1. Introduction
The Appalachian Mountains are one of the world’s oldest ranges, and they host an exceptionally diverse biota. The range extends from Alabama to southeastern Canada, and encompasses a wealth of natural communities. The Appalachian fauna has evolved over millennia of climatic fluctuations, with many elements believed to have persisted over tens of millions of years (e.g., Plethodontid salamanders; [
1]). Lineages have diversified and adapted in response to these fluctuations, alternately retreating to and expanding from scattered refugia [
2]. In the southern Appalachians and the Blue Ridge region, this is reflected in numerous short range endemic taxa, with distributions less than 1000 km
2 [
3]. Some groups, especially small arthropods, such as Coleoptera [
4] and Collembola [
5], have very high diversity in the area and yet have received little taxonomic attention.
One of the most distinctive environments in the southern Appalachians is the high elevation Red spruce-Fraser fir (
Picea rubens Sarg. &
Abies fraseri (Pursh) Poir.) forest belt. These sky island forests are found only in the highest portions of southern Appalachia, in eastern Tennessee, western North Carolina, and southwestern Virginia, where elevations exceed 5500 ft. (1700 m) (see
Figure 1). Widespread and more contiguous during glacial advances, these isolated forests now persist on a few dozen scattered peaks. These forests host numerous endemic arthropods (including
Trechus ground beetles [
6],
Geostiba rove beetles [
7], and
Adelopsis fungus beetles [
8,
9],
Dasycerus beetles, [
10], the collembolan genus
Intricatonura [
11], and many others). Genetic diversity within many such lineages (e.g.,
Hypochilus pococki [
12], and the federally endangered mygalomorph spider
Microhexura montivaga Crosby & Bishop [
13]) has been shown to be high, revealing another dimension of cryptic diversity. This rich, restricted fauna is increasingly imperiled by threats from climate change [
14,
15,
16] and invasive species [
17,
18].
Springtails, or the hexapod class Collembola, are important detritivores, contributing to decomposition of organic debris on the forest floor [
19,
20]. Their common name refers to a spring-operated jumping mechanism possessed by most, comprising an abdominal furca and retinaculum. The scientific name refers to the collophore, which is a unique abdominal appendage that characterizes all Collembola. Collembola are ubiquitous in leaf litter environments on forest floors worldwide [
21], and the high elevations of the southern Appalachians are no exception [
5,
22]. Despite their small size, Collembola often have wide distributional ranges, species sometimes spanning continents or occurring across, for example, the Nearctic and Palearctic regions [
23,
24]. Whether this results from high dispersal rates through unknown mechanisms, or simply from coarse and inadequate taxonomic resolution is very unclear, but would suggest relatively low rates of endemism in otherwise distinctive faunas, like high Appalachia.
Here, we explore the taxonomic and genetic diversity within two orders of Collembola, the Symphypleona and Neelipleona, in the higher elevations of southern Appalachia. Members of these orders appear to have fused body segments, giving them a globular shape. These minute arthropods have received scant attention in the southeastern US, let alone in any specific subregions like high Appalachia. Bernard & Felderhoff [
5] provided a brief review of the Collembola fauna of Great Smoky Mts National Park, but this did not focus on higher elevations, did not include a species list, and made only passing mention of Symphypleona or Neelipleona. Wray [
25] did provide a species list for the Great Smoky Mountains, but the taxonomy has changed considerably since then, and numerous potentially occurring species have been described or separated out since (e.g., [
26,
27,
28,
29,
30,
31,
32,
33,
34]). Direct information on the Symphypleona of the region can otherwise only be gleaned from general references in Christiansen & Bellinger’s [
22] Collembola of North America, most records in which are not resolved below the state or county level. Resources available nevertheless suggest as many as 67 described Symphypleona and Neelipleona species potentially occurring in the southern Appalachians (Dukes & Caterino, unpub.)
By applying an intensive COI metabarcoding approach (e.g., [
35]), we simultaneously assess species level diversity of globular Collembola in southern high Appalachia, attempting to identify specimens by their barcodes, and species coherence, assessing the degree to which morphologically delimited species correspond to ones suggested by genetic data. Although COI can provide only preliminary insight into cryptic species diversity [
36,
37], species delimited on the basis of COI sequences can provide initial hypotheses for further testing using other genes and novel morphological characters (e.g., [
38]), as well as helping to delimit evolutionarily significant units for conservation management [
39,
40].
2. Materials and Methods
As part of a larger inventory of leaf litter inhabiting arthropods, litter samples were obtained from 26 high elevation localities (>3300 ft or 1000 m) across 5 states (Virginia, North Carolina, Tennessee, South Carolina, Georgia) over the years 2018–2021 (see
Figure 1 for general localities and
Table 1 for details on each site). We visited most sites on two different dates, roughly in spring and fall timeframes. On each visit we took at least 3 leaf litter samples by sifting. Litter in most spruce-fir sites consists of deep needle litter, with minor components of deciduous leaves and fine woody debris. Litter was sifted down to the soil surface (or to a depth where litter was so decayed as to be indistinguishable from soil, where the interface was not a hard boundary), over an area of approximately one square meter, through an 8 mm mesh, until a bag of approximately 6 L was filled. Precise GPS coordinates were captured for each sample. Samples were processed in the lab using Berlese-Tullgren funnels, running subsamples until thoroughly dry, approximately 12 h per batch. Specimens were collected directly into 100% ethanol, and moved to −20 C storage after each subsample was complete. Springtail specimens were removed from bulk samples and sorted to morphospecies.
The analyses here include 204 Symphypleona and Neelipleona sequences plus an outgroup
Isotoma (Isotomidae) sequence. These represent one individual of each morphospecies from 41 sampling events (41 sets of samples from a given site/date). Full collecting data for each specimen extracted are available in
Table S1. Multiple individuals of a putative morphospecies were only included from a site if they were collected on different dates. Specimens were tentatively identified using Christiansen & Bellinger’s ‘Collembola of North America’ [
22] and through comparisons to specimen photographs online that had been identified by specialists (e.g., collembola.org; [
41]). Each specimen was imaged, subdivided or punctured to permit tissue digestion, and placed in a separate well in a 96-well plate. Images of morphospecies are archived on our lab Flickr page (
https://flickr.com/photos/183480085@N02/albums/72157720213462655; accessed on 16 May 2022), identifiable by morphospecies code (site.visit.###, as given in
Table S1). Tissues were digested with lysis buffer and proteinase K (Omega BioTek, Norcross, GA, USA), then the liquid fraction was removed to a new plate, with the voucher remains saved for archiving. The digested tissue mixture was extracted using Omega BioTek’s MagBind HDQ Blood and Tissue kit on a Hamilton Microlab Star automated liquid handling system, eluting with 150 μL elution buffer.
Following digestion, remains of extracted specimens were recombined with any non-extracted body parts, labelled, assigned unique CUAC (Clemson University Arthropod Collection) identifiers, and curated into the CUAC. Unextracted representatives of morphospecies, if any, remain in bulk order-level samples, and are also permanently vouchered in the CUAC, as are unsorted residues (containing additional representatives of hyperabundant taxa, principally Acari and Collembola).
These analyses include sequences from three separate sequencing approaches. For one plate of extracts we amplified a 658 base pair region of the cytochrome oxidase one (COI) mitochondrial ‘barcoding’ gene using primers LCO1490 and HCO2198 (GGTCAACAAATCATAAAGATATTGG & TAAACTTCAGGGTGACCAAAAAATCA, respectively; [
42]). These PCR products were run on an agarose gel to assess amplification success and sent for clean-up and Sanger sequencing to Psomagen (Rockville, MD, USA); amplicons were sequenced in both directions. This produced 64 of the sequences used here. The other specimens were sequenced using next generation platforms as ‘mini-barcodes’, a 421 bp fragment of the mitochondrial COI gene using the primers BF2-BR2 (GCHCCHGAYATRGCHTTYCC & TCDGGRTGNCCRAARAAYCA, respectively; [
43]), corresponding to the downstream two-thirds of the standard barcoding region. Each well was tagged with a unique combination of forward and reverse 9 bp indexes, synthesized as part of the primer by Eurofins Genomics (Louisville, KY, USA). These indexes were derived from a list provided by Meier et al. [
44], to allow multiplexed next-generation sequencing. All PCRs were conducted in 12.5 μL volumes (5.6 μL water, 1.25 μL Taq buffer, 1.25 μL dNTP mix [2.5 mM each], 0.4 μL MgCl [50 mM], 1.5 μL each primer, 0.05 μL Platinum Taq polymerase, 1 μL DNA template, with a 95 C initial denaturation for 5 min, followed by 35 cycles of 94 C (30 s), 50 C (30 s), 72 C (30 s), and a 5 min 72 C final extension on an Eppendorf Gradient Mastercycler.
For Illumina library preparation, PCR products were combined and purified using Omega Bio-Tek’s Mag-Bind Total Pure NGS Kit, in a ratio of 0.7:1 (enriching for fragments > 300 bp). Illumina adapters and sequencing primers were ligated to PCR products using New England BioLab’s Blunt/TA Ligase Master Mix. The amplicon + adapter library was again purified using Mag-Bind Total Pure NGS, and subsequently quantified using a Qubit fluorometer. This final library was sequenced on an Illumina MiSeq using a v.3 2 × 300 paired-end kit. Nanopore libraries were prepared using the ligation sequencing kit LSK-112 (Oxford Nanopore Technologies, Oxford, UK) and sequenced on a MinION using a v10.4 flowcell.
Sanger sequences were edited in Geneious (v8.1.8) by combining forward and reverse reads, confirming basecalls, and exporting as text. Illumina reads were processed with bbtools software package (
https://jgi.doe.gov/data-and-tools/bbtools/; v38.87 [
45]; accessed on 10 February 2022) to merge paired read ends, remove PhiX reads, trim Illumina adapters, filter reads for the correct size, remove reads with quality score < 30, cluster sequences by similarity allowing 5 mismatches (~1%) and generate a final matrix in FASTA format. Nanopore reads were basecalled using the ‘super-accurate’ algorithm of Guppy (v6.1.2), then demultiplexed using ONTbarcoder v0.1.9 [
46], with minimum coverage set at 5. FASTA files from all sequencing methods were trimmed to match the shorter 421 bp BF2-BR2 fragment, combined, and aligned with the online version of Mafft v7 [
47] using the auto strategy.
Phylogenetic reconstructions were performed using Maximum likelihood (ML) and Bayesian inference (BI) methods, providing trees for assessment as to species coherence and identity. The ML analysis was done with W-IQ-TREE v2.0 [
48,
49], available at
http://iqtree.cibiv.univie.ac.at (accessed on 9 August 2022). This program was used also to determine the best substitution model for our data. We set a perturbation strength of 0.4. and an IQ-TREE stopping rule value at 200. Branch support is based on an ultrafast bootstrap analysis [
50], run with 1000 bootstrap replicates with a minimum correlation coefficient of 0.99. Bayesian analysis was performed using BEAST v1.10.4 [
51], with a dataset including no outgroups, a relaxed lognormal molecular clock and a birth-death incomplete sampling speciation tree prior [
52]. Given the absence of an adequate fossil record to calibrate a molecular clock for our data, we used an estimated substitution rate for COI of 0.0169 ± 0.0019 [
53], which has been the most commonly used in Collembola studies [
54,
55,
56,
57]. The analysis was run for 30 × 10
6 generations sampling every 30,000, and repeated independently three times to assess the consistency of the results. We used Tracer v1.7 [
58] to determine that effective sample sizes (ESS) of the generated statistics were higher than 200. Finally, we built a maximum clade credibility (MCC) tree using TreeAnnotator v1.10.4, excluding the first 2000 trees as burn-in, that was midpoint rooted in FigTree v1.4.0 (
http://tree.bio.ed.ac.uk/software/figtree/; accessed on 5 September 2022).
For automated species delimitation we used five different single-locus delimitation methods. Automatic Barcode Gap Discovery (ABGD; [
59]) and Assemble Species by Automatic Partitioning (ASAP; [
60]) are both based on the characterization of barcode gaps from pairwise genetic distances. Two different implementations of the Poisson Tree Process (PTP) method [
61] were also used, Bayesian PTP (bPTP) and multi-rate PTP (mPTP) [
62]; this method is based on the detection of transitions in branching rates on a phylogenetic tree according to speciation and coalescent models. This is also true for the other method used, the General Mixed Yule-Coalescent model (GMYC) [
63], that uses an ultrametric tree to estimate those rate transitions. ABGD analysis was performed using the web version (available at
https://bioinfo.mnhn.fr/abi/public/abgd/abgdweb.html; accessed on 16 May 2022) using Jukes-Cantor (JC69) genetic distances and setting a prior maximum divergence of intraspecific diversity (P) from 0.005 to 0.15, with a relative gap width (X) of 1 and a number of bins of 20. ASAP was run using the web version (available at
https://bioinfo.mnhn.fr/abi/public/asap/asapweb.html; accessed on 16 May 2022), with Kimura 2-parameter [
64] distances and a split probability of 0.01. For both PTP analyses we used our ML tree as input. We used the bPTP web server (available at
https://species.h-its.org; accessed on 16 May 2022), running the analysis for 5 × 10
5 generations, removing the outgroup from the tree and using a thinning of 500 and a burn-in of 0.2. The mPTP analysis was performed using the web server (available at
https://mptp.h-its.org/#/tree; accessed on 16 May 2022), removing the outgroup from the tree. The GMYC web server (available at
https://species.h-its.org/gmyc/; accessed on 16 May 2022) used the BEAST ultrametric tree as input, running both single and multi-threshold methods (see [
65]). We examined clades corresponding to morphospecies to assess diversity across sampling sites. Any sequences for which we did not have an a priori identification were searched on the Barcoding of Life database and on GenBank through BlastN for tentative matches. However, this did not identify any additional sequences.
3. Results and Discussion
The Bayesian tree showing hypothesized species delimitations is shown in
Figure 2 (outgroup removed), with the more readily identifiable species indicated. Our dated tree is available as
Figure S1. Automated single-locus species delimitation methods yielded diverse results for our dataset. The most conservative method was mPTP, recovering a total of 43 distinct symphypleonan and neelipleonan species. By contrast, the greatest subdivision is observed with bPTP suggesting up to 90 putative species. GMYC resulted in 74 species using the single-threshold method and 77 with the multi-threshold method; the latter can show over-split in its results [
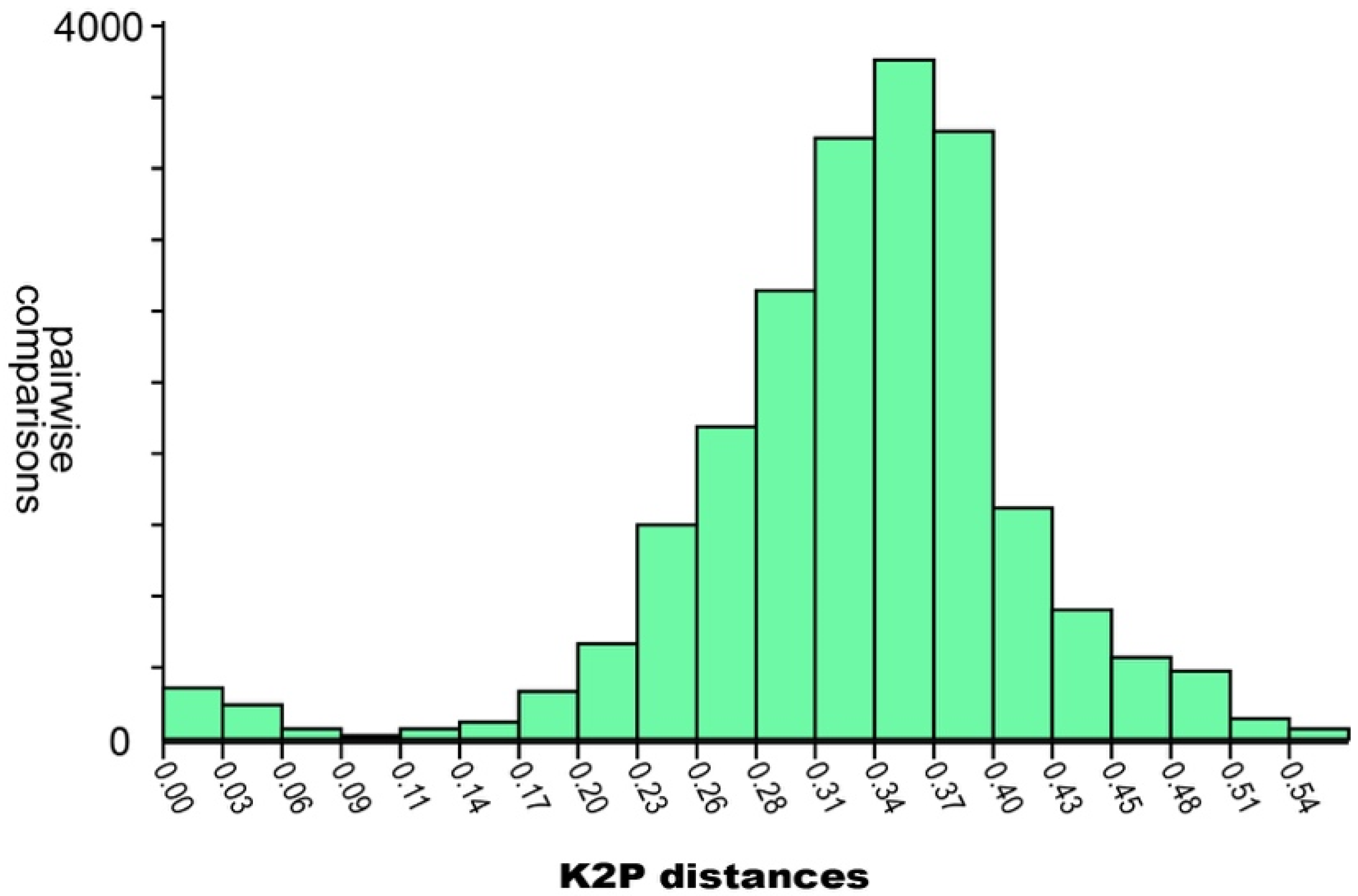
65], so we discuss the single-threshold results. As for the “barcode gap” methods, ABCD suggested the existence of 77 species in our dataset, while the best partition in ASAP resulted in a total of 82 species. The best estimated intra-/interspecific distance threshold estimated by ASAP was at 7% (K2P-corrected distance). A histogram showing the distribution of all pairwise distances is included as
Figure 3. All distances cited below are K2P distances, and are interpreted relative to this hypothesized threshold.
These results would seem to be a significant overestimate in at least some of the methods. Our own morphological identifications and morphospecies sorting would have suggested a more modest 25–30 species. Under all automated methods, some apparently morphologically uniform species were separated into multiple species. For example,
Ptenothrix atra was split into at least 9 species. However, levels of divergence were also consistently very high for COI, reaching 18% uncorrected (22% corrected) distance between Grandfather Mt and other localities for
P. atra, for example. Relatively high distances have been reported in other intraspecific studies of springtails [
66,
67,
68,
69]; Porco [
66], for example, considering 14% (K2P) distance to represent a conservative intraspecific cutoff. Clearly rates of mitochondrial evolution are much higher in globular Collembola than in other arthropods commonly examined in the barcoding literature (e.g., [
70,
71,
72,
73]). However, it is nonetheless worth considering that, where highly divergent and strongly supported intraspecific lineages show geographical coherence, there may be considerable cryptic species diversity in the fauna. We would not consider COI alone sufficient basis for concluding that cryptic species were present, but it is certainly a hypothesis worth examining further with additional data.
Second, attempts to identify any of these sequences via DNA barcodes using a variety of algorithms against BOLD and GenBank databases failed completely. Between poor representation of these groups (and litter arthropods in general; Recuero & Caterino, in prep.) in public databases, and extremely high degrees of COI divergence within and across lineages, our Symphypleona and Neelipleona sequences were not sufficiently close to any publicly available sequences to strongly support identifications at any taxonomic level-no sequence was less than 5% different from anything available, and many very incorrect (to arthropod order) possibilities were only a few percent more distant. Submission of these sequences will aid in future efforts, but these problems are likely to plague litter and soil arthropod identification for the foreseeable future, until major investments are made in establishing comprehensive reference sequences across arthropod taxa and geographic areas.
3.1. Neelipleona
Neelidae
Neelidae were represented by seven individuals, all morphologically identified as
Neelides Caroli (
Figure 4A). These were resolved into two widely separated clades that are nearly 40% divergent. While this group is too poorly represented in our data to reach any serious conclusions about biogeographic relationships or taxonomy, delimitation analyses suggest anywhere from 2 to 5 species, with at least two of them approximately sympatric in the central Great Smoky Mountains (the ‘Hwy’ clade and one or more lineages from Clingmans Dome and Big Cataloochee Mt.) The larger lineage, which includes the latter individuals, comprises three highly divergent (>20%) lineages, with these two having as their closest relatives other individuals from distant localities, Brasstown Bald and Grandfather Mt, respectively. The third lineage includes only a single individual from Mt. Rogers in the northeast, and it seems reasonable to hypothesize that all three of these represent distinct species, giving a total of 4. Only two species of
Neelides have been reported from southern Appalachia,
N. dianae Christiansen and Bellinger, and
N. minutus (Folsom) [
22], although even they recognized the possibility that the latter might be a complex of species. Unfortunately, none of these vouchers is adequately preserved to seriously assess identities based on specific morphological characters. More material and comparisons with type specimens will be necessary to sort out just how many species we’ve sampled, and whether they correspond to described ones or not.
3.2. Symphypleona
3.2.1. Dicyrtomidae
We recovered at least five morphological species of Dicyrtomidae, Dicyrtoma hageni (Folsom) (f. frontalis), Calvatomina rossi (Wray), Ptenothrix atra (Linnaeus), P. renateae Snider, and P. marmorata (Packard). These are estimated to represent between 5 (mPTP) and 33 (bPTP) species by delimitation analyses. The lower estimate lumps several well-differentiated morphospecies together and can be disregarded, while all the other estimates finely subdivide each of the species of Ptenothrix. The reality certainly lies somewhere in between.
Dicyrtoma hageni (
Figure 4B) was found only in localities southwest of the French Broad River, from Mount Kephart in the Smokies south to Brasstown Bald in north Georgia. These localities fall into three highly distinct genetic clades, and are reconstructed as three species by 4 of 5 delimitation analyses. The fact that we observe some sympatry (‘Hwy’ localities in the Great Smoky Mts) among these very divergent clades lends support the idea that there are indeed multiple cryptic species present. One of these clades includes only our southernmost localities, spanning north Georgia and South Carolina, none of which have any spruce-fir component. This suggests that some ecological differentiation might also have occurred.
Ptenothrix renateae (
Figure 4E) was described from north Georgia and lower elevations of South Carolina, while our records extend this northward into the Great Balsam Mts. of southwestern North Carolina. bPTP results suggest that each of the six localities for
P. renateae represents a distinct species, which would initially seem to be an overestimate, as they cover no more than 100 linear km. However, no delimitation analyses support fewer than four species, and divergences among them mostly exceed ASAP’s estimated threshold (ranging to 21% between the Richland Balsam and Black Balsam Knob populations, for example).
Ptenothrix atra (
Figure 4C) was found over a broad area from the higher parts of the Smokies (Big Cataloochee Mt.) in the west to Mt. Rogers in the northeast. However, these represent two deeply divergent clades that are not resolved as each other’s closest relatives. Delimitation analyses subdivide these into 9 or more species. These larger clades are broadly sympatric, sharing a couple of localities (Browning Knob and Roan High Knob) where individuals are >21% divergent. So there seems clear evidence that multiple cryptic species are present. Even within each larger
P. atra clade some structuring may be significant. For example, two examples from Clingmans Dome are resolved in highly divergent lineages (almost 13% between CD.A.176 and CD.B.448) with individuals from other localities interspersed. So, in this case, more than two cryptic species seems a reasonable hypothesis.
Individuals identified as
P. marmorata (
Figure 4D) fell out in two clades within a paraphyletic set of
P. atra lineages, mostly from the southwestern portion of our sampling region, though one immature individual from Celo Knob in the Black Mts also resolved among these. Delimitation analyses would suggest that every locality sampled represented a distinct species, and divergences across the two major lineages is comparable to those in
P. atra, well above estimated intraspecific thresholds.
Specimens identified as
Calvatomina rossi (
Figure 4F) were found from Sassafras Mt. SC to the Roan Highlands along the NC/TN border, and represent the first records for the region, having previously only been reported from Florida [
29], Massachusetts, and Illinois [
20]. All localities are lumped as one species by mPTP but divided into 4 by all other delimitation analyses. There is not obvious geographic signal in the relationships apart from identity between two Roan Mt localities (GRB and RHK).
3.2.2. Arrhopalitidae
Arrhopalitidae (
Figure 4G,H) is represented by 14 specimens, resolved into two independent lineages, with some apparently misplaced
Sminthurus (Sminthuridae) close to one. All should represent the genus
Arrhopalites (or the genus
Pygmarrhopalites Vargovitsh, the status of which has been disputed by [
74]), which contains 25 poorly defined nearctic species, with perhaps half of these expected to occur in the southern Appalachians. Delimitation analyses suggest that our sequences represent between 6 and 8 distinct species, and divergences among the lineages corresponding with geography would seem to support the higher end of this range. However, none are well-enough sampled to conclude much now. One larger cluster of 7 sequences (GRB.A.060 to BCat.A.130) comprises all darker blueish specimens with rather distinctive lighter patterning, but even across two subgroups here (e.g., GRB.A.388 vs. WT.A.060), divergences reach nearly 30%. Otherwise, darker and lighter rust-colored forms are intermingled on several very long branches. This family will need much more focused attention.
3.2.3. Sminthurididae
The family Sminthurididae was represented by 22 specimens of
Sminthurides (one of which is possibly an immature
Sphaeridia;
Figure 5C). These included two individuals of the Holarctic
Sminthurides malmgreni, from Brasstown Bald, GA, and Grassy Ridge Bald in the Roan Highlands, clustered together though about 8% divergent from each other, and hypothesized as distinct by just 2 of 5 delimitation techniques. This clade was far from the remaining
Sminthurides, which mostly corresponded to
Sminthurides hyogramme (
Figure 5A), a distinctive species with blue stripes and a bright white lateral spot, among the most common Symphypleona encountered. These spanned our entire sampling range, from Sassafras Mt., SC in the south to Mt. Rogers, VA in the north (and Brasstown Bald in the west, though that individual did not sequence successfully). There is considerable variation in specifics of color pattern among these, some of which may be meaningful. Darker individuals, especially with a darker head, from Sassafras Mt (
Figure 5B) and Grandfather Mt (GrM.A.077) cluster together, conceivably representing
Sminthurides macnamarai Folsom & Mills (as described in [
22]). There is also a clade of several individuals with a complete ventral stripe below the white lateral spot (CD.B.449, MK.B.406, MK.A.094, BCat.A.128). The latter, however, also represents a series of relatively proximate localities in the Great Smoky Mts., nested among clusters of more typical coloration, so the significance for possible specific-level differentiation is not yet clear. A singleton from Roan High Knob (RHK.A.383) has a dark body and white head, and is >25% divergent from any others. Delimitation analyses subdivide this
S. hyogramme clade into anywhere from 9 to 13 species. We would suggest the total is more likely in the range of 3–5 species based on morphological variation. However, further molecular data will be needed to test whether more truly cryptic species are present.
3.2.4. Katiannidae
Katiannidae are represented by at least 4 species in three genera. A total of 11 individuals of the genus
Vesicephalus (
Figure 5D) were sequenced from ten localities, spanning our whole sampled range from the Grayson Highlands in the northeast to Brasstown Bald in the southwest. Delimitation analyses are unanimous in resolving these into exactly 2 species. Though represented by only a single individual, the Brasstown Bald specimen (BBld.B.454) has dark eyes and a fairly distinctive color pattern, and is more than 20% divergent from all others. It is possible that our samples represent
V. longisetis (Guthrie) and
V. crossleyi Snider. However, the type of the former is poorly preserved, and described differences between the two are of dubious value [
75]. Christiansen & Bellinger [
22] report both species from the region, but also suggest that what they considered
V. longisetis could represent multiple species. Further work will be needed to conclusively identify these as either of the described or possibly (the Brasstown Bald specimen) undescribed.
The monotypic genus
Katiannina (
Figure 5E) was represented by 9 specimens from 7 peaks, resolving into two deeply divergent lineages (separated by ~20%). These almost certainly represent at least two species, and the one lineage with just two individuals from quite distant localities (‘Hwy’, near Newfound Gap in Great Smoky Mountains National Park and Big Bald, northeast of the Asheville Depression) is itself subdivided by most delimitation analyses. In the larger clade there is no obvious geographic structuring among populations or lineages, with an individual from Clingmans Dome very similar to one from the Roan Highlands, and the rather proximate Big Bald and Roan Highlands representatives (only 35 km apart, both on same side of French Broad River valley) are separated as far as possible in the species’ cluster. Whether any of these represents the sole described species
K. macgillivrayi (Banks), described from New York, is questionable, as all are simply pale orange and lack the ‘black stripe […] from the eye running back to the anal tubercle’ originally described (though that may be a condition of a feeding instar rather than a morphological character of the species) [
76]. Another form known from lowland South Carolina that we have not yet sequenced seems different still, predominantly reddish with distinct, bright white dorsal spots. The genus clearly needs further taxonomic attention.
Fifteen individuals of three morphologically distinctive forms of
Sminthurinus were sampled, though these did not all resolve together in the tree. Three individuals of a species near
S. conchyliatus Snider (spanning Rabun Cliffs, GA to Mount Mitchell, NC;
Figure 5F) were resolved together, differing by at most 8.4%, only barely above the ASAP estimated threshold. These were sister to a lineage of 5 individuals of a species near
S. minutus MacGillivray (
Figure 5G), all collected from NE of the French Broad River valley, most from the Roan Highlands. These did cluster tightly together (<1% difference) relative to the one individual from Whitetop Mt. in Virginia (~17%). These (including the Whitetop specimen) seem likely to be distinct species from
S. minutus, all possessing a distinctive, complete white cap between the eyes, and a mostly dark-colored head, where in
S. minutus two separate white spots seem always to be present, separated by an orange wedge, with the rest of the head relatively light-colored. Christiansen & Bellinger [
22] considered
S. minutus to be a potential synonym of
S. quadrimaculatus (Ryder), but we agree with their admitted possibility that these represent a species cluster in need of subdivision. Lastly, we obtained 7 sequences for
Sminthurinus henshawi, 5 striped individuals representing what has been termed a form ‘
similitortus’ (
Figure 5H), and 2, a deeply divergent monophyletic sister to those, the form ‘
aureus’, lacking longitudinal blue stripes (
Figure 5I). The two ‘
aureus’ individuals, both from the Great Balsam Mts. (RB and BBK) differ by less than 1%. The ‘
similitortus’ types form two highly divergent clusters (>25%), one from the Roan Highlands (2 individuals < 1% different) and one with individuals from the Black Mts (Big Tom and Celo Knob, 6% different) and Big Bald (~4.5% from either of the Black Mts. individuals). Big Bald is about equidistant from either the Blacks or the Roan Highlands, so its much closer relationship to the former is surprising.
3.2.5. Sminthuridae
Finally, the family Sminthuridae, which formerly contained nearly all Symphypleona species (e.g., [
77], is represented here by three genera. As many as 7 or 8 described species of
Sminthurus should occur in the region, which are said to be largely indistinguishable based on external color patterns. Our first of 3 lineages (‘clade A’) of these (resolved as sister to the larger
Arrhopalites clade) contains 4 individuals, mostly distinctively patterned (aside from one immature) with dark blue and strongly contrasting white dorsal stripes (
Figure 6A). These range from Clingmans Dome in the west to Mount Mitchell in the northeast (and probably includes very similar as-yet-unsequenced specimens from Mt Rogers and Whitetop even further north). Delimitation results subdividing these into 2 or 3 distinct species are difficult to evaluate, though distances among lineages do exceed estimated thresholds. In the other major lineage (‘Clade B’), a tight cluster of three individuals (MHy.A.119, GRB.A.059, and GRB.A.412; (
Figure 6B) seems to represent a distinct species, as supported by all delimitation analyses. Unfortunately 2 of these individuals are immature, so it is impossible to assess meaningful morphological consistencies among them. The other lineage in clade B includes 17 individuals ranging across the region from Brasstown Bald to Mt. Rogers. These exhibit remarkably low divergences among those considered here (<5%), and almost certainly do represent a single species. Assessing its morphology to attempt to identify it is complicated by the surprisingly high incidence of immatures. Mature individuals (like Sass.A.339) have most of the body dark blue, with numerous, small, obscure lighter spots, and a round, distinctively green cheek patch below the eye (
Figure 6C). However, this can be clearly seen in only a handful of specimens. It is possible that this corresponds to
S. bivittatus Snider, in which a ‘gena with dark green polygons forming rosettes’ is described.
A single individual of
Sphyrotheca minnesotensis (
Figure 6G) was found at Cowee Bald, a non-spruce-fir peak at a slightly lower elevation (~1500 m). Despite its name, this species has previously been recorded through much of the eastern Nearctic, from Minnesota to Ontario to Louisiana, though never specifically from higher parts of Appalachia.
Neosminthurus represents the last, very commonly collected genus. Specimens have relatively short antennae, and a lightly debris-cloaked appearance, often appearing to have retained parts of previous molts on the body. There are three named species in the region, and it’s likely that all are represented among our more than 50 individuals sequenced, though delimitation analyses suggest between 6 and 10 species. Resolution of exactly what clades corresponds to what species is not entirely straightforward. The largest lineage, spanning the whole range from Huckleberry Knob, NC (HKnb.B.378) and Sassafras Mt., SC (Sass.A.313), to Whitetop Mt, VA (WT.A.063), comprises mottled individuals with each antennomere apically darkened (
Figure 6F), characters that correspond well to
Neosminthurus bakeri Snider. Divergences within this cluster are relatively shallow, no more than about 2%. Another large clade (almost divided into north and south clades, apart from the ‘misplaced’ Celo Knob CK.B.400 individual) consists of individuals exhibiting generally white heads in mature individuals (e.g., BCat.A.132 and CB.095; (
Figure 6E). Divergences between these subgroups are around 13%, though they are nearly as high within, particularly in the north group, at least comparing Mount Mitchell with the Roan Highlands group. One small clade from northeast of the Asheville depression (GRB.A.383, RHK.A.407, etc.) corresponds to
N. clavatus, with voucher specimens exhibiting the diagnostically flattened dorsal setae. These individuals are all dark, head and body. Members of a smaller clade from further north, (MRg.B.074 + WT.A.064) also have the flattened clavate setae, but have light colored heads, suggesting homoplasy in dorsal setal morphology. Lastly, a larger clade of 12 individuals found only south of the Asheville depression (HKnb.A.101, Hwy.A.180, etc.;
Figure 6D) are entirely dark, body and head, like
N. clavatus. However, these have narrower, cylindrical dorsal setae, and would not be assignable to that species.
N. bakeri is the only described species in the region that lacks ‘clavate’ body setae, but given the deep divergences, lack of monophyly and broadly sympatric distributions of the clades exhibiting that morphology, it is clear that there is more than one species involved. Most likely the ‘all dark’ clade from western North Carolina and north Georgia represents something undescribed, as does the ‘white head’ lineage (or lineages). However, further work will be needed to test this possibility.
4. Conclusions
This work represents a significant step forward in the integrative systematic study of globular Collembola in the southeastern US. At the simplest level, we have broadened the known distributions for a number of poorly documented species, and begun to reveal some meaningful biogeographic patterns in some. More significantly, these data reveal extraordinary levels of intraspecific diversity in nearly all unambiguously identifiable species, as has become typical in Collembola intraspecific work [
37,
67,
68,
69,
78,
79,
80], indicating long residence times for these in the region, and high potential for the presence of cryptic species. While we do not see much basis for the high levels of splitting that most automated delimitation methods suggested, almost all morphologically well-defined species contain highly divergent, geographically coherent clades that would qualify at least as evolutionarily significant units (ESUs) and as candidates for separate conservation consideration. More comprehensive sampling of different genomic regions, particularly nuclear genes, and inclusion of more individuals from within and beyond this region will be necessary before conclusions about cryptic endemics can be supported.
The attempted use of DNA barcodes to identify globular Collembola failed completely; none of our sequences was a close enough match in any public database to confidently support an identification. The closest matches were often correct to family, and occasionally even to species. However, even in such cases the similarities were never greater than 90%, often with not much worse matches (~82–85%) being to members of different genera, families, or even incorrect hexapod orders (Coleoptera, Hemiptera, and others). Clearly these databases have a long way to go in representation of more obscure animal groups to be up to the task of molecular identification.
Regarding the workflow presented here, methods for high-throughput generation of sequence data for studying arthropod biodiversity have been evolving rapidly. The data analyzed here represent a mix of ‘traditional’ Sanger sequencing techniques and two next-generation approaches, Illumina and Nanopore sequencing. After using all these, we agree with Srivathsan et al. [
46] in endorsing the Nanopore approach for its relative ease and cost-effectiveness. Despite its allegedly higher error rates, the high degree of replication and ability to sort through these with software such as ONT barcoder effectively neutralizes this concern, and not needing to devote significant flow cell space to control sequences (as required on Illumina’s MiSeq with low diversity libraries) ensure that yields are maximized. One additional consideration, associated with our use of 96 well plates and liquid handling robots for parts of the DNA extraction procedure, is the loss of numerous (nearly half) of the voucher specimens. This problem is probably worse for Collembola than for any other arthropod group we’ve worked with due to their very thin cuticle and their becoming completely transparent during digestion. Single tube extractions, though much slower, would probably recover a greater proportion of voucher specimens. Regardless, we would emphasize the importance of photographing specimens of minute arthropods before extracting them assuming relatively high rates of destruction and loss.
Symphypleona and Neelipleona represent a diverse and, for their sizes, quite charismatic group of litter arthropods. Their relative neglect by the broader community can only be attributed to their minuteness and limited taxonomic resources. Large scale biodiversity assessments using molecular methods stand to revolutionize our understanding of this and other dark taxa [
81,
82], and we hope that this contribution helps underscore the potential. Threatened areas and faunas, like the high elevations of southern Appalachia, include large numbers of such species that through ignorance risk extinction before we’re even aware of their existence. They desperately deserve the attention of conservation biologists, but careful taxonomic revision, accurate species delimitation, and useable identification resources are necessary before they can be practically considered in such planning.














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}