occurrence of MGEs, histidine kinases and transcriptional regulators. We can speculate that the interaction between MGEs and environmental sensing factors could play an essential role in the adaptation to the extreme conditions in the desert soil. However, those genes have been described as the most prevalent in nature [8], which was also observed in our data. In that way, the co-occurrence of MGEs, histidine kinases and transcriptional regulators could be an artifact of their high abundance.
Besides, we observed an enrichment of MGEs co-occurring with genes associated with energy production, intracellular trafficking, secretion, vesicular transport and DNA restriction at the Hyperarid sites. Comparing the total abundance of genes coding for energy production, we found the opposite trend. Whereas the total of genes coding for enzymes involved in energy production were enriched at the Arid sites, the fraction of those genes close to MGEs was significantly enriched at the Hyperarid site. This indicates that these genes could be selected by MGEs at the Hyperarid sites, probably to overcome the low availability of nutrients. For example, the gene coding for dihydrolipoyl dehydrogenase was the most abundant gene related to energy production co-occurring with MGEs. This enzyme, involved in the TCA cycle, has been recognized as a central step in the carbohydrate metabolism in the Nabim Desert [4]. The authors of this study suggested that nutrients acquisition (nitrogen, phosphorus and sulfur assimilation and carbon fixation) rather than the stress response has a central role in the transcriptional activity. We also observed the co-occurrence of MGEs with enzymes involved in nitrogen assimilation (nitrite reductases, formamidase and nitroreductase) and alternative carbon metabolism and detoxification (aldo-keto reductases and chlorite dismutase) [56]. Chlorite dismutase is often found in perchlorate- or chlorate-reducing bacteria, also co-occurring with MGEs [57]. Hyperarid Atacama Desert soils contain perchlorate salts and the presence of this enzyme close to MGEs can indicate a selection and possible advantage due to the use of alternative nutrient sources [58].
Furthermore, predicted Restriction-modification (RM) systems co-occurring with MGEs were more abundant at the Hyperarid sites. RM systems are commonly found on potential MGEs as plasmids, prophages, integrons and transposons [14,59], which is an indication of their high mobility. RM systems are involved in recombination [60], genome rearrangement [61,62], differential gene expression [63,64] and nutrition in viruses [65], which are all putative strategies to increase survival and environmental adaptation. Besides, it was hypothesized that the release of cellular proteins after cell lysis, produced by post-segregational killing, could be exploited as nutrients by the cells retaining the RM systems [65,66]. This strategy could be relevant in nutrient-poor soils such as desert soil. Nonetheless, this mechanism needs more investigation.
4.3. Stress Response, Heavy Metal and Antimicrobial Resistance Genes Co-Occurring with MGEs
Interaction between MGEs and stress response genes has been described. For instance, the co-occurrence of an insertion sequence within the primary regulator of gene expression under stress conditions (rpoS), presumably increases nutrient scavenging [13]. Further, SOS response and HGT events induce each other and it could be considered as a mechanism for increasing bacterial genome plasticity [16]. In the present study, we detected several stress response genes associated with cold-shock, heat-shock and osmotic pressure, though only a small fraction was co-occurring with MGEs. The stress response is highly transcribed in potentially active microbial communities from desert soils but it seems that nutrient acquisition is a more critical factor [4]. Even so, rpoE co-occurring with MGEs was the most abundant gene identified with GhostKOALA in the present study. This gene has an essential role in the stress response of the cell envelope [67]. Osmotic pressure is one of the major threats in deserts soils due to the low water content. This could be the foremost reason for the selection of rpoE in the microbial communities of the Atacama Desert. Contrary to what was expected, stress response genes were not enriched at the most arid soils of the desert. Instead, MGEs were more abundant at the most arid soils, which could also suggest an essential role of these elements in such environments.
Antimicrobial and heavy metal resistance genes are relatively low abundant but a fraction has the potential for mobilization in the Atacama Desert. Resistance to antimicrobials and heavy metals has been found in extreme environments like Antarctica [68,69], hot deserts [70] and high-altitude wetlands [71]. However, it seems that heavy metal resistance is a more enriched functional trait compared with antimicrobial resistance in the Atacama Desert soils. This can be explained by the chemical nature of the soils (high in metals) and mining activities in the region [72] and the probable minimum input of antibiotic from human sources. Nevertheless, AMRs co-occurring with MGEs are rarely found in soils [73]. Additionally, SOS response to stress has been associated with the mobilization of antimicrobial resistance [71,74]. In this way, antimicrobial- and heavy metal resistance genes could be selected in the Atacama most arid soils due to the stressing conditions as UV-irradiation and desiccation.
5. Conclusions
Mobile genetic elements were enriched at the hyperarid core of the Atacama Desert, while stress response genes were not, which could suggest a more prominent role of MGEs in the microbiome adaptation to desert soils due to the selection of those elements. Further, MGEs were rarely found close to stress response genes. Instead, MGEs were co-occurring with several genes related to general functional traits as energy production, enriched at the hyperarid sites. Besides, the most dominant bacterial groups in the Atacama Desert soils, Actinobacteria, also harboured most of the MGEs. Overall, the present study suggests that MGEs could play an important role in the adaptation to the conditions present in desert soils by the possible modulation of housekeeping genes instead of the direct stress response. However, because we cannot state that the primary role of MGEs is to mobilize the different housekeeping genes, a more parsimonious explanation is that they also participate in gene regulation.
Supplementary Materials
The following are available online at https://www.mdpi.com/1424-2818/11/11/205/s1, Supplementary file S1 containing: Table S1. Chemical-physical characterization of soils from the Atacama Desert; Table S2. Taxonomic annotation of the non-assembled reads; Table S3. Nonpareil index and coverage; Table S4. Reads and contigs quality; Figure S1. The relative abundance of the most abundant COG name/description found in the total functional annotation. Table S5. The relative abundance of the most dominant phylum found in three regions of the Atacama Desert, Table S6. The relative abundance of MGE; Table S7. The relative abundance of the most abundant transposase families; Figure S2. The negative correlation between the nonpareil diversity index and the total relative abundance of the MGEs; Figure S3. Taxonomic annotation of the total MGEs found; Table S8. The most abundant COG of the significant different categories found between the soils; Table S9. Significantly different COGs between the three soils. DESeq2 < 0.05; Figure S4. Average fold change between the fraction of genes co-occurring with MGEs per each category and the average fraction of genes co-occurring with MGEs within all the COG categories; Figure S5. The most abundant COG protein name/description co-occurring with MGEs relative to the total of genes from the same COG protein name/description. Figure S6. Stress response genes and restriction-like genes predicted with GhostKOALA and KEEG; Figure S7. Most abundant COGs related to DNA restriction found co-occurring with MGEs. Supplementary file S2: In-home scripts for the detection of co-occurrence between MGEs and other genes.
Author Contributions
Conceptualization, J.S.S., M.S. and G.V.; formal analysis, J.S.S.; funding acquisition, D.S.-M. and M.S.; investigation, J.S.S., A.A. and D.S.-M.; software, G.V.; supervision, G.V.; visualization, J.S.S.; writing—original draft, J.S.S.; writing—review & editing, A.A., D.S.-M., M.S. and G.V.
Funding
This research was funded by the European Research Council (ERC), grant number 339231.
Acknowledgments
We are thankful to Helmholtz Graduate School Environmental Health (HELENA) for providing funding to J.S.S. and the ERC Advanced Grant HOME (#339231) to financially support this study.
Conflicts of Interest
The authors declare no conflict of interest.
References
- De Wit, R.; Bouvier, T. ‘Everything is everywhere, but, the environment selects’; What did Baas Becking and Beijerinck really say? Environ. Microbiol. 2006, 8, 755–758. [Google Scholar] [CrossRef] [PubMed]
- Rothschild, L.J.; Mancinelli, R.L. Life in extreme environments. Nature 2001, 409, 1092–1101. [Google Scholar] [CrossRef] [PubMed]
- Schulze-Makuch, D.; Wagner, D.; Kounaves, S.P.; Mangelsdorf, K.; Devine, K.G.; de Vera, J.P.; Schmitt-Kopplin, P.; Grossart, H.P.; Parro, V.; Kaupenjohann, M.; et al. Transitory microbial habitat in the hyperarid Atacama Desert. Proc. Natl. Acad. Sci. USA 2018, 115, 2670–2675. [Google Scholar] [CrossRef] [PubMed]
- Leon-Sobrino, C.; Ramond, J.B.; Maggs-Kolling, G.; Cowan, D.A. Nutrient acquisition, rather than stress response over diel cycles, drives microbial transcription in a hyper-arid Namib Desert soil. Front. Microbiol. 2019, 10, 1054. [Google Scholar] [CrossRef] [PubMed]
- Li, S.J.; Hua, Z.S.; Huang, L.N.; Li, J.; Shi, S.H.; Chen, L.X.; Kuang, J.L.; Liu, J.; Shu, W.S. Microbial communities evolve faster in extreme environments. Sci. Rep. 2014, 4, 6205. [Google Scholar] [CrossRef]
- Frost, L.S.; Leplae, R.; Summers, A.O.; Toussaint, A. Mobile genetic elements: The agents of open source evolution. Nat. Rev. Microbiol. 2005, 3, 722–732. [Google Scholar] [CrossRef]
- Orgel, L.E.; Crick, F.H. Selfish DNA: The ultimate parasite. Nature 1980, 284, 604–607. [Google Scholar] [CrossRef]
- Aziz, R.K.; Breitbart, M.; Edwards, R.A. Transposases are the most abundant, most ubiquitous genes in nature. Nucleic Acids Res. 2010, 38, 4207–4217. [Google Scholar] [CrossRef]
- Koonin, E.V. Viruses and mobile elements as drivers of evolutionary transitions. Philos. Trans. R. Soc. B 2016, 371, 20150442. [Google Scholar] [CrossRef]
- Partridge, S.R.; Kwong, S.M.; Firth, N.; Jensen, S.O. Mobile genetic elements associated with antimicrobial resistance. Clin. Microbiol. Rev. 2018, 31, e00088-17. [Google Scholar] [CrossRef]
- Stokes, H.W.; Gillings, M.R. Gene flow, mobile genetic elements and the recruitment of antibiotic resistance genes into Gram-negative pathogens. FEMS Microbiol. Rev. 2011, 35, 790–819. [Google Scholar] [CrossRef] [PubMed]
- Van Hoek, A.H.A.M.; Mevius, D.; Guerra, B.; Mullany, P.; Roberts, A.P.; Aarts, H.J.M. Acquired antibiotic resistance genes: An overview. Front. Microbiol. 2011, 2, 203. [Google Scholar] [CrossRef] [PubMed]
- Vandecraen, J.; Chandler, M.; Aertsen, A.; van Houdt, R. The impact of insertion sequences on bacterial genome plasticity and adaptability. Crit. Rev. Microbiol. 2017, 43, 709–730. [Google Scholar] [CrossRef]
- Oliveira, P.H.; Touchon, M.; Rocha, E.P. The Interplay of restriction-modification systems with mobile genetic elements and their prokaryotic hosts. Nucleic Acids Res. 2014, 42, 10618–10631. [Google Scholar] [CrossRef] [PubMed]
- Furuta, Y.; Kobayashi, I. Restriction-modification systems as mobile epigenetic elements. In Bacterial Integrative Mobile Genetic Elements; Landes Bioscience: Austin, TX, USA, 2011. [Google Scholar]
- Baharoglu, Z.; Mazel, D. SOS, the formidable strategy of bacteria against aggressions. FEMS Microbiol. Rev. 2014, 38, 1126–1145. [Google Scholar] [CrossRef] [PubMed]
- Narayan, A.; Jain, K.; Shah, A.R.; Madamwar, D. An efficient and cost-effective method for DNA extraction from athalassohaline soil using a newly formulated cell extraction buffer. 3 Biotech 2016, 6, 62. [Google Scholar] [CrossRef]
- Töwe, S.; Wallisch, S.; Bannert, A.; Fischer, D.; Hai, B.; Haesler, F.; Kleineidam, K.; Schloter, M. Improved protocol for the simultaneous extraction and column-based separation of DNA and RNA from different soils. J. Microbiol. Methods 2011, 84, 406–412. [Google Scholar] [CrossRef]
- Schubert, M.; Lindgreen, S.; Orlando, L. AdapterRemoval v2: Rapid adapter trimming, identification, and read merging. BMC Res. Notes 2016, 9, 88. [Google Scholar] [CrossRef]
- Schmieder, R.; Edwards, R. Fast identification and removal of sequence contamination from genomic and metagenomic datasets. PLoS ONE 2011, 6, e17288. [Google Scholar] [CrossRef]
- Menzel, P.; Ng, K.L.; Krogh, A. Fast and sensitive taxonomic classification for metagenomics with Kaiju. Nat. Commun. 2016, 7, 11257. [Google Scholar] [CrossRef]
- Rodriguez-r, L.M.; Konstantinidis, K.T. Nonpareil: A redundancy-based approach to assess the level of coverage in metagenomic datasets. Bioinformatics 2014, 30, 629–635. [Google Scholar] [CrossRef] [PubMed]
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.; Dvorkin, M.; Kulikov, A.S.; Lesin, V.M.; Nikolenko, S.I.; Pham, S.; Prjibelski, A.D.; et al. SPAdes: A new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 2012, 19, 455–477. [Google Scholar] [CrossRef] [PubMed]
- Finn, R.D.; Coggill, P.; Eberhardt, R.Y.; Eddy, S.R.; Mistry, J.; Mitchell, A.L.; Potter, S.C.; Punta, M.; Qureshi, M.; Sangrador-Vegas, A.; et al. The Pfam protein families database: Towards a more sustainable future. Nucleic Acids Res. 2016, 44, D279–D285. [Google Scholar] [CrossRef] [PubMed]
- Riadi, G.; Medina-Moenne, C.; Holmes, D.S. TnpPred: Aweb service for the robust prediction of prokaryotic transposases. Comp. Funct. Genom. 2012, 2012, 678761. [Google Scholar] [CrossRef]
- Mistry, J.; Finn, R.D.; Eddy, S.R.; Bateman, A.; Punta, M. Challenges in homology search: HMMER3 and convergent evolution of coiled-coil regions. Nucleic Acids Res. 2013, 41, e121. [Google Scholar] [CrossRef]
- Jensen, L.J.; Julien, P.; Kuhn, M.; von Mering, C.; Muller, J.; Doerks, T.; Bork, P. eggNOG: Automated construction and annotation of orthologous groups of genes. Nucleic Acids Res. 2008, 36, D250–D254. [Google Scholar] [CrossRef]
- Buchfink, B.; Xie, C.; Huson, D.H. Fast and sensitive protein alignment using diamond. Nat. Methods 2015, 12, 59–60. [Google Scholar] [CrossRef]
- Kanehisa, M.; Sato, Y.; Morishima, K. BlastKOALA and GhostKOALA: KEGG tools for functional characterization of genome and metagenome sequences. J. Mol. Biol. 2016, 428, 726–731. [Google Scholar] [CrossRef]
- Jia, B.F.; Raphenya, A.R.; Alcock, B.; Waglechner, N.; Guo, P.Y.; Tsang, K.K.; Lago, B.A.; Dave, B.M.; Pereira, S.; Sharma, A.N.; et al. CARD 2017: Expansion and model-centric curation of the comprehensive antibiotic resistance database. Nucleic Acids Res. 2017, 45, D566–D573. [Google Scholar] [CrossRef]
- Pal, C.; Bengtsson-Palme, J.; Rensing, C.; Kristiansson, E.; Larsson, D.G.J. BacMet: Antibacterial biocide and metal resistance genes database. Nucleic Acids Res. 2014, 42, D737–D743. [Google Scholar] [CrossRef]
- Le, P.T.; Makhalanyane, T.P.; Guerrero, L.D.; Vikram, S.; van de Peer, Y.; Cowan, D.A. Comparative metagenomic analysis reveals mechanisms for stress response in hypoliths from extreme hyperarid deserts. Genome Biol. Evol. 2016, 8, 2737–2747. [Google Scholar] [CrossRef] [PubMed]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2013. [Google Scholar]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed]
- Field, A.; Miles, J.; Field, Z. Discovering Statistics Using R; SAGE Publications Ltd.: London, UK, 2012; p. 992. [Google Scholar]
- Wilcox, R.R. Understanding and Applying Basic Statistical Methods Using R; John Wiley & Sons: Hoboken, NJ, USA, 2017. [Google Scholar]
- Parks, D.H.; Tyson, G.W.; Hugenholtz, P.; Beiko, R.G. Stamp: Statistical analysis of taxonomic and functional profiles. Bioinformatics 2014, 30, 3123–3124. [Google Scholar] [CrossRef] [PubMed]
- Muziasari, W.I.; Parnanen, K.; Johnson, T.A.; Lyra, C.; Karkman, A.; Stedtfeld, R.D.; Tamminen, M.; Tiedje, J.M.; Virta, M. Aquaculture changes the profile of antibiotic resistance and mobile genetic element associated genes in Baltic Sea sediments. FEMS Microbiol. Ecol. 2016, 92, fiw052. [Google Scholar] [CrossRef]
- Beaber, J.W.; Hochhut, B.; Waldor, M.K. SOS response promotes horizontal dissemination of antibiotic resistance genes. Nature 2004, 427, 72–74. [Google Scholar] [CrossRef]
- Saenz, J.S.; Marques, T.V.; Barone, R.S.C.; Cyrino, J.E.P.; Kublik, S.; Nesme, J.; Schloter, M.; Rath, S.; Vestergaard, G. Oral administration of antibiotics increased the potential mobility of bacterial resistance genes in the gut of the fish Piaractus mesopotamicus. Microbiome 2019, 7, 24. [Google Scholar] [CrossRef]
- Makarova, K.S.; Aravind, L.; Wolf, Y.I.; Tatusov, R.L.; Minton, K.W.; Koonin, E.V.; Daly, M.J. Genome of the extremely radiation-resistant bacterium Deinococcus radiodurans viewed from the perspective of comparative genomics. Microbiol. Mol. Biol. Rev. 2001, 65, 44–79. [Google Scholar] [CrossRef]
- Brazelton, W.J.; Baross, J.A. Abundant transposases encoded by the metagenome of a hydrothermal chimney biofilm. ISME J. 2009, 3, 1420–1424. [Google Scholar] [CrossRef]
- Vigil-Stenman, T.; Ininbergs, K.; Bergman, B.; Ekman, M. High abundance and expression of transposases in bacteria from the Baltic Sea. ISME J. 2017, 11, 2611–2623. [Google Scholar] [CrossRef]
- Lebre, P.H.; De Maayer, P.; Cowan, D.A. Xerotolerant Bacteria: Surviving through a Dry Spell. Nat. Rev. Microbiol. 2017, 15, 285–296. [Google Scholar] [CrossRef]
- Montero-Calasanz Mdel, C.; Goker, M.; Broughton, W.J.; Cattaneo, A.; Favet, J.; Potter, G.; Rohde, M.; Sproer, C.; Schumann, P.; Klenk, H.P.; et al. Geodermatophilus tzadiensis Sp. Nov., a Uv Radiation-Resistant Bacterium Isolated from Sand of the Saharan Desert. Syst. Appl. Microbiol. 2013, 36, 177–182. [Google Scholar] [CrossRef] [PubMed]
- Azua-Bustos, A.; Gonzalez-Silva, C.; Mancilla, R.A.; Salas, L.; Gomez-Silva, B.; McKay, C.P.; Vicuna, R. Hypolithic cyanobacteria supported mainly by fog in the coastal range of the Atacama Desert. Microb. Ecol. 2011, 61, 568–581. [Google Scholar] [CrossRef] [PubMed]
- Makhalanyane, T.P.; Valverde, A.; Gunnigle, E.; Frossard, A.; Ramond, J.B.; Cowan, D.A. Microbial ecology of hot desert edaphic systems. FEMS Microbiol. Rev. 2015, 39, 203–221. [Google Scholar] [CrossRef] [PubMed]
- Lacap-Bugler, D.C.; Lee, K.K.; Archer, S.; Gillman, L.N.; Lau, M.C.Y.; Leuzinger, S.; Lee, C.K.; Maki, T.; McKay, C.P.; Perrott, J.K.; et al. Global diversity of desert hypolithic cyanobacteria. Front. Microbiol. 2017, 8, 867. [Google Scholar] [CrossRef]
- Jain, R.; Rivera, M.C.; Lake, J.A. Horizontal gene transfer among genomes: The complexity hypothesis. Proc. Natl. Acad. Sci. USA 1999, 96, 3801–3806. [Google Scholar] [CrossRef]
- Diaz-Maldonado, H.; Gomez, M.J.; Moreno-Paz, M.; san Martin-Uriz, P.; Amils, R.; Parro, V.; de Saro, F.J.L. Transposase interaction with the β sliding clamp: Effects on insertion sequence proliferation and transposition rate. Sci. Rep. 2015, 5, 13329. [Google Scholar] [CrossRef]
- Zaratiegui, M. Cross-regulation between transposable elements and host DNA replication. Viruses 2017, 9, 57. [Google Scholar] [CrossRef]
- Luo, W.T.; Xu, Z.F.; Riber, L.; Hansen, L.H.; Sorensen, S.J. Diverse gene functions in a soil mobilome. Soil Biol. Biochem. 2016, 101, 175–183. [Google Scholar] [CrossRef]
- Martins-Pinheiro, M.; Galhardo, R.S.; Lage, C.; Lima-Bessa, K.M.; Aires, K.A.; Menck, C.F.M. Different patterns of evolution for duplicated DNA repair genes in bacteria of the Xanthomonadales group. BMC Evol. Biol. 2004, 4, 29. [Google Scholar] [CrossRef]
- Rodrigue, A.; Quentin, Y.; Lazdunski, A.; Mejean, V.; Foglino, M. Two-component systems in Pseudomonas aeruginosa: Why so many? Trends Microbiol. 2000, 8, 498–504. [Google Scholar] [CrossRef]
- Alm, E.; Huang, K.; Arkin, A. The evolution of two-component systems in bacteria reveals different strategies for niche adaptation. PLoS Comput. Biol. 2006, 2, 1329–1342. [Google Scholar] [CrossRef] [PubMed]
- Ellis, E.M. Microbial aldo-keto reductases. FEMS Microbiol. Lett. 2002, 216, 123–131. [Google Scholar] [CrossRef] [PubMed]
- Mlynek, G.; Sjoblom, B.; Kostan, J.; Fureder, S.; Maixner, F.; Gysel, K.; Furtmuller, P.G.; Obinger, C.; Wagner, M.; Daims, H.; et al. Unexpected diversity of chlorite dismutases: A catalytically efficient dimeric enzyme from Nitrobacter winogradsky. J. Bacteriol. 2011, 193, 2408–2417. [Google Scholar] [CrossRef] [PubMed]
- Calderon, R.; Palma, P.; Parker, D.; Molina, M.; Godoy, F.A.; Escudey, M. Perchlorate levels in soil and waters from the Atacama Desert. Arch. Environ. Contam. Toxicol. 2014, 66, 155–161. [Google Scholar] [CrossRef] [PubMed]
- Mruk, I.; Kobayashi, I. To be or not to be: Regulation of restriction-modification systems and other toxin-antitoxin systems. Nucleic Acids Res. 2014, 42, 70–86. [Google Scholar] [CrossRef] [PubMed]
- Murray, N.E. 2001 Fred Griffith review lecture. Immigration control of DNA in bacteria: Self versus non-self. Microbiology 2002, 148, 3–20. [Google Scholar] [CrossRef] [PubMed]
- Furuta, Y.; Abe, K.; Kobayashi, I. Genome comparison and context analysis reveals putative mobile forms of restriction-modification systems and related rearrangements. Nucleic Acids Res. 2010, 38, 2428–2443. [Google Scholar] [CrossRef]
- Asakura, Y.; Kojima, H.; Kobayashi, I. Evolutionary genome engineering using a restriction-modification system. Nucleic Acids Res. 2011, 39, 9034–9046. [Google Scholar] [CrossRef]
- Srikhanta, Y.N.; Maguire, T.L.; Stacey, K.J.; Grimmond, S.M.; Jennings, M.P. The phasevarion: A genetic system controlling coordinated, random switching of expression of multiple genes. Proc. Natl. Acad. Sci. USA 2005, 102, 5547–5551. [Google Scholar] [CrossRef]
- Hallet, B. Playing Dr Jekyll and Mr Hyde: Combined mechanisms of phase variation in bacteria. Curr. Opin. Microbiol. 2001, 4, 570–581. [Google Scholar] [CrossRef]
- Vasu, K.; Nagaraja, V. Diverse functions of restriction-modification systems in addition to cellular defense. Microbiol. Mol. Biol Rev. 2013, 77, 53–72. [Google Scholar] [CrossRef] [PubMed]
- Asakura, Y.; Kobayashi, I. From damaged genome to cell surface: Transcriptome changes during bacterial cell death triggered by loss of a restriction-modification gene complex. Nucleic Acids Res. 2009, 37, 3021–3031. [Google Scholar] [CrossRef] [PubMed]
- Jordan, S.; Hutchings, M.I.; Mascher, T. Cell envelope stress response in Gram-positive bacteria. FEMS Microbiol. Rev. 2008, 32, 107–146. [Google Scholar] [CrossRef] [PubMed]
- Romaniuk, K.; Ciok, A.; Decewicz, P.; Uhrynowski, W.; Budzik, K.; Nieckarz, M.; Pawlowska, J.; Zdanowski, M.K.; Bartosik, D.; Dziewit, L. Insight into heavy metal resistome of soil psychrotolerant bacteria originating from King George Island (Antarctica). Polar Biol. 2018, 41, 1319–1333. [Google Scholar] [CrossRef]
- Van Goethem, M.W.; Pierneef, R.; Bezuidt, O.K.I.; de Peer, Y.V.; Cowan, D.A.; Makhalanyane, T.P. A reservoir of ‘historical’ antibiotic resistance genes in remote pristine Antarctic soils. Microbiome 2018, 6, 40. [Google Scholar] [CrossRef]
- Noronha, M.F.; Lacerda, G.V.; Gilbert, J.A.; de Oliveira, V.M. Taxonomic and functional patterns across soil microbial communities of global biomes. Sci. Total Environ. 2017, 609, 1064–1074. [Google Scholar] [CrossRef]
- Dib, J.; Motok, J.; Zenoff, V.F.; Ordonez, O.; Farias, M.E. Occurrence of resistance to antibiotics, UV-B, and arsenic in bacteria isolated from extreme environments in high-altitude (above 4400 m) Andean wetlands. Curr. Microbiol. 2008, 56, 510–517. [Google Scholar] [CrossRef]
- Tapia, J.; Gonzalez, R.; Townley, B.; Oliveros, V.; Alvarez, F.; Aguilar, G.; Menzies, A.; Calderon, M. Geology and geochemistry of the Atacama Desert. Antonie Leeuwenhoek 2018, 111, 1273–1291. [Google Scholar] [CrossRef]
- Forsberg, K.J.; Patel, S.; Gibson, M.K.; Lauber, C.L.; Knight, R.; Fierer, N.; Dantas, G. Bacterial phylogeny structures soil resistomes across habitats. Nature 2014, 509, 612–616. [Google Scholar] [CrossRef]
- Poole, K. Bacterial stress responses as determinants of antimicrobial resistance. J. Antimicrob. Chemother. 2012, 67, 2069–2089. [Google Scholar] [CrossRef]







© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).