
Temporal Variation in the Genetic Composition of an Endangered Marsupial Reflects Reintroduction History

 , , and
, , and
Abstract
:1. Introduction
2. Materials and Methods
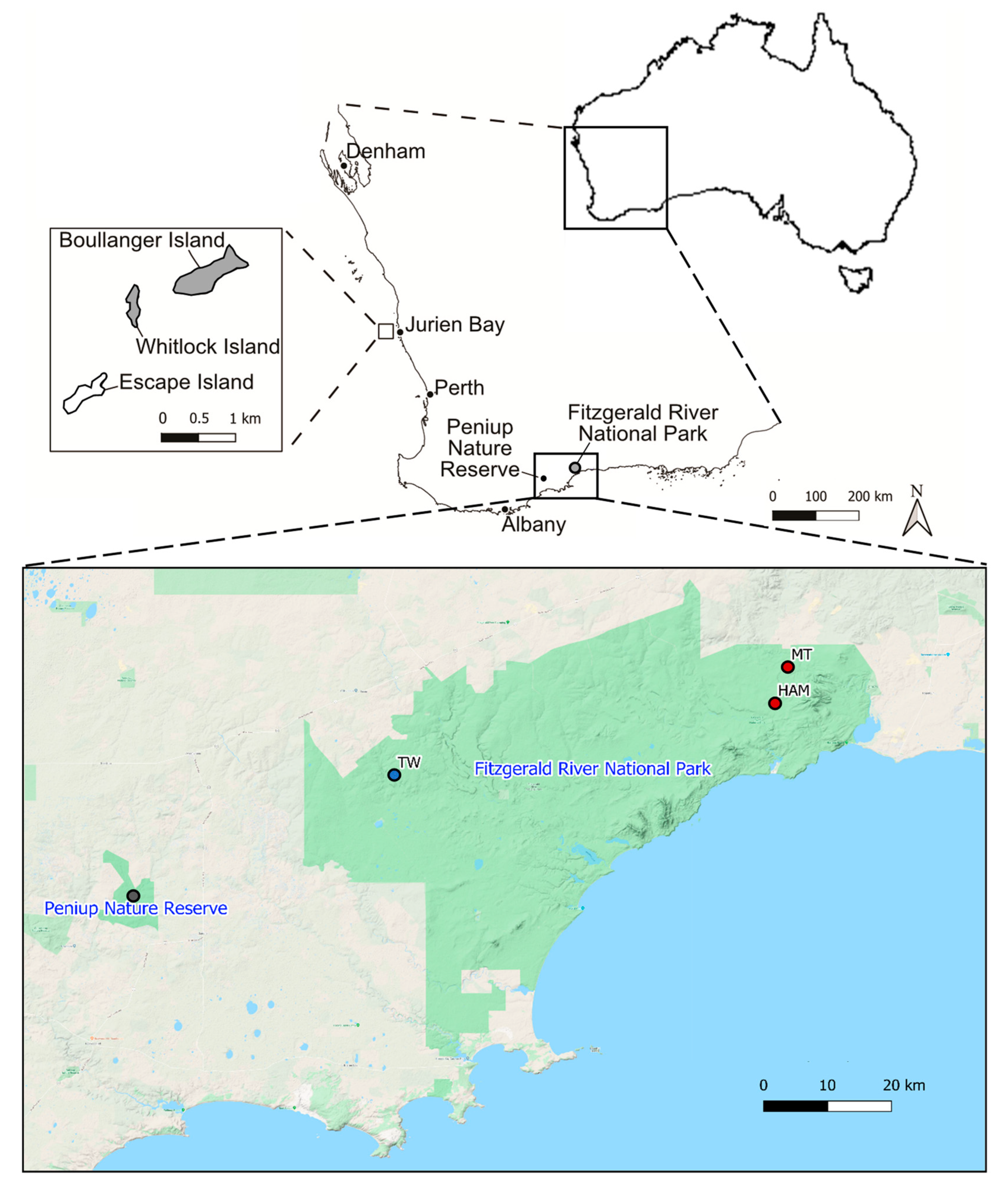
2.1. Sample Collection
2.2. Data Analysis
3. Results
3.1. Effects of Reintroduction on Genetic Variability
3.2. Population Structure of Captive and Reintroduced Populations
3.3. Genetic Relatedness Comparisons
4. Discussion
4.1. Genetic Consequences of Mixing Subpopulations
4.2. Consequences of Admixture on Population Structure
4.3. Genetic Mixing and Relatedness
4.4. Conservation Applications
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Woinarski, J.C.Z.; Burbidge, A.A.; Harrison, P. Ongoing unraveling of a continental fauna: Decline and extinction of Australian mammals since European settlement. Proc. Natl. Acad. Sci. USA 2015, 112, 4531–4540. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- IUCN/SSC. Guidelines for Reintroductions and Other Conservation Translocations. Version 1.0; IUCN Species Survival Commission: Gland, Switzerland, 2013; pp. 1–24. Available online: https://portals.iucn.org/library/sites/library/files/documents/2013-009.pdf (accessed on 27 May 2021).
- Fischer, J.; Lindenmayer, D. An assessment of the published results of animal relocations. Biol. Conserv. 2000, 96, 1–11. [Google Scholar] [CrossRef]
- Wolf, C.M.; Griffith, B.; Reed, C.; Temple, S.A. Avian and Mammalian Translocations: Update and Reanalysis of 1987 Survey Data. Conserv. Biol. 1996, 10, 1142–1154. [Google Scholar] [CrossRef]
- Wolf, C.M.; Garland, T.; Griffith, B. Predictors of avian and mammalian translocation success: Reanalysis with phylogenetically independent contrasts. Biol. Conserv. 1998, 86, 243–255. [Google Scholar] [CrossRef]
- Short, J. The Characteristics and Success of Vertebrate Translocations within Australia: A Progress Report to Department of Agriculture, Fisheries and Forestry; Wildlife Research and Management: Kalamunda, WA, Australia, 2009. [Google Scholar]
- Schwartz, M.K.; Luikart, G.; Waples, R.S. Genetic monitoring as a promising tool for conservation and management. Trends Ecol. Evol. 2007, 22, 25–33. [Google Scholar] [CrossRef] [Green Version]
- Ottewell, K.; Dunlop, J.; Thomas, N.; Morris, K.; Coates, D.; Byrne, M. Evaluating success of translocations in maintaining genetic diversity in a threatened mammal. Biol. Conserv. 2014, 171, 209–219. [Google Scholar] [CrossRef] [Green Version]
- Kennington, W.J.; Hevroy, T.H.; Johnson, M.S. Long-term genetic monitoring reveals contrasting changes in the genetic composition of newly established populations of the intertidal snail Bembicium vittatum. Mol. Ecol. 2012, 21, 3489–3500. [Google Scholar] [CrossRef]
- Robert, A. Captive breeding genetics and reintroduction success. Biol. Conserv. 2009, 142, 2915–2922. [Google Scholar] [CrossRef]
- Jamieson, I.G. Founder Effects, Inbreeding, and Loss of Genetic Diversity in Four Avian Reintroduction Programs. Conserv. Biol. 2010, 25, 115–123. [Google Scholar] [CrossRef]
- Hundertmark, K.; Van Daele, L.J. Founder effect and bottleneck signatures in an introduced, insular population of elk. Conserv. Genet. 2009, 11, 139–147. [Google Scholar] [CrossRef]
- Broders, H.G.; Mahoney, S.P.; A Montevecchi, W.; Davidson, W.S. Population genetic structure and the effect of founder events on the genetic variability of moose, Alces alces, in Canada. Mol. Ecol. 1999, 8, 1309–1315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Biebach, I.; Keller, L.F. A strong genetic footprint of the re-introduction history of Alpine ibex (Capra ibex ibex). Mol. Ecol. 2009, 18, 5046–5058. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, J.F.; English, S.; Goodall-Copestake, W.; Wang, J.; Walling, C.A.; Bateman, A.W.; Flower, T.P.; Sutcliffe, R.L.; Samson, J.; Thavarajah, N.K.; et al. Inbreeding and inbreeding depression of early life traits in a cooperative mammal. Mol. Ecol. 2012, 21, 2788–2804. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grueber, C.E.; Laws, R.J.; Nakagawa, S.; Jamieson, I.G. Inbreeding Depression Accumulation across Life-History Stages of the Endangered Takahe. Conserv. Biol. 2010, 24, 1617–1625. [Google Scholar] [CrossRef] [PubMed]
- Madsen, T.; Stille, B.; Shine, R. Inbreeding depression in an isolated population of adders Vipera berus. Biol. Conserv. 1996, 75, 113–118. [Google Scholar] [CrossRef]
- Laikre, L.; Ryman, N. Inbreeding Depression in a Captive Wolf (Canis lupus) Population. Conserv. Biol. 1991, 5, 33–40. [Google Scholar] [CrossRef]
- Bilski, D.R.; Pie, M.R.; Passos, F. Variable inbreeding effects across life-history stages in a captive carnivorous mammal population. Anim. Conserv. 2013, 16, 633–640. [Google Scholar] [CrossRef]
- Swinnerton, K.J.; Groombridge, J.J.; Jones, C.G.; Burn, R.W.; Mungroo, Y. Inbreeding depression and founder diversity among captive and free-living populations of the endangered pink pigeon Columba mayeri. Anim. Conserv. 2004, 7, 353–364. [Google Scholar] [CrossRef]
- Ransler, F.A.; Quinn, T.W.; Oyler-McCance, S.J. Genetic consequences of trumpeter swan (Cygnus buccinator) reintroductions. Conserv. Genet. 2011, 12, 257–268. [Google Scholar] [CrossRef]
- Binks, R.M.; Kennington, W.J.; Johnson, M.S. Rapid evolutionary responses in a translocated population of intertidal snail (Bembicium vittatum) utilise variation from different source populations. Conserv. Genet. 2007, 8, 1421–1429. [Google Scholar] [CrossRef]
- Thavornkanlapachai, R.; Mills, H.R.; Ottewell, K.; Dunlop, J.; Sims, C.; Morris, K.; Donaldson, F.; Kennington, W.J. Mixing Genetically and Morphologically Distinct Populations in Translocations: Asymmetrical Introgression in A Newly Established Population of the Boodie (Bettongia lesueur). Genes 2019, 10, 729. [Google Scholar] [CrossRef] [Green Version]
- Biebach, I.; Keller, L.F. Genetic variation depends more on admixture than number of founders in reintroduced Alpine ibex populations. Biol. Conserv. 2012, 147, 197–203. [Google Scholar] [CrossRef]
- Sigg, D.; Goldizen, A.; Pople, A. The importance of mating system in translocation programs: Reproductive success of released male bridled nailtail wallabies. Biol. Conserv. 2005, 123, 289–300. [Google Scholar] [CrossRef]
- Rick, K.; Ottewell, K.; Lohr, C.; Thavornkanlapachai, R.; Byrne, M.; Kennington, W.J. Population Genomics of Bettongia lesueur: Admixing Increases Genetic Diversity with no Evidence of Outbreeding Depression. Genes 2019, 10, 851. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Allendorf, F.W.; Luikart, G. Conservation and the Genetics of Populations; Blackwell Publishing: Carlton, Australia, 2007. [Google Scholar]
- Hedrick, P.W. Genetics of Populations, 2nd ed.; Jones and Bartlett publishers, Inc.: London, UK, 2000. [Google Scholar]
- Moro, D. Translocation of captive-bred dibblers Parantechinus apicalis (Marsupialia: Dasyuridae) to Escape Island, Western Australia. Biol. Conserv. 2003, 111, 305–315. [Google Scholar] [CrossRef]
- Miller, S.; Bencini, R.; Mills, H.; Moro, D. Food availability for the dibbler (Parantechinus apicalis) on Boullanger and Whitlock Islands, Western Australia. Wildl. Res. 2003, 30, 649–654. [Google Scholar] [CrossRef]
- Bencini, R.; McCulloch, C.; Mills, H.R.; Start, A.N. Habitat and diet of the dibbler (Parantechinus apicalis) on two islands in Jurien Bay, Western Australia. Wildl. Res. 2001, 28, 465–468. [Google Scholar] [CrossRef]
- Woolley, P.A. Dibbler. In The Mammals of Australia, 3rd ed.; Van Dyck, S., Strahan, R., Eds.; New Holland: Sydney, Australia, 2008; p. 39. [Google Scholar]
- Friend, T. Dibbler (Parantechinus apicalis) Recovery Plan. July 2003-June 2013; Department of Conservation and Land Management: Perth, Australia, 2003. [Google Scholar]
- Baynes, A. The mammal of Shark Bay, Western Australia. In Research in Shark Bay. Report of the France-Australe Expedition Committee; Berry, P.F., Bradshaw, S.D., Wilson, B.R., Eds.; Western Australian Museum: Perth, Australia, 1990; pp. 313–325. [Google Scholar]
- Baynes, A. The original mammal fauna of the Nullarbor and southern peripheral regions: Evidence from skeletal remains in superficial cave deposits. In A Biological Survey of the Nullarbor Region South and Western Australia in 1984; McKenzie, N.L., Robinson, A.C., Eds.; S.A. Department of Environment and Planning, Adelaide and W.A. Department of Conservation and Land Management, Perth: Adelaide, Australia, 1987; pp. 401–402. [Google Scholar]
- Morcombe, M.K. The rediscovery after 83 years of the Dibbler Antechinus apicalis (Marsupialia, Dasyuridae). West. Aust. Nat. 1967, 10, 103–111. [Google Scholar]
- Fuller, P.J.; Burbidge, A.A. Discovery of the Dibbler, Parantechinus apicalis, on islands at Jurien Bay. West. Aust. Nat. 1987, 16, 177–181. [Google Scholar]
- Chapman, A.D.; Newbey, K.R. A Biological Survey of the Fitzgerald Area, Western Australia; Department of Conservation and Land Management: Perth, Australia, 1995; Volume 3, pp. 1–258. [Google Scholar]
- Mills, H.; Bradshaw, F.; Lambert, C.; Bradshaw, S.; Bencini, R. Reproduction in the marsupial dibbler, Parantechinus apicalis; differences between island and mainland populations. Gen. Comp. Endocrinol. 2012, 178, 347–354. [Google Scholar] [CrossRef]
- Mills, H.R.; Moro, D.; Spencer, P.B.S. Conservation significance of island versus mainland populations: A case study of dibblers (Parantechinus apicalis) in Western Australia. Anim. Conserv. 2004, 7, 387–395. [Google Scholar] [CrossRef]
- Friend, T.; Collins, P. Male die-off in the dibbler: No evidence in mainland populations (Abstract). In Proceedings of the Australian Mammal Society: 51st Scientific Meeting Program and Abstracts, Albany, WA, Australia, 4–8 July 2005; p. 30. [Google Scholar]
- Aisya, Z.; White, D.; Thavornkanlapachai, R.; Rick, K.; Mitchell, N.J. Using PVA and captive breeding to balance trade-offs in the rescue of the island dibbler onto a new island ark. Sci. Rep. under review.
- Burbidge, A.A.; Woinarski, J. Parantechinus apicalis. The IUCN Red List of Threatened Species 2016: E.T16138A21944584. Available online: https://www.iucnredlist.org/species/16138/21944584 (accessed on 21 April 2021).
- Woinarski, J.; Burbidge, A.; Harrison, P. Action Plan for Australian Mammals 2012. In Action Plan for Australian Mammals 2012; CSIRO Publishing: Melbourne, Victoria, Australia, 2014; pp. 97–100. [Google Scholar]
- Thavornkanlapachai, R.; Kennington, W.J.; Ottewell, K.; Friend, J.A.; Mills, H.R. Dispersal, philopatry and population genetic structure of the mainland dibbler, Parantechinus apicalis. Conserv. Genet. 2019, 20, 1087–1099. [Google Scholar] [CrossRef]
- Sunnucks, P.; Hales, D.F. Numerous transposed sequences of mitochondrial cytochrome oxidase I-II in aphids of the genus Sitobion (Hemiptera: Aphididae). Mol. Biol. Evol. 1996, 13, 510–524. [Google Scholar] [CrossRef]
- Mills, H.R.; Spencer, P.B.S. Polymorphic microsatellites identified in an endangered dasyurid marsupial, the dibbler (Parantechinus apicalis). Mol. Ecol. Notes 2003, 3, 218–220. [Google Scholar] [CrossRef]
- Spencer, P.B.S.; Cardoso, M.; How, R.A.; Williams, J.; Bunce, M.; Schmitt, L.H. Cross-species amplification at microsatellite loci in Australian quolls including the description of five new markers from the Chuditch (Dasyurus geoffroii). Mol. Ecol. Notes 2007, 7, 1100–1103. [Google Scholar] [CrossRef]
- Firestone, K.B. Isolation and characterization of microsatellites from carnivorous marsupials (Dasyuridae: Marsupialia). Mol. Ecol. 1999, 8, 1084–1086. [Google Scholar] [CrossRef]
- Jones, M.E.; Paetkau, D.; Geffen, E.; Moritz, C. Microsatellites for the Tasmanian devil (Sarcophilus laniarius). Mol. Ecol. Notes 2003, 3, 277–279. [Google Scholar] [CrossRef]
- Banks, S.C.; Ward, S.J.; Lindenmayer, D.B.; Finlayson, G.R.; Lawson, S.J.; Taylor, A.C. The effects of habitat fragmentation on the social kin structure and mating system of the agile antechinus, Antechinus agilis. Mol. Ecol. 2005, 14, 1789–1801. [Google Scholar] [CrossRef] [PubMed]
- Kraaijeveld-Smit, F.J.L.; Ward, S.J.; Temple-Smith, P.D.; Paetkau, D. Factors influencing paternity success in Antechinus agilis: Last-male sperm precedence, timing of mating and genetic compatibility. J. Evol. Biol. 2002, 15, 100–107. [Google Scholar] [CrossRef]
- Pompanon, F.; Bonin, A.; Bellemain, E.; Taberlet, P. Genotyping errors: Causes, consequences and solutions. Nat. Rev. Genet. 2005, 6, 847–859. [Google Scholar] [CrossRef]
- Van Oosterhout, C.; Hutchinson, W.; Derek, P.; Wills, M.; Shipley, P. MICRO-CHECKER: Software for identifying and cor-recting genotyping errors in microsatellite data. Mol. Ecol. Notes 2004, 4, 535–538. [Google Scholar] [CrossRef]
- Rice, W.R. Analyzing tables of statistical tests. Evolution 1989, 43, 223–225. [Google Scholar] [CrossRef] [PubMed]
- Weir, B.S.; Cockerham, C.C. Estimating F-Statistics for the analysis of population structure. Evolution 1984, 38, 1358–1370. [Google Scholar] [PubMed]
- Goudet, J. FSTAT, a Program to Estimate and Test Gene Diversities and Fixation Indices Version 2.9.3. Available online: https://www.semanticscholar.org/paper/FSTAT%2C-a-program-to-estimate-and-test-gene-and-from-Goudet/5dc6a83189eb3f49d3887d2d28ada3bf1ebabd57 (accessed on 25 September 2017).
- Peakall, R.; Smouse, P.E. GenAlEx 6.5: Genetic analysis in Excel. Population genetic software for teaching and research—An update. Bioinformatics 2012, 28, 2537–2539. [Google Scholar] [CrossRef] [Green Version]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2018. [Google Scholar]
- Waples, R.S.; Do, C. ldne: A program for estimating effective population size from data on linkage disequilibrium. Mol. Ecol. Resour. 2008, 8, 753–756. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Waples, R.S.; Do, C. Linkage disequilibrium estimates of contemporary N e using highly variable genetic markers: A largely untapped resource for applied conservation and evolution. Evol. Appl. 2010, 3, 244–262. [Google Scholar] [CrossRef]
- Piry, S.; Luikart, G.; Cornuet, J.-M. Computer note. BOTTLENECK: A computer program for detecting recent reductions in the effective size using allele frequency data. J. Hered. 1999, 90, 502–503. [Google Scholar] [CrossRef]
- Luikart, G.; Cornuet, J.-M. Empirical Evaluation of a Test for Identifying Recently Bottlenecked Populations from Allele Frequency Data. Conserv. Biol. 2008, 12, 228–237. [Google Scholar] [CrossRef]
- Luikart, G.; Allendorf, F.W.; Cornuet, J.-M.; Sherwin, W.B. Distortion of allele frequency distributions provides a test for recent population bottlenecks. J. Hered. 1998, 89, 238–247. [Google Scholar] [CrossRef]
- Jombart, T. Adegenet: A R package for the multivariate analysis of genetic markers. Bioinformatics 2008, 24, 1403–1405. [Google Scholar] [CrossRef] [Green Version]
- Jombart, T.; Devillard, S.; Balloux, F. Discriminant analysis of principal components: A new method for the analysis of genetically structured populations. BMC Genet. 2010, 11, 94. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lynch, M.; Ritland, K. Estimation of Pairwise Relatedness with Molecular Markers. Genetics 1999, 152, 1753–1766. [Google Scholar] [CrossRef] [PubMed]
- Ramstad, K.M.; Colbourne, R.M.; Robertson, H.A.; Allendorf, F.W.; Daugherty, C.H. Genetic consequences of a century of protection: Serial founder events and survival of the little spotted kiwi (Apteryx owenii). Proc. R. Soc. B Boil. Sci. 2013, 280, 20130576. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Allendorf, F.W. Genetic drift and the loss of alleles versus heterozygosity. Zoo Biol. 1986, 5, 181–190. [Google Scholar] [CrossRef]
- Leberg, P.L. Effects of Population Bottlenecks on Genetic Diversity as Measured by Allozyme Electrophoresis. Evolution 1992, 46, 477–494. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nei, M.; Maruyama, T.; Chakraborty, R. The Bottleneck Effect and Genetic Variability in Populations. Evolution 1975, 29, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Deyoung, R.W.; DeMarais, S.; Honeycutt, R.L.; Rooney, A.; Gonzales, R.A.; Gee, K.L. Genetic consequences of white-tailed deer (Odocoileus virginianus) restoration in Mississippi. Mol. Ecol. 2003, 12, 3237–3252. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zenger, K.R.; Richardson, B.J.; Vachot-Griffin, A.-M. A rapid population expansion retains genetic diversity within European rabbits in Australia. Mol. Ecol. 2003, 12, 789–794. [Google Scholar] [CrossRef] [PubMed]
- Kolbe, J.J.; Glor, R.E.; Schettino, L.R.; Lara, A.C.; Larson, A.; Losos, J.B. Multiple Sources, Admixture, and Genetic Variation in Introduced Anolis Lizard Populations. Conserv. Biol. 2007, 21, 1612–1625. [Google Scholar] [CrossRef]
- Williams, B.W.; Scribner, K.T. Effects of multiple founder populations on spatial genetic structure of reintroduced American martens. Mol. Ecol. 2010, 19, 227–240. [Google Scholar] [CrossRef] [PubMed]
- Hedrick, P.W.; Miller, P.S.; Geffen, E.; Wayne, R. Genetic evaluation of the three captive mexican wolf lineages. Zoo Biol. 1997, 16, 47–69. [Google Scholar] [CrossRef]
- Raisin, C.; Frantz, A.C.; Kundu, S.; Greenwood, A.G.; Jones, C.G.; Zuël, N.; Groombridge, J.J. Genetic consequences of intensive conservation management for the Mauritius parakeet. Conserv. Genet. 2012, 13, 707–715. [Google Scholar] [CrossRef]
- Lemer, S.; Planes, S. Translocation of wild populations: Conservation implications for the genetic diversity of the black-lipped pearl oyster Pinctada margaritifera. Mol. Ecol. 2012, 21, 2949–2962. [Google Scholar] [CrossRef]
- Mitchell, A.A.; Lau, J.; Chemnick, L.G.; Thompson, E.A.; Alberts, A.C.; Ryder, O.A.; Gerber, G.P. Using microsatellite diversity in wild Anegada iguanas (Cyclura pinguis) to establish relatedness in a captive breeding group of this critically endangered species. Conserv. Genet. 2011, 12, 771–781. [Google Scholar] [CrossRef]
- Prober, S.M.; Byrne, M.; McLean, E.H.; Steane, D.A.; Potts, B.M.; Vaillancourt, R.E.; Stock, W.D. Climate-adjusted provenancing: A strategy for climate-resilient ecological restoration. Front. Ecol. Evol. 2015, 3, 1–5. [Google Scholar] [CrossRef] [Green Version]
- Weeks, A.R.; Moro, D.; Thavornkanlapachai, R.; Taylor, H.R.; White, N.E.; Weiser, E.L.; Heinze, D. Conserving and enhancing genetic diversity in translocation programs. In Advances in Reintroduction Biology of Australian and New Zealand Fauna; Armstrong, D., Hayward, M.W., Moro, D., Seddon, P.J., Eds.; CSIRO Publishing: Clayton South, Victoria, Australia, 2015; pp. 127–140. [Google Scholar]
- Jonson, J. Ecological restoration of cleared agricultural land in Gondwana Link: Lifting the bar at ‘Peniup’. Ecol. Manag. Restor. 2010, 11, 16–26. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Year | Source Populations | Founders | Captive | Released | Peniup | |||
|---|---|---|---|---|---|---|---|---|
| West | East | |||||||
| TW | HAM | MT | West | East | ||||
| 2000 | 11 | 7 | 8 | |||||
| 2001 | 2 | 1 | 3 | 41 | 41 | |||
| 2002 | 3 | 39 | 46 | 5 | ||||
| 2003 | 4 | 3 | 36 | 43 | 14 | |||
| 2004 | 18 | 3 | 6 | 3 | 43 | |||
| 2005 | 10 | 45 | 5 | 3 | ||||
| 2006 | 17 | 15 | 6 | 11 | ||||
| 2007 | 22 | 7 | 5 | 3 | ||||
| 2008 | 18 | 19 | 24 | |||||
| 2009 | 5 | 3 | 20 | 34 | 7 | |||
| 2010 | 17 | 37 | 41 | 14 | ||||
| 2011 | 1 | 2 | 21 | |||||
| 2012 | 19 | 15 | ||||||
| Year of Release | Sex | Age (Year) | Total | |||
|---|---|---|---|---|---|---|
| <1 | 1 | 2 | 3 | |||
| 2001 | Female | 16 | 3 | [1] | [1] | 21 |
| Male | 14 | 4 | [2] | 20 | ||
| 2002 | Female | 18 | 2 | [3] | 23 | |
| Male | 16 | 5 | [2] | 23 | ||
| 2003 | Female | 16 | 1 | 3 | 20 | |
| Male | 20 | 2 | 1 | 23 | ||
| 2006 | Female | 4 | 4 | |||
| Male | 2 | 2 | ||||
| 2008 | Female | 5 | 1 [1] | 2 [1] | 10 | |
| Male | 8 | 3 [2] | 1 | 14 | ||
| 2009 | Female | 4 | 2 | [1] | 3 [1] | 11 |
| Male | 14 | 3 | 2 [1] | 2 [1] | 23 | |
| 2010 | Female | 22 | 1 | 1 | 24 | |
| Male | 15 | 1 | 1 | 17 | ||
| Total | 174 | 23 | 24 | 14 | 235 | |
| Population | N | A | Au | A(rare) | Ar | H | FIS | GD | Ne | Ne Range | Bottleneck |
|---|---|---|---|---|---|---|---|---|---|---|---|
| East | |||||||||||
| 2005 | 40.2 ± 2.3 | 113 | 0.3 ± 0.2 | 1.2 ± 0.3 | 4.2 ± 0.3 | 0.64 ± 0.05 | 0.11 * | 0 | NA | NA | N |
| 2006 | 16.3 ± 0.3 | 103 | 0.1 ± 0.1 | 0.9 ± 0.4 | 4.3 ± 0.4 | 0.63 ± 0.06 | −0.02 | 0 | 15.0 | 11.1–21.4 | N |
| 2007 | 20.4 ± 0.3 | 108 | 0.1 ± 0.1 | 1.3 ± 0.4 | 4.3 ± 0.4 | 0.64 ± 0.05 | 0.02 | 0 | 42.9 | 26.9–90.3 | N |
| 2008 | 17.3 ± 0.2 | 93 | 0 | 0.5 ± 0.2 | 4.0 ± 0.3 | 0.62 ± 0.05 | 0.04 | 0 | 15.2 | 11.4–21.2 | Y |
| 2010 | 15.5 ± 0.5 | 96 | 0 | 1.0 ± 0.2 | 4.1 ± 0.3 | 0.64 ± 0.06 | 0.00 | 0 | NA | NA | N |
| 2012 | 19.0 ± 0.1 | 95 | 0 | 0.6 ± 0.2 | 3.9 ± 0.3 | 0.64 ± 0.05 | −0.04 | 0 | 9.4 | 7.5–11.6 | N |
| Overall | 141.3 ± 3.3 | 133 | 0.9 ± 0.3 | 2.4 ± 2.8 | 4.2 ± 0.4 | 0.64 ± 0.05 | 0.05 * | 1 | 74.1 | 52.5–110.9 | N |
| West | |||||||||||
| 2000 | 6.7 ± 0.5 | 83 | 0.1 ± 0.1 | 0.1 ± 0.1 | NA | 0.64 ± 0.06 | 0.13 | 0 | NA | NA | - |
| 2004 | 16.8 ± 0.2 | 97 | 0.1 ± 0.1 | 0.6 ± 0.3 | 4.1 ± 0.4 | 0.64 ± 0.05 | 0.00 | 0 | 100.2 | 35.6–∞ | N |
| 2005 | 9.9 ± 0.1 | 79 | 0 | 0 | 3.6 ± 0.4 | 0.61 ± 0.05 | 0.03 | 0 | 42.3 | 18.8–∞ | N |
| Overall | 42.9 ± 0.9 | 112 | 0.4 ± 0.2 | 1.5 ± 1.6 | 4.0 ± 0.4 | 0.63 ± 0.05 | 0.03 | 0 | 54.1 | 36.7–91.4 | N |
| Captive founders | 24.7 ± 0.3 | 114 | 0 | 1.6 ± 2.2 | 4.3 ± 0.4 | 0.64 ± 0.05 | 0.03 | 0 | 69.7 | 40.0–204.1 | N |
| Captive population | |||||||||||
| 2001 | 41.0 ± 0.0 | 96 | 0 | 0.6 ± 0.3 | 3.9 ± 0.4 | 0.60 ± 0.05 | 0.01 | 14 | 5.6 | 4.0–7.0 | N |
| 2002 | 38.2 ± 0.3 | 90 | 0 | 0.7 ± 0.3 | 3.6 ± 0.3 | 0.58 ± 0.05 | −0.08 | 7 | 5.3 | 3.8–6.9 | N |
| 2003 | 35.8 ± 0.1 | 83 | 0 | 0.5 ± 0.2 | 3.5 ± 0.3 | 0.57 ± 0.05 | −0.06 | 7 | 6.0 | 3.9–8.0 | N |
| 2006 | 15.0 ± 0.0 | 78 | 0 | 0.5 ± 0.2 | 3.4 ± 0.3 | 0.55 ± 0.05 | −0.07 | 0 | 2.1 | 1.9–2.5 | Y |
| 2008 | 18.9 ± 0.1 | 84 | 0 | 0.2 ± 0.1 | 3.7 ± 0.4 | 0.58 ± 0.06 | −0.10 | 3 | 2.0 | 1.8–2.3 | Y |
| 2009 | 18.7 ± 0.4 | 80 | 0 | 0.3 ± 0.1 | 3.5 ± 0.3 | 0.56 ± 0.06 | −0.03 | 1 | 4.2 | 3.0–6.1 | Y |
| 2010 | 36.9 ± 0.1 | 97 | 0 | 0.8 ± 0.3 | 3.8 ± 0.3 | 0.60 ± 0.05 | −0.02 | 2 | 9.8 | 8.1–11.7 | N |
| Overall | 220.4 ± 0.6 | 113 | 0 | 1.6 ± 2.6 | 4.0 ± 0.3 | 0.61 ± 0.05 | −0.01 | 50 | 24.5 | 21.0–28.5 | N |
| Reintroduced population | |||||||||||
| 2003 | 13.7 ± 0.1 | 67 | 0 | 0.4 ± 0.1 | 3.0 ± 0.2 | 0.51 ± 0.05 | 0.00 | 0 | 4.0 | 2.6–7.8 | N |
| 2004 | 41.8 ± 0.4 | 85 | 0 | 0.7 ± 0.2 | 3.5 ± 0.3 | 0.57 ± 0.05 | −0.05 | 0 | 10.8 | 8.7–13.3 | N |
| 2006 | 9.0 ± 0.3 | 58 | 0.1 ± 0.1 | 0.1 ± 0.1 | 2.8 ± 0.2 | 0.48 ± 0.05 | −0.08 | 0 | 16.7 | 6.5–1890.3 | Y |
| 2010 | 12.5 ± 0.3 | 75 | 0 | 0.6 ± 0.2 | 3.4 ± 0.2 | 0.59 ± 0.05 | −0.03 | 0 | 5.4 | 3.0–8.8 | N |
| 2011 | 21.0 ± 0.0 | 83 | 0 | 0.7 ± 0.2 | 3.5 ± 0.3 | 0.59 ± 0.05 | −0.10 | 0 | 4.2 | 3.1–5.7 | N |
| 2012 | 15.0 ± 0.0 | 77 | 0 | 0.4 ± 0.1 | 3.5 ± 0.3 | 0.60 ± 0.05 | −0.03 | 0 | 8.2 | 5.6–11.8 | Y |
| Overall | 131.6 ± 1 | 103 | 0.1 ± 0.1 | 1.3 ± 1.2 | 3.7 ± 0.3 | 0.60 ± 0.05 | 0.01 | 13 | 16.7 | 14.5–19.1 | N |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Thavornkanlapachai, R.; Mills, H.R.; Ottewell, K.; Friend, J.A.; Kennington, W.J. Temporal Variation in the Genetic Composition of an Endangered Marsupial Reflects Reintroduction History. Diversity 2021, 13, 257. https://doi.org/10.3390/d13060257
Thavornkanlapachai R, Mills HR, Ottewell K, Friend JA, Kennington WJ. Temporal Variation in the Genetic Composition of an Endangered Marsupial Reflects Reintroduction History. Diversity. 2021; 13(6):257. https://doi.org/10.3390/d13060257
Chicago/Turabian StyleThavornkanlapachai, Rujiporn, Harriet R. Mills, Kym Ottewell, J. Anthony Friend, and W. Jason Kennington. 2021. "Temporal Variation in the Genetic Composition of an Endangered Marsupial Reflects Reintroduction History" Diversity 13, no. 6: 257. https://doi.org/10.3390/d13060257
APA StyleThavornkanlapachai, R., Mills, H. R., Ottewell, K., Friend, J. A., & Kennington, W. J. (2021). Temporal Variation in the Genetic Composition of an Endangered Marsupial Reflects Reintroduction History. Diversity, 13(6), 257. https://doi.org/10.3390/d13060257