
Genetic Differentiation among Subspecies of Banksia nivea (Proteaceae) Associated with Expansion and Habitat Specialization


Abstract
:1. Introduction
2. Materials and Methods
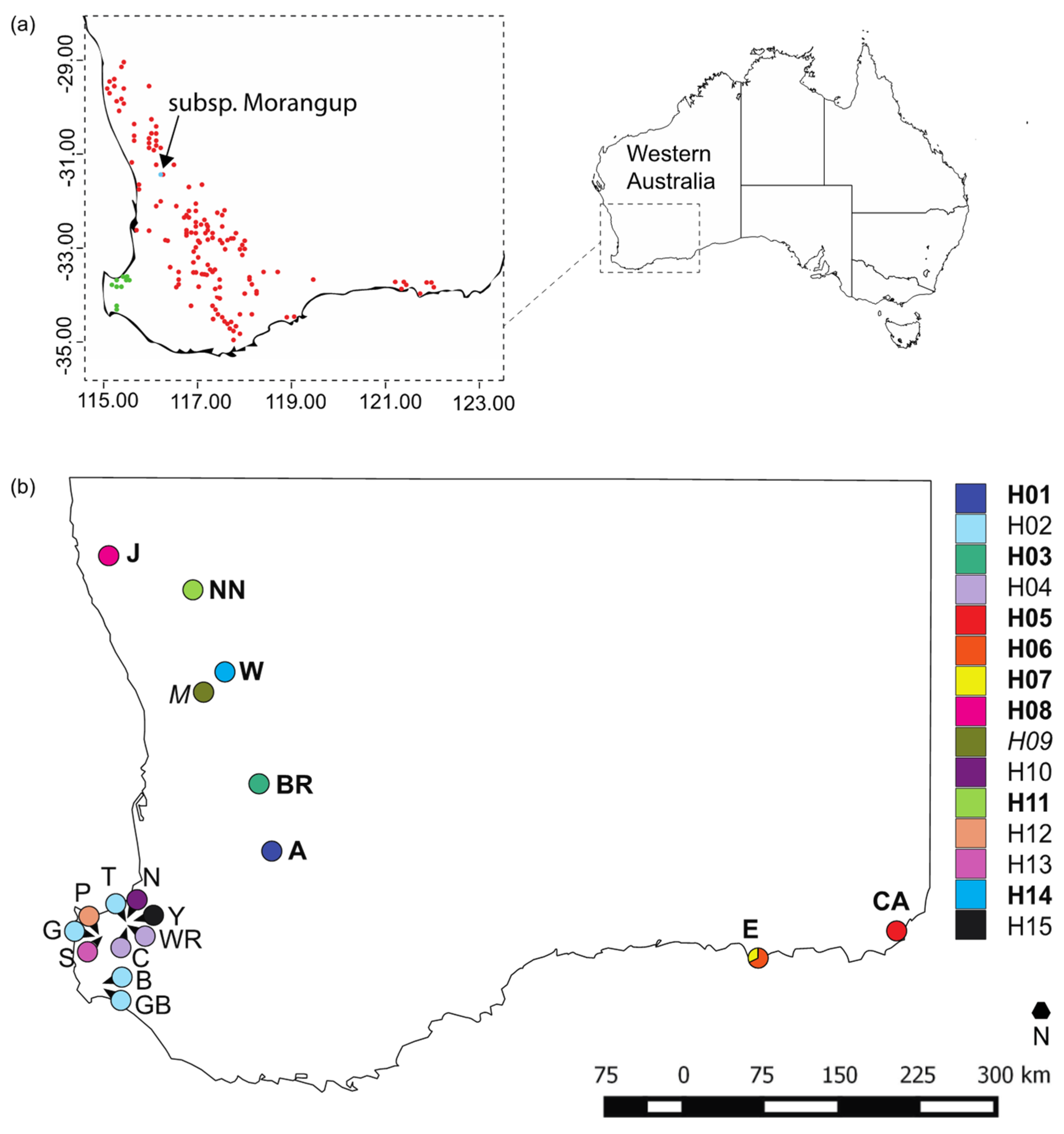
2.1. Sampling and Genotyping
2.2. Chloroplast DNA Diversity and Relatedness
2.3. Nuclear SSR Diversity and Structure
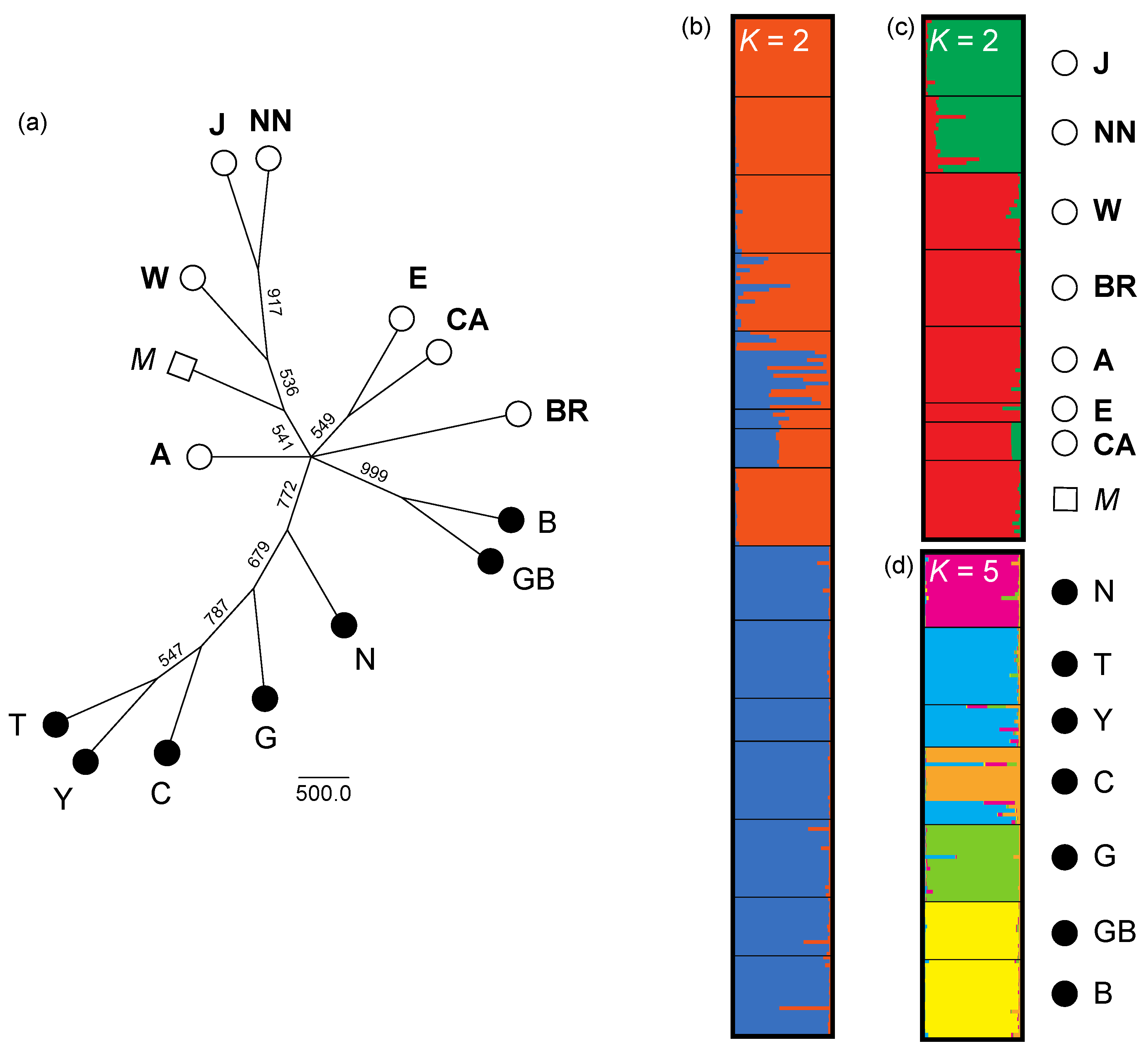
3. Results
3.1. Chloroplast Diversity and Divergence
3.2. Nuclear Diversity and Structure
4. Discussion
4.1. Distinction between Subspecies
4.2. Biogeographic Expansion
4.3. Contemporary Genetic Diversity and Population Structure
5. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Coates, D.J.; Byrne, M.; Moritz, C. Genetic diversity and conservation units: Dealing with the species-population continuum in the age of genomics. Front. Ecol. Evol. 2018, 6, 165. [Google Scholar] [CrossRef] [Green Version]
- Mayr, E. Systematics and the Origin of Species; Columbia University Press: New York, NY, USA, 1942. [Google Scholar]
- Grant, V. Plant Speciation; Columbia University Press: New York, NY, USA, 1981. [Google Scholar]
- Hamilton, C.; Reichard, S. Current practice in the use of subspecies, variety, and forma in the classification of wild plants. Taxon 2019, 41, 485–498. [Google Scholar] [CrossRef]
- Duminil, J.; Kenfack, D.; Viscosi, V.; Grumiau, L.; Hardy, O.J. Testing species delimitation in sympatric species complexes: The case of an African tropical tree, Carapa spp. (Meliaceae). Mol. Phylogenet. Evol. 2012, 62, 275–285. [Google Scholar] [CrossRef] [PubMed]
- Christmas, M.J.; Biffin, E.; Lowe, A.J. Measuring genome-wide genetic variation to reassess subspecies classifications in Dodonaea viscosa (Sapindaceae). Aust. J. Bot. 2018, 66, 287–297. [Google Scholar] [CrossRef] [Green Version]
- Ellstrand, N.C. Is gene flow the most important evolutionary force in plants? Am. J. Bot. 2014, 101, 737–753. [Google Scholar] [CrossRef]
- Wang, I.J.; Bradburd, G.S. Isolation by environment. Mol. Ecol. 2014, 23, 5649–5662. [Google Scholar] [CrossRef]
- Zhang, Y.-H.; Wang, I.J.; Comes, H.P.; Peng, H.; Qiu, Y.-X. Contributions of historical and contemporary geographic and environmental factors to phylogeographic structure in a Tertiary relict species, Emmenopterys henryi (Rubiaceae). Sci. Rep. 2016, 6, 24041. [Google Scholar] [CrossRef] [Green Version]
- Robins, T.P.; Binks, R.M.; Byrne, M.; Hopper, S.D. Contrasting patterns of population divergence on young and old landscapes in Banksia seminuda (Proteaceae), with evidence for recognition of subspecies. Biol. J. Linn. Soc. 2021, 133, 449–463. [Google Scholar] [CrossRef]
- Moore, A.J.; Merges, D.; Kadereit, J.W. The origin of the Serpentine endemic Minuartia larcifolia subsp. ophiolitica by vicariance and competitive exclusion. Mol. Ecol. 2013, 22, 2218–2231. [Google Scholar] [CrossRef]
- Nistelberger, H.M.; Tapper, S.L.; Coates, D.J.; McArthur, S.L.; Byrne, M. As old as the hills: Pliocene palaeogeographical processes influence patterns of genetic structure in the widespread, common shrub Banksia sessilis. Ecol. Evol. 2021, 11, 1069–1082. [Google Scholar] [CrossRef]
- Byrne, M.; Murphy, D.J. The origins and evolutionary history of xerophytic vegetation in Australia. Aust. J. Bot. 2020, 68, 195–207. [Google Scholar] [CrossRef]
- Coates, D.J. Defining conservation units in a rich and fragmented flora: Implications for the management of genetic resources and evolutionary processes in south-west Australian plants. Aust. J. Bot. 2000, 48, 329–339. [Google Scholar] [CrossRef]
- Byrne, M.; Coates, D.; Forest, F.; Hopper, S.; Krauss, S.; Sniderman, J.; Thiele, K. A diverse flora—species and genetic relationships. In Plant Life on the Sandplains in Southwest Australia, a Global Biodiversity Hotspot; Lambers, H., Ed.; University of Western Australia Publishing: Crawley, WA, USA, 2014; pp. 81–99. [Google Scholar]
- Coates, D.J.; McArthur, S.L.; Byrne, M. Significant genetic diversity loss following pathogen driven population extinction in the rare endemic Banksia brownii (Proteaceae). Biol. Conserv. 2015, 192, 353–360. [Google Scholar] [CrossRef]
- Sampson, J.F.; Byrne, M.; Yates, C.J.; Gibson, N.; Thavornkanlapachai, R.; Stankowski, S.; Macdonald, B.; Bennett, I. Contemporary pollen-mediated gene immigration reflects the historical isolation of a rare, animal-pollinated shrub in a fragmented landscape. Heredity 2014, 112, 172–181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cavanagh, A.K.; Pieroni, M. The Dryandras; Australian Plants Society (SGAP Victoria) Inc.; Wildflower Society of Western Australia Inc.: Melbourne, Australia, 2006. [Google Scholar]
- Cardillo, M.; Pratt, R. Evolution of a hotspot genus: Geographic variation in speciation and extinction rates in Banksia (Proteaceae). BMC Evol. Biol. 2013, 13, 155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thavornkanlapachai, R.; Byrne, M.; Yates, C.J.; Ladd, P.G. Degree of fragmentation and population size do not adversely affect reproductive success of a rare shrub species, Banksia nivea (Proteaceae), in a naturally fragmented community. Bot. J. Linn. Soc. 2019, 191, 261–273. [Google Scholar] [CrossRef]
- George, A. New taxa and a new infrageneric classification in Dryandra R.Br. (Proteaceae: Grevilleoideae). Nuytsia 1996, 10, 313–408. [Google Scholar]
- Brown, A.; Thomson-Dans, C.M.N. Western Australia’s Threatened Flora; Brown, A., Thomson-Dans, C.M.N., Eds.; Department of Conservation and Land Management: Perth, Australia, 1998. [Google Scholar]
- Thavornkanlapachai, R.; Ladd, P.G.; Byrne, M. Population density and size influence pollen dispersal pattern and mating system of the predominantly outcrossed Banksia nivea (Proteaceae) in a threatened ecological community. Biol. J. Linn. Soc. 2018, 124, 492–503. [Google Scholar] [CrossRef]
- FloraBase-the Western Australian Flora. Available online: https://florabase.dpaw.wa.gov.au/browse/profile/32205 (accessed on 11 June 2021).
- Broadhurst, L.; Breed, M.; Lowe, A.; Bragg, J.; Catullo, R.; Coates, D.; Encinas-Viso, F.; Gellie, N.; James, E.; Krauss, S.; et al. Genetic diversity and structure of the Australian flora. Divers. Distrib. 2017, 23, 41–52. [Google Scholar] [CrossRef] [Green Version]
- Byrne, M.; Parrish, T.L.; Moran, G.F. Nuclear RFLP diversity in Eucalyptus nitens. Heredity 1998, 81, 225–233. [Google Scholar] [CrossRef]
- Byrne, M.; Hankinson, M. Testing the variability of chloroplast sequences for plant phylogeography. Mol. Ecol. Resour. 2012, 60, 569–574. [Google Scholar] [CrossRef]
- Maddison, W.P.; Maddison, D.R. Mesquite: A Modular System for Evolutionary Analysis. Available online: http://mesquiteproject.org (accessed on 2 October 2017).
- Librado, P.; Rozas, J. DnaSP v.5: A software for comprehensive analysis of DNA polymorphism data. Bioinformatics 2009, 25, 1451–1452. [Google Scholar] [CrossRef] [Green Version]
- Atlas of Living Australia. Available online: https://bie.ala.org.au/species/https://id.biodiversity.org.au/node/apni/2894027 (accessed on 16 June 2021).
- Millar, M.A.; Byrne, M. Characterization of polymorphic microsatellite DNA markers in Banksia nivea, formerly Dryandra nivea. Mol. Ecol. Resour. 2008, 8, 1393–1394. [Google Scholar] [CrossRef] [PubMed]
- van Oosterhout, C.; Hutchinson, W.F.; Wills, D.P.M.; Shipley, P. MICRO-CHECKER: Software for identifying and correcting genotyping errors in microsatellite data. Mol. Ecol. Notes 2004, 4, 535–538. [Google Scholar] [CrossRef]
- Rousset, F. GENEPOP’007: A complete reimplementation of the GENEPOP software for Windows and Linux. Mol. Ecol. Resour. 2008, 8, 103–106. [Google Scholar] [CrossRef]
- Excoffier, L.; Lischer, H. Arlequin Suite Ver 3.5: A new series of programs to perform population genetic analyses under Linux and Windows. Mol. Ecol. Resour. 2010, 10, 464–467. [Google Scholar] [CrossRef]
- Pons, O.; Petit, R.J. Measuring and testing genetic differentiation with ordered versus unordered alleles. Genetics 1996, 144, 1238–1245. [Google Scholar] [CrossRef]
- Tajima, F. Statistical methods for testing the neutral mutation hypothesis by DNA polymorphism. Genetics 1989, 123, 585–595. [Google Scholar] [CrossRef]
- Fu, Y.X. Statistical tests of neutrality of mutations against population growth, hitchhiking and background selection. Genetics 1997, 147, 915–925. [Google Scholar] [CrossRef]
- Bandelt, H.-J.; Forster, P.; Rohl, A. median-joining networks for inferring intraspecific phylogenies. Mol. Biol. Evol. 1999, 16, 37–48. [Google Scholar] [CrossRef]
- Peakall, R.; Smouse, P.E. GenALEx 6.5: Genetic analysis in Excel. Population genetic software for teaching and research-an update. Bioinformatics 2012, 28, 2537–2539. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kalinowski, S.J. HP-RARE 1.0: A computer program for performing rarefaction on measures of allelic richness. Mol. Ecol. Notes 2005, 5, 187–189. [Google Scholar] [CrossRef]
- Chapuis, M.P.; Estoup, A. Microsatellite null alleles and estimation of population differentiation. Mol. Biol. Evol. 2007, 24, 621–631. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Felsenstein, J. PHYLIP—phylogeny inference package (Version 3.2). Cladistics 1989, 5, 164–166. [Google Scholar]
- Dieringer, D.; Schlötterer, C. Microsatellite Analyser: A platform independent analysis tool for large microsatellite data sets. Mol. Ecol. Notes 2003, 3, 167–169. [Google Scholar] [CrossRef]
- Pritchard, J.K.; Stephens, M.; Donnelly, P. Inference of population structure using multilocus genotype data. Genetics 2000, 155, 945–959. [Google Scholar] [CrossRef] [PubMed]
- Evanno, G.; Regnaut, S.; Goudet, J. Detecting the number of clusters of individuals using the software STRUCTURE: A simulation study. Mol. Ecol. 2005, 14, 2611–2620. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kopelman, N.M.; Mayzel, J.; Jakobsson, M.; Rosenberg, N.A.; Mayrose, I. Clumpak: A program for identifying clustering modes and packaging population structure inferences across K. Mol. Ecol. Resour. 2015, 15, 1179–1191. [Google Scholar] [CrossRef] [Green Version]
- Wang, J. The computer program Structure for assigning individuals to populations: Easy to use but easier to misuse. Mol. Ecol. Resour. 2017, 17, 981–990. [Google Scholar] [CrossRef]
- Broadhurst, L.; Byrne, M.; Craven, L.; Lepschi, B. Genetic congruence with new species boundaries in the Melaleuca uncinata complex (Myrtaceae). Aust. J. Bot. 2004, 52, 729–737. [Google Scholar] [CrossRef]
- Nistelberger, H.; Gibson, N.; Macdonald, B.; Tapper, S.-L.; Byrne, M. Phylogeographic evidence for two mesic refugia in a biodiversity hotspot. Heredity 2014, 113, 454–463. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sampson, J.F.; Hankinson, M.; McArthur, S.; Tapper, S.; Langley, M.; Gibson, N.; Yates, C.; Byrne, M. Long-term “islands” in the landscape: Low gene flow, effective population size and genetic divergence in the shrub Hakea oldfieldii (Proteaceae). Bot. J. Linn. Soc. 2015, 179, 319–334. [Google Scholar] [CrossRef] [Green Version]
- Byrne, M. Evidence for multiple refugia at different time scales during Pleistocene climatic oscillations in southern Australia inferred from phylogeography. Quat. Sci. Rev. 2008, 27, 2576–2585. [Google Scholar] [CrossRef]
- Byrne, M.; Steane, D.A.; Joseph, L.; Yeates, D.K.; Jordan, G.J.; Crayn, D.; Aplin, K.; Cantrill, D.J.; Cook, L.G.; Crisp, M.D.; et al. Decline of a biome: Evolution, contraction, fragmentation, extinction and invasion of the Australian mesic zone biota. J. Biogeogr. 2011, 38, 1635–1656. [Google Scholar] [CrossRef] [Green Version]
- Byrne, M.; Yeates, D.K.; Joseph, L.; Kearney, M.; Bowler, J.; Williams, M.A.J.; Cooper, S.; Donnellan, S.C.; Keogh, J.S.; Leys, R.; et al. Birth of a biome: Insights into the assembly and maintenance of the Australian arid zone Biota. Mol. Ecol. 2008, 17, 4398–4417. [Google Scholar] [CrossRef] [PubMed]
- Llorens, T.M.; Tapper, S.-L.; Coates, D.J.; McArthur, S.; Hankinson, M.; Byrne, M. Does population distribution matter? Influence of a patchy versus continuous distribution on genetic patterns in a wind-pollinated shrub. J. Biogeogr. 2016, 44, 361–374. [Google Scholar] [CrossRef]
- Sampson, J.; Tapper, S.; Coates, D.; Hankinson, M.; McArthur, S.; Byrne, M. Persistence with episodic range expansion from the early Pleistocene: The distribution of genetic variation in the forest tree Corymbia calophylla (Myrtaceae) in south-Western Australia. Biol. J. Linn. Soc. 2018, 123, 545–560. [Google Scholar] [CrossRef]
- Loveless, M.; Hamrick, J. Ecological determinants of genetic structure in plant populations. Annu. Rev. Ecol. Evol. Syst. 1984, 15, 65–96. [Google Scholar] [CrossRef]
- Nybom, H. Comparison of different nuclear DNA markers for estimating intraspecific genetic diversity in plants. Mol. Ecol. 2004, 13, 1143–1155. [Google Scholar] [CrossRef]
- Duminil, J.; Fineschi, S.; Hampe, A.; Jordano, P.; Salvini, D.; Vendramin, G.G.; Petit, R.J.; The, S.; Naturalist, A.; May, N. Can population genetic structure be predicted from life-history traits? Am. Nat. 2014, 169, 662–672. [Google Scholar] [CrossRef]
- Gitzendanner, M.A.; Soltis, P.S. Patterns of genetic variation in rare and widespread plant congeners. Am. J. Bot. 2000, 87, 783–792. [Google Scholar] [CrossRef] [PubMed]
- Ellstrand, N.C.; Elam, D.R. Population genetic consequences of small population size: Implications for plant conservation. Annu. Rev. Ecol. Evol. Syst. 1993, 24, 217–242. [Google Scholar] [CrossRef]
- Ford, H.A.; Paton, D.C.; Forde, N. Birds as pollinators of Australian plants. N. Z. J. Bot. 1979, 17, 509–519. [Google Scholar] [CrossRef] [Green Version]
- Wooller, R.D.; Russell, E.M.; Renfree, M.B.; Towers, P.A. A comparison of seasonal changes in the pollen loads of nectarivorous marsupials [Tarsipes] and birds [honeyeaters]. Wildl. Res. 1983, 10, 311–317. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
| Subspecies/Pop | N | A | AR | HO | UHe | F |
|---|---|---|---|---|---|---|
| Subsp. nivea | ||||||
| J | 19.900 (0.100) | 3.900 (0.994) | 2.080 (0.273) | 0.351 (0.081) | 0.374 (0.083) | 0.036 (0.045) |
| NN | 19.600 (0.400) | 5.800 (0.663) | 3.070 (0.294) | 0.498 (0.085) | 0.627 (0.081) | 0.185 (0.087) * |
| W | 18.100 (0.737) | 6.700 (0.831) | 3.370 (0.273) | 0.566 (0.070) | 0.697 (0.058) | 0.173 (0.072) * |
| BR | 19.100 (0.605) | 6.700 (1.146) | 3.120 (0.344) | 0.435 (0.085) | 0.625 (0.068) | 0.248 (0.118) * |
| A | 17.900 (0.623) | 8.300 (1.126) | 3.640 (0.340) | 0.462 (0.073) | 0.714 (0.069) | 0.281 (0.105) * |
| E | 3.300 (0.578) | 2.300 (0.496) | 12.630 (0.285) | 0.363 (0.098) | 0.409 (0.103) | −0.046 (0.098) |
| CA | 10.000 (0.000) | 3.500 (0.734) | 2.300 (0.367) | 0.350 (0.107) | 0.404 (0.102) | 0.101 (0.098) |
| Mean | 15.414 (0.107) | 5.314 (0.092) | 4.316 (0.015) | 0.432 (0.005) | 0.550 (0.007) | 0.140 (0.009) * |
| Subsp. Morangup | ||||||
| M | 19.100 (0.605) | 5.300 (0.932) | 2.840 (0.321) | 0.433 (0.089) | 0.574 (0.083) | 0.185 (0.117) |
| Subsp. uliginosa | ||||||
| N | 18.900 (0.100) | 3.800 (0.533) | 2.210 (0.215) | 0.401 (0.064) | 0.441 (0.073) | 0.018 (0.084) |
| T | 17.600 (0.933) | 4.100 (0.640) | 2.620 (0.283) | 0.510 (0.071) | 0.548 (0.074) | 0.009 (0.070) |
| Y | 10.400 (0.306) | 3.000 (0.365) | 2.080 (0.267) | 0.335 (0.072) | 0.366 (0.086) | −0.036 (0.076) |
| C | 18.000 (1.022) | 4.700 (0.667) | 2.810 (0.239) | 0.472 (0.066) | 0.593 (0.058) | 0.164 (0.097) |
| G | 14.800 (2.489) | 3.400 (0.653) | 8.330 (0.245) | 0.452 (0.077) | 0.498 (0.086) | 0.029 (0.047) |
| GB | 11.400 (0.909) | 4.100 (0.605) | 2.650 (0.279) | 0.448 (0.065) | 0.553 (0.074) | 0.107 (0.071) |
| B | 15.800 (1.245) | 4.300 (0.517) | 2.740 (0.264) | 0.404 (0.051) | 0.582 (0.067) | 0.195 (0.116) |
| Mean | 15.271 (0.292) | 3.914 (0.040) | 3.349 (0.009) | 0.432 (0.003) | 0.511 (0.004) | 0.069 (0.008) |
| Total mean | 15.593 (0.441) | 4.660 (0.227) | 3.766 (0.744) | 0.432 (0.020) | 0.534 (0.021) | 0.117 (0.024) |
| Total | Subsp. nivea | Subsp. uliginosa | Subsp. Morangup | |
|---|---|---|---|---|
| Populations (n) | 18 | 7 | 10 | 1 |
| Haplotypes (n) | 15 | 8 | 6 | 1 |
| Haplotype diversity HD | 0.920 (0.020) | 0.910 (0.026) | 0.786 (0.053) | 0 |
| Nucleotide diversity π | 0.003 (0.002) | 0.005 (0.003) | 0.001 (0.001) | 0 |
| Population differentiation (unordered) GST | 0.961 (0.039) | 0.905 (0.095) | 1 | - |
| Population differentiation (ordered) NST | 0.992 (0.008) | 0.982 (0.018) | 1 | - |
| Phylogenetic structure (NST > GST) | NS | p < 0.01 | NS | - |
| Tajima’s D | −0.56 p = 0.33 | 0.16 p = 0.61 | 0.14 p = 0.59 | - |
| Fu’s Fs | 1.13 p = 0.70 | 4.67 p = 0.98 | 1.62 p = 0.77 | - |
| Demographic expansion | SSD 0.010, | SSD 0.034, | SSD 0.257, | - |
| p = 0.61 | p = 0.12 | p = 0.38 | ||
| HRag 0.021, | HRag 0.045, | HRag 0.093, | - | |
| p = 0.43 | p = 0.17 | p = 0.25 | ||
| Spatial expansion | SSD 0.009, | SSD 0.032, | SSD 0.022, | - |
| p = 0.74 | p = 0.20 | p = 0.50 | ||
| HRag 0.021, | HRag 0.045, | HRag 0.093, | - | |
| p = 0.56 | p = 0.26 | p = 0.52 |
| Source of Variation | d.f. | SS | Variance Component | % Variation |
|---|---|---|---|---|
| Chloroplast haplotypes | ||||
| Among subspecies | 2 | 27.6 | 0.569 | 23.42 |
| Among populations within subspecies | 15 | 83.2 | 1.842 | 75.82 |
| Within populations | 36 | 0.7 | 0.019 | 0.76 |
| Nuclear microsatellites | ||||
| Among subspecies | 2 | 127.3 | 0.126 | 3.0 |
| Among populations within subspecies | 12 | 481.5 | 1.100 | 26.7 |
| Within populations | 505 | 1463.2 | 2.897 | 70.3 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sampson, J.; Byrne, M. Genetic Differentiation among Subspecies of Banksia nivea (Proteaceae) Associated with Expansion and Habitat Specialization. Diversity 2022, 14, 98. https://doi.org/10.3390/d14020098
Sampson J, Byrne M. Genetic Differentiation among Subspecies of Banksia nivea (Proteaceae) Associated with Expansion and Habitat Specialization. Diversity. 2022; 14(2):98. https://doi.org/10.3390/d14020098
Chicago/Turabian StyleSampson, Jane, and Margaret Byrne. 2022. "Genetic Differentiation among Subspecies of Banksia nivea (Proteaceae) Associated with Expansion and Habitat Specialization" Diversity 14, no. 2: 98. https://doi.org/10.3390/d14020098
APA StyleSampson, J., & Byrne, M. (2022). Genetic Differentiation among Subspecies of Banksia nivea (Proteaceae) Associated with Expansion and Habitat Specialization. Diversity, 14(2), 98. https://doi.org/10.3390/d14020098