
Assessing the Cultivability of Bacteria and Fungi from Arable Crop Residues Using Metabarcoding Data as a Reference


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
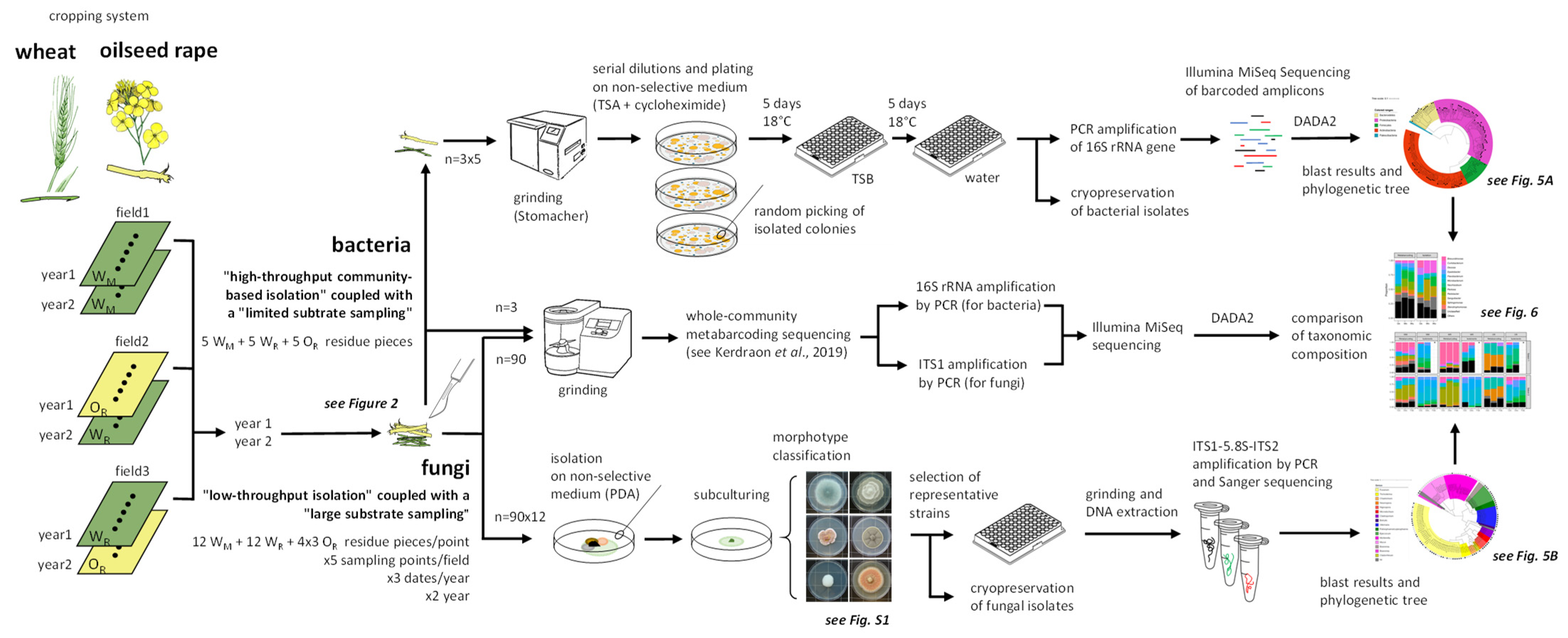
2.1. Overall Strategy
2.2. Culture-Dependent Molecular Identification of Residue-Associated Bacteria
2.2.1. Isolation of the Bacterial Fraction
2.2.2. High-Throughput Partial Sequencing of the 16S rRNA Gene
2.2.3. Sequence Analyses
2.3. Culture-Dependent Molecular Identification of Residue-Associated Fungi
2.3.1. Isolation of the Fungal Fraction
2.3.2. Molecular Characterization of the Fungal Collection by Sanger Sequencing
2.3.3. Phylogenetic Tree for the ITS Region of Fungal Strains
3. Results
3.1. Bacterial Culture Collection
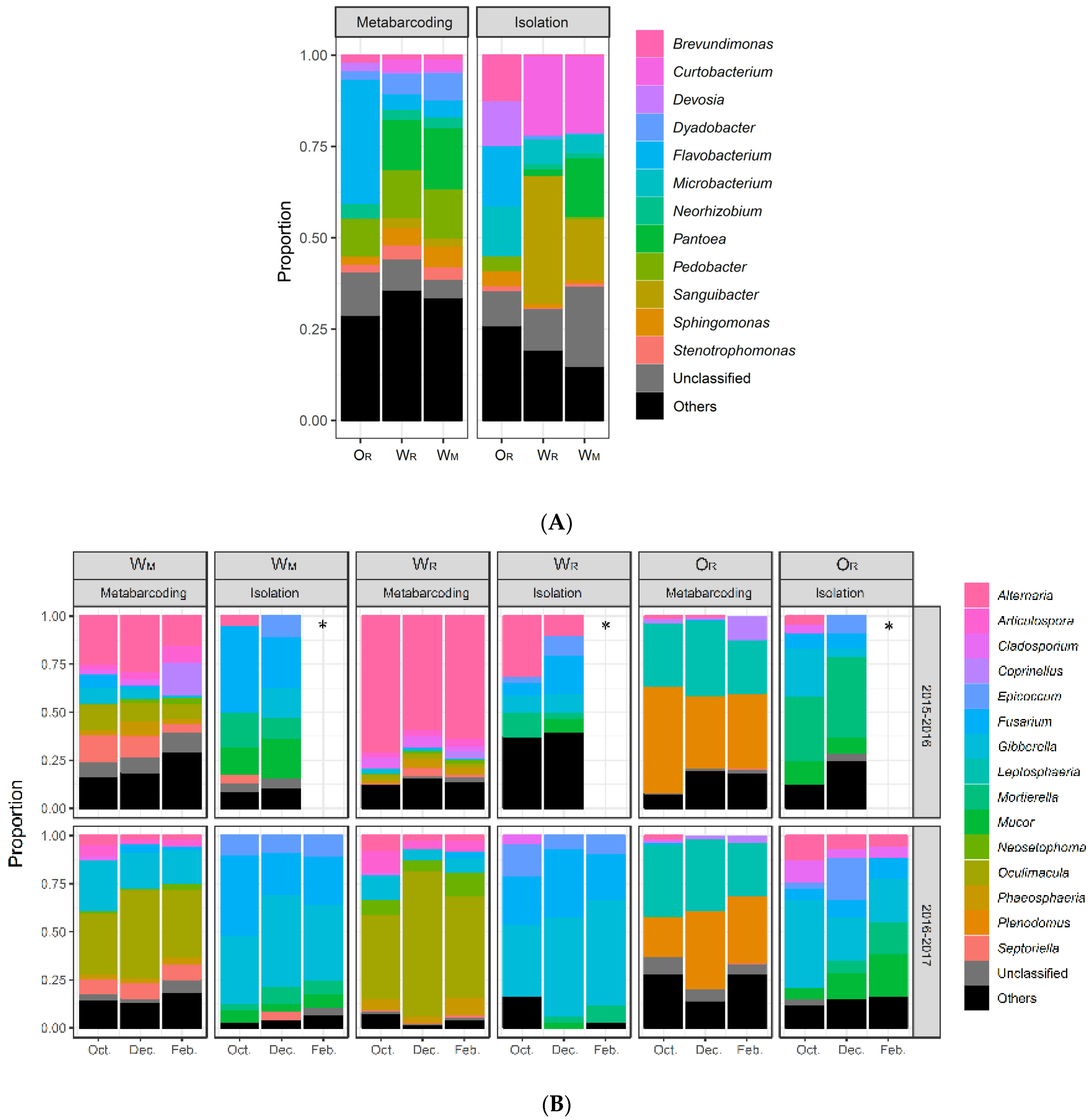
3.2. Comparison of Bacterial Culture-Collection and Metabarcoding Data
3.3. Fungal Culture Collection
3.4. Comparison of Fungal Culture Collection and Metabarcoding Data
4. Discussion
4.1. Complementarity of the Two Approaches
4.1.1. The Two Approaches Yield Consistent Results, Particularly for Bacteria
4.1.2. The Two Approaches Provide Complementary Information, Particularly for Fungi
4.2. Some Methodological Considerations
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Smil, V. Crop residues: Agriculture’s largest harvest crop residues incorporate more than half of the world’s agricultural phytomass. BioScience 1999, 49, 299–308. [Google Scholar] [CrossRef] [Green Version]
- Kerdraon, L.; Laval, V.; Suffert, F. Microbiomes and pathogen survival in crop residues, an ecotone between plant and soil. Phytobiomes J. 2019, 3, 246–255. [Google Scholar] [CrossRef] [Green Version]
- Pascault, N.; Cécillon, L.; Mathieu, O.; Hénault, C.; Sarr, A.; Lévêque, J.; Farcy, P.; Ranjard, L.; Maron, P.-A. In situ dynamics of microbial communities during decomposition of wheat, rape, and alfalfa residues. Microb. Ecol. 2010, 60, 816–828. [Google Scholar] [CrossRef] [PubMed]
- Cobo-Díaz, J.F.; Baroncelli, R.; Le Floch, G.; Picot, A. Combined metabarcoding and co-occurrence network analysis to profile the bacterial, fungal and Fusarium communities and their interactions in maize stalks. Front. Microbiol. 2019, 10, 261. [Google Scholar] [CrossRef] [Green Version]
- Kerdraon, L.; Balesdent, M.-H.; Barret, M.; Laval, V.; Suffert, F. Crop residues in wheat-oilseed rape rotation system: A pivotal, shifting platform for microbial meetings. Microb. Ecol. 2019, 77, 931–945. [Google Scholar] [CrossRef]
- Yang, H.; Ye, W.; Ma, J.; Zeng, D.; Rong, Z.; Xu, M.; Wang, Y.; Zhen, X. Endophytic fungal communities associated with field-grown soybean roots and seeds in the Huang-Huai region of China. PeerJ 2018, 6, e4713. [Google Scholar] [CrossRef] [Green Version]
- Øvreås, L.; Torsvik, V. Microbial diversity and community structure in two different agricultural soil communities. Microb. Ecol. 1998, 36, 303–315. [Google Scholar] [CrossRef]
- Fath, B.D.; Scharler, U.M.; Ulanowicz, R.E.; Hannone, B. Ecological network analysis: Network construction. Ecol. Model. 2007, 208, 49–55. [Google Scholar] [CrossRef]
- Daniel, R. The soil metagenome–A rich resource for the discovery of novel natural products. Curr. Opin. Biotechnol. 2004, 15, 199–204. [Google Scholar] [CrossRef] [PubMed]
- Comby, M.; Gacoin, M.; Robineau, M.; Rabenoelina, F.; Ptas, S.; Dupont, J.; Profizi, C.; Baillieul, F. Screening of wheat endophytes as biological control agents against Fusarium head blight using two different In Vitro tests. Microbiol. Res. 2017, 202, 11–20. [Google Scholar] [CrossRef]
- Tuan Hamzah, T.N.; Lee, S.H.; Hidayat, A.; Terhem, R.; Faridah-Hanum, I.; Mohamed, R. Diversity and characterization of endophytic fungi isolated from the tropical mangrove species, Rhizophora mucronata, and identification of potential antagonists against the soil-borne fungus, Fusarium solani. Front. Microbiol. 2018, 25, 1707. [Google Scholar] [CrossRef] [PubMed]
- Lewis, W.H.; Tahon, G.; Geesink, P.; Sousa, D.Z.; Ettema, T.J.G. Innovations to culturing the uncultured microbial majority. Nat. Rev. Microbiol. 2020. [Google Scholar] [CrossRef] [PubMed]
- Wintzingerode, F.V.; Göbel, U.B.; Stackebrandt, E. Determination of microbial diversity in environmental samples: Pitfalls of PCR-based rRNA analysis. FEMS Microbiol. Rev. 1997, 21, 213–229. [Google Scholar] [CrossRef]
- Armanhi, J.S.L.; de Souza, R.S.C.; de Brito Damasceno, N.; de Araújo, L.M.; Imperial, J.; Arruda, P. A community-based culture collection for targeting novel plant growth-promoting bacteria from the sugarcane microbiome. Front. Plant Sci. 2018, 8, 2191. [Google Scholar] [CrossRef] [Green Version]
- Müller, T.; Ruppel, S. Progress in cultivation-independent phyllosphere microbiology. FEMS Microbiol. Ecol. 2014, 87, 2–17. [Google Scholar] [CrossRef]
- Ritz, K. The Plate Debate: Cultivable communities have no utility in contemporary environmental microbial ecology. FEMS Microbiol. Ecol. 2007, 60, 358–362. [Google Scholar] [CrossRef] [Green Version]
- Rastogi, G.; Tech, J.J.; Coaker, G.L.; Leveau, J.H.J. APCR-based toolbox for the culture-independent quantification of total bacterial abundances in plant environments. J. Microbiol. Meth. 2010, 83, 127–132. [Google Scholar] [CrossRef]
- Stiefel, P.; Zambelli, T.; Vorholt, J.A. Isolation of optically targeted single bacteria using FluidFM applied to aerobic anoxygenic phototrophs from the phyllosphere. Appl. Environ. Microbiol. 2013, 79, 4895–4905. [Google Scholar] [CrossRef] [PubMed]
- Klenke, F.; Scholler, M. Pflanzenparasitische Kleinpilze: Bestimmungsbuch für Brand-, Rost-, Mehltau-, Flagellatenpilze und Wucherlingsverwandte in Deutschland, Österreich, der Schweiz und Südtirol; Springer: Berlin, Germany, 2015. [Google Scholar]
- Durán, P.; Thiergart, T.; Garrido-Oter, R.; Agler, M.; Kemen, E.; Schulze-Lefert, P.; Hacquard, S. Microbial interkingdom interactions in roots promote Arabidopsis survival. Cell 2018, 175, 973–983. [Google Scholar] [CrossRef]
- Abayasekara, L.M.; Perera, J.; Chandrasekharan, V.; Gnanam, V.S.; Udunuwara, N.A.; Liyanage, D.S.; Bulathsinhala, N.E.; Adikary, S.; Aluthmuhandiram, J.V.S.; Thanaseelan, C.S.; et al. Detection of bacterial pathogens from clinical specimens using conventional microbial culture and 16S metagenomics: A comparative study. BMC Infect. Dis. 2017, 17, 631. [Google Scholar] [CrossRef]
- Hilton, S.K.; Castro-Nallar, E.; Pérez-Losada, M.; Toma, I.; McCaffrey, T.A.; Hoffman, E.P.; Siegel, M.O.; Simon, G.L.; Johnson, W.E.; Crandall, K.A. Metataxonomic and metagenomic approaches vs. culture-dependent techniques for clinical pathology. Front. Microbiol. 2016, 7, 484. [Google Scholar] [CrossRef]
- Zapka, C.; Leff, J.; Henley, J.; Tittl, J.; De Nardo, E.; Butler, M.; Griggs, R.; Fierer, N.; Edmonds-Wilson, S. Comparison of standard culture-dependent method to culture-independent method for evaluation of hygiene effects on the hand microbiome. mBio 2017, 8, e00093-17. [Google Scholar] [CrossRef] [Green Version]
- Carraro, L.; Maifreni, M.; Bartolomeoli, I.; Martino, M.E.; Novelli, E.; Frigo, F.; Marino, M.; Cardazzo, B. Comparison of culture-dependent and -independent methods for bacterial community monitoring during Montasio cheese manufacturing. Res. Microbiol. 2011, 162, 231–239. [Google Scholar] [CrossRef]
- Armanhi, J.S.L.; de Souza, R.S.C.; de Araújo, L.M.; Okura, V.K.; Mieczkowski, P.A.; Imperial, J.; Arruda, P. Multiplex amplicon sequencing for microbe identification in community-based culture collections. Sci. Rep. 2016, 12, 29543. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Armani, A.; Giusti, A.; Guardone, L.; Castigliego, L.; Gianfaldoni, D.; Guidi, A. Universal primers used for species identification of foodstuff of animal origin: Effects of oligonucleotide tails on PCR amplification and sequencing performance. Food Anal. Methods 2016, 9, 1199–1209. [Google Scholar] [CrossRef]
- Zhang, J.; Liu, Y.X.; Guo, X.; Qin, Y.; Garrido-Oter, R.; Schulze-Lefert, P.; Bai, Y. High-throughput cultivation and identification of bacteria from the plant root microbiota. Nat. Protocol. 2021, 16, 988–1012. [Google Scholar] [CrossRef] [PubMed]
- Ben Chobba, I.; Elleuch, A.; Ayadi, I.; Khannous, L.; Namsi, A.; Cerqueira, F.; Gharsallah, N.; Vallaeys, T. Fungal diversity in adult date palm (Phoenix dactylifera L.) revealed by culture-dependent and culture-independent approaches. J. Zhejiang Univ. Sci. B 2013, 14, 1084–1099. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Comby, M.; Lacoste, S.; Baillieul, F.; Profizi, C.; Dupont, J. Spatial and temporal variation of cultivable communities of co-occurring endophytes and pathogens in wheat. Front. Microbiol. 2016, 7, 403. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dissanayake, A.J.; Purahong, W.; Wubet, T.; Hyde, K.D.; Zhang, W.; Xu, H.; Zhang, G.; Fu, C.; Liu, M.; Xing, Q.; et al. Direct comparison of culture-dependent and culture-independent molecular approaches reveal the diversity of fungal endophytic communities in stems of grapevine (Vitis vinifera). Fung Div. 2018, 90, 85–107. [Google Scholar] [CrossRef]
- Thiergart, T.; Zgadzaj, R.; Bozsóki, Z.; Garrido-Oter, R.; Radutoiu, S.; Schulze-Lefert, P. Lotus japonicus symbiosis genes impact microbial interactions between symbionts and multikingdom commensal communities. mBio 2019, 10, e01833-19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suffert, F.; Sache, I. Relative importance of different types of inoculum to the establishment of Mycosphaerella graminicola in wheat crops in north-west Europe. Plant Pathol. 2011, 60, 878–889. [Google Scholar] [CrossRef]
- Fitt, B.D.L.; Brun, H.; Barbetti, M.J.; Rimmer, S.R. World-wide importance of phoma stem canker (Leptosphaeria maculans and L. biglobosa) on oilseed rape (Brassica napus). In Sustainable Strategies for Managing Brassica napus (Oilseed rape) Resistance to Leptosphaeria maculans (phoma stem canker); Fitt, B.D.L., Evans, N., Howlett, B.J., Cooke, B.M., Eds.; Springer: Berlin, Germany, 2006; pp. 3–15. [Google Scholar]
- Kerdraon, L.; Barret, M.; Laval, V.; Suffert, F. Differential dynamics of microbial community networks help identify microorganisms interacting with residue-borne pathogens: The case of Zymoseptoria tritici in wheat. Microbiome 2019, 7, 125. [Google Scholar] [CrossRef] [PubMed]
- Kerdraon, L.; Barret, M.; Balesdent, M.; Suffert, F.; Laval, V. Impact of a resistance gene against a fungal pathogen on the plant host residue microbiome: The case of the Leptosphaeria maculans-Brassica napus pathosystem. Mol. Plant Pathol. 2020, 21, 1545–1558. [Google Scholar] [CrossRef] [PubMed]
- Großkopf, T.; Soyer, O.S. Synthetic microbial communities. Curr. Opin. Microbiol. 2014, 18, 72–77. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Navrátilová, D.; Tláskalová, P.; Kohout, P.; Dřevojan, P.; Fajmon, K.; Chytrý, M.; Baldrian, P. Diversity of fungi and bacteria in species-rich grasslands increases with plant diversity in shoots but not in roots and soil. FEMS Microb. Ecol. 2019, 95, fiy208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tan, X.; Kan, L.; Su, Z.; Liu, X.; Zhang, L. The composition and diversity of soil bacterial and fungal communities along an urban-to-rural gradient in South China. Forests 2019, 10, 797. [Google Scholar] [CrossRef] [Green Version]
- Bahram, M.; Netherway, T.; Frioux, C.; Ferretti, P.; Coelho, L.P.; Geisen, S.; Bork, P.; Hildebrand, F. Metagenomic assessment of the global diversity and distribution of bacteria and fungi. Environ. Microb. 2020, 23, 316–326. [Google Scholar] [CrossRef]
- Caporaso, J.G.; Lauber, C.L.; Walters, W.A.; Berg-Lyons, D.; Lozupone, C.A.; Turnbaugh, P.J.; Fierer, N.; Knight, R. Global patterns of 16S rRNA diversity at a depth of millions of sequences per sample. Proc. Natl. Acad. Sci. USA 2011, 108, 4516–4522. [Google Scholar] [CrossRef] [Green Version]
- Martin, M. CUTADAPT removes adapter sequences from high-throughput sequencing reads. EMBnet. J. 2011, 17, 1. [Google Scholar] [CrossRef]
- Callahan, B.J.; McMurdie, P.J.; Rosen, M.J.; Han, A.W.; Johnson, A.J.A.; Holmes, S.P. DADA2: High-resolution sample inference from Illumina amplicon data. Nat. Methods 2016, 13, 581–583. [Google Scholar] [CrossRef] [Green Version]
- Wang, Q.; Garrity, G.M.; Tiedge, J.M.; Cole, J.R. Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl. Environ. Microbiol. 2007, 73, 5264–5267. [Google Scholar] [CrossRef] [Green Version]
- Davis, N.M.; Proctor, D.M.; Holmes, S.P.; Relman, D.A.; Callahan, B.J. Simple statistical identification and removal of contaminant sequences in marker-gene and metagenomics data. Microbiome 2018, 6, 226. [Google Scholar] [CrossRef] [Green Version]
- Schliep, K.P. phangorn: Phylogenetic analysis in R. Bioinformatics 2011, 27, 592–593. [Google Scholar] [CrossRef] [Green Version]
- Wright, E.S. DECIPHER: Harnessing local sequence context to improve protein multiple sequence alignment. BMC Bioinform. 2015, 16, 322. [Google Scholar] [CrossRef] [Green Version]
- Gardes, M.; Bruns, T.D. ITS primers with enhanced specificity for basidiomycetes-application to the identification of mycorrhizae and rusts. Mol. Ecol. 1993, 2, 113–118. [Google Scholar] [CrossRef] [PubMed]
- White, T.J.; Bruns, T.; Lee, S.; Taylor, J. Amplification and direct sequencing of fungal ribosomal RNA genes for phylogenetics. PCR Protoc. Guide Methods Appl. 1990, 18, 315–322. [Google Scholar]
- Burland, T.G. DNASTAR’s Lasergene sequence analysis software. Methods Mol. Biol. 2000, 132, 71–91. [Google Scholar]
- Huerta-Cepas, J.; Serra, F.; Bork, P. ETE 3: Reconstruction, analysis, and visualization of phylogenomic data. Mol. Biol. Evol. 2016, 33, 1635–1638. [Google Scholar] [CrossRef] [Green Version]
- Katoh, K.; Standley, D.M. MAFFT Multiple Sequence Alignment Software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [Green Version]
- Price, M.N.; Dehal, P.S.; Arkin, A.P. FastTree: Computing large minimum evolution trees with profiles instead of a distance matrix. Mol. Biol. Evol. 2009, 26, 1641–1650. [Google Scholar] [CrossRef]
- Letunic, I.; Bork, P. Interactive Tree Of Life (iTOL) v4: Recent updates and new developments. Nucleic Acids Res. 2019, 47, 256–259. [Google Scholar] [CrossRef] [Green Version]
- Vera, D.I.; Murray, T.D. Occurrence and survival of apothecia of the eyespot pathogens Oculimacula acuformis and O. yallundae on wheat stubble in the U.S. Pacific Northwest. Plant Dis. 2016, 100, 991–995. [Google Scholar] [CrossRef] [Green Version]
- Baldani, J.I.; Rouws, L.; Magalhães Cruz, L.; Lopes Olivares, F.; Schmid, M.; Hartmann, A. The Family Oxalobacteraceae. In The Prokaryotes-Alphaproteobacteria and Betaproteobacteria; Rosenberg, E., DeLong, E.F., Lory, S., Stackebrandt, E., Thompson, F., Eds.; Springer: Berlin, Germany, 2014; pp. 919–974. [Google Scholar]
- Nishioka, T.; Elsharkawy, M.M.; Suga, H.; Kageyama, K.; Hyakumachi, M.; Shimizu, M. Development of culture medium for the isolation of Flavobacterium and Chryseobacterium from rhizosphere soil. Microbes Environ. 2016, 31, 104–110. [Google Scholar] [CrossRef] [Green Version]
- Rinke, C.; Schwientek, P.; Sczyrba, A.; Ivanova, N.N.; Anderson, I.J.; Cheng, J.F.; Darling, A.; Malfatti, S.; Swan, B.K.; Gies, E.A.; et al. Insights into the phylogeny and coding potential of microbial dark matter. Nature 2013, 499, 431–437. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Filippidou, S.; Junier, T.; Wunderlin, T.; Lo, C.C.; Li, P.E.; Chain, P.S.; Junier, P. Under-detection of endospore-forming Firmicutes in metagenomic data. Comput. Struct. Biotech. J. 2015, 13, 299–306. [Google Scholar] [CrossRef] [Green Version]
- Evtushenko, L.I.; Takeuchi, M.T. The Family Microbacteriaceae. In The Prokaryotes-Alphaproteobacteria and Betaproteobacteria; Rosenberg, E., DeLong, E.F., Lory, S., Stackebrandt, E., Thompson, F., Eds.; Springer: Berlin, Germany, 2017; pp. 1020–1098. [Google Scholar]
- Hamad, I.; Ranque, S.; Azhar, E.I.; Yasir, M.; Jiman-Fatani, A.A.; Tissot-Dupont, H.; Raoult, D.; Bittar, F. Culturomics and amplicon-based metagenomic approaches for the study of fungal population in human gut microbiota. Sci. Rep. 2017, 7, 16788. [Google Scholar] [CrossRef]
- Krehenwinkel, H.; Pomerantz, A.; Henderson, J.B.; Kennedy, S.R.; Lim, J.Y.; Swamy, V.; Shoobridge, J.D.; Graham, N.; Patel, N.H.; Gillespie, R.G.; et al. Nanopore sequencing of long ribosomal DNA amplicons enables portable and simple biodiversity assessments with high phylogenetic resolution across broad taxonomic scale. GigaScience 2019, 8, giz006. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heeger, F.; Bourne, E.C.; Baschien, C.; Yurkov, A.; Bunk, B.; Spröer, C.; Overmann, J.; Mazzoni, C.J.; Monaghan, M.T. Long-read DNA metabarcoding of ribosomal RNA in the analysis of fungi from aquatic environments. Mol. Ecol. Resour. 2018, 18, 1500–1514. [Google Scholar] [CrossRef]
- Raja, H.A.; Miller, A.N.; Pearce, C.J.; Oberlies, N.H. Fungal identification using molecular tools: A primer for the natural products research community. J. Nat. Prod. 2017, 80, 756–770. [Google Scholar] [CrossRef]
- Callahan, B.J.; Wong, J.; Heiner, C.; Oh, S.; Theriot, C.M.; Gulati, A.S.; McGill, S.K.; Dougherty, M.K. High-throughput amplicon sequencing of the full-length 16S rRNA gene with single-nucleotide resolution. Nucleic Acids Res. 2019, 47, e103. [Google Scholar] [CrossRef] [Green Version]
- Barret, M.; Briand, M.; Bonneau, S.; Préveaux, A.; Valière, S.; Bouchez, O.; Hunault, G.; Simoneau, P.; Jacques, M.-A. Emergence shapes the structure of the seed microbiota. Appl. Environ. Microbiol. 2015, 81, 1257–1266. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- El-Tarabily, K.A.; Sivasithamparam, K. Potential of yeasts as biocontrol agents of soil-borne fungal plant pathogens and as plant growth promoters. Mycoscience 2006, 47, 25–35. [Google Scholar] [CrossRef]
- Sergaki, C.; Lagunas, B.; Lidbury, I.; Gifford, M.L.; Schäfer, P. Challenges and approaches in microbiome research: From fundamental to applied. Front. Plant Sci. 2018, 9, 1205. [Google Scholar] [CrossRef]
- Liu, Y.X.; Qin, Y.; Bai, Y. Reductionist synthetic community approaches in root microbiome research. Curr. Opin. Microbiol. 2019, 49, 97–102. [Google Scholar] [CrossRef]
- Zhuang, L.; Li, Y.; Wang, Z.; Yu, Y.; Zhang, N.; Yang, C.; Zeng, Q.; Wang, Q. Synthetic community with six Pseudomonas strains screened from garlic rhizosphere microbiome promotes plant growth. Microb. Biotech. 2020, 14, 488–502. [Google Scholar] [CrossRef] [PubMed]
- Martiny, A.C. High proportions of bacteria are culturable across major biomes. ISME 2019, 13, 2125–2128. [Google Scholar] [CrossRef]
- Steen, A.D.; Crits-Christoph, A.; Carini, P.; DeAngelis, K.M.; Fierer, N.; Lloyd, K.G.; Thrash, J.C. High proportions of bacteria and archaea across most biomes remain uncultured. ISME J. 2019, 13, 3126–3130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bai, Y.; Müller, D.B.; Srinivas, G.; Garrido-Oter, R.; Potthoff, E.; Rott, M.; Dombrowski, N.; Münch, P.C.; Spaepen, S.; Remus-Emsermann, M.; et al. Functional overlap of the Arabidopsis leaf and root microbiota. Nature 2015, 528, 364–369. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Laval, V.; Kerdraon, L.; Barret, M.; Liabot, A.-L.; Marais, C.; Boudier, B.; Balesdent, M.-H.; Fischer-Le Saux, M.; Suffert, F. Assessing the Cultivability of Bacteria and Fungi from Arable Crop Residues Using Metabarcoding Data as a Reference. Diversity 2021, 13, 404. https://doi.org/10.3390/d13090404
Laval V, Kerdraon L, Barret M, Liabot A-L, Marais C, Boudier B, Balesdent M-H, Fischer-Le Saux M, Suffert F. Assessing the Cultivability of Bacteria and Fungi from Arable Crop Residues Using Metabarcoding Data as a Reference. Diversity. 2021; 13(9):404. https://doi.org/10.3390/d13090404
Chicago/Turabian StyleLaval, Valérie, Lydie Kerdraon, Matthieu Barret, Anne-Lise Liabot, Coralie Marais, Benjamin Boudier, Marie-Hélène Balesdent, Marion Fischer-Le Saux, and Frédéric Suffert. 2021. "Assessing the Cultivability of Bacteria and Fungi from Arable Crop Residues Using Metabarcoding Data as a Reference" Diversity 13, no. 9: 404. https://doi.org/10.3390/d13090404