
Dynamic Shifts in the Root Microbiota of Cultivated Paphiopedilum armeniacum during Different Stages of Growth


Abstract
:1. Introduction
2. Materials and Methods
2.1. Sample Collection and Surface Sterilization
2.2. DNA Extraction and PCR Amplification
2.3. Statistical Analyses
2.4. Covariation Network Analysis of Core Bacterial Genera and Mycorrhizal Fungi
2.5. Comparative Analysis of Tulasnella in Paphiopedilum armeniacum
3. Results
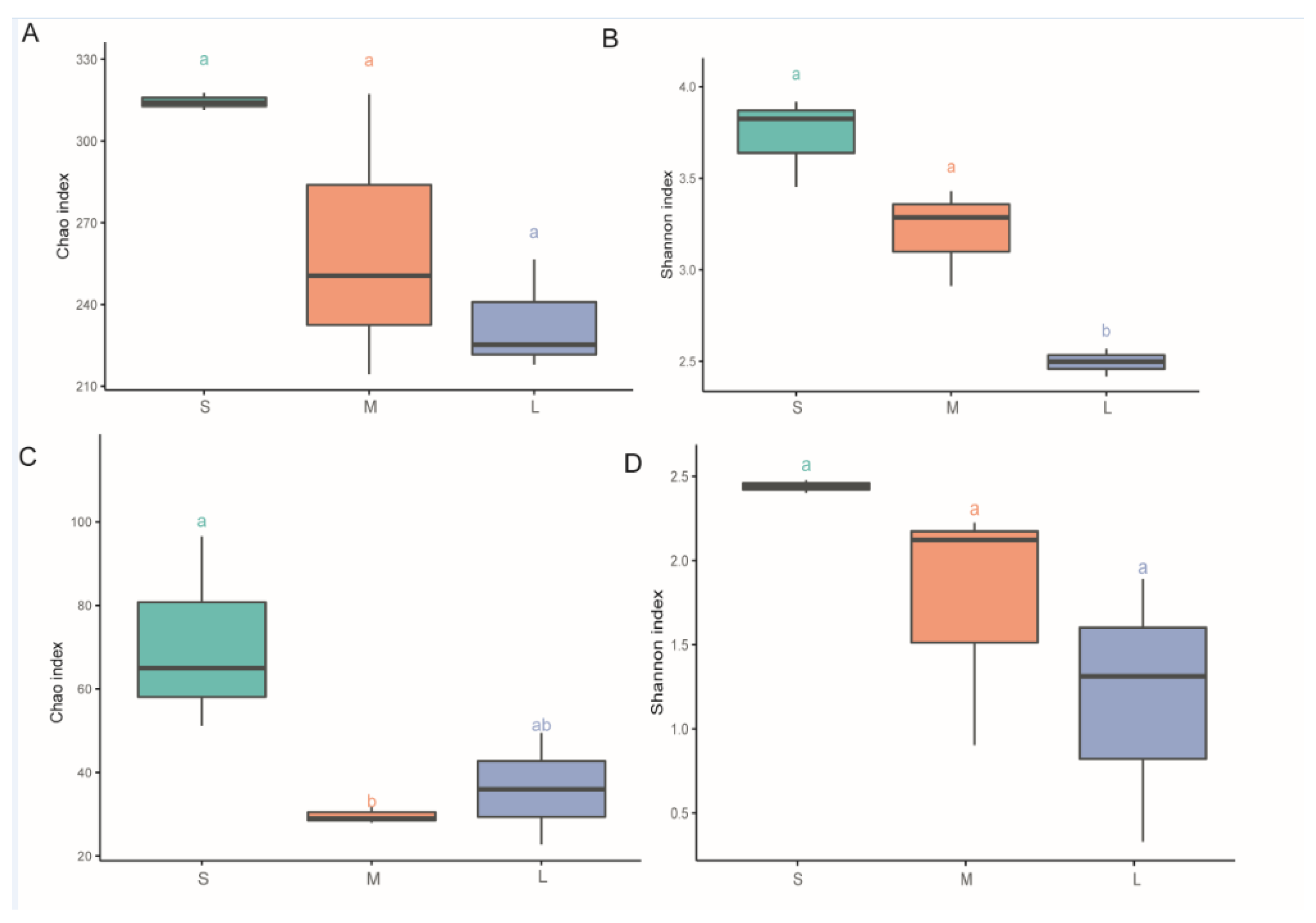
3.1. Diversity Analysis in Different Samples
3.2. Taxonomic Assignment of the Microbial Community Composition
3.3. Covariation Network Analysis of Core Bacterial Genera and Mycorrhizal Fungi
3.4. Comparison of the Mycorrhizal Fungi of P. armeniacum in Xingyi Greenhouse and in Other Habitats
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Cribb, P. The Genus Paphiopedilum, 2nd ed.; Natural History Publications: Kota Kinabalu, Malaysia, 1998; pp. 254–260. [Google Scholar]
- Wu, Z.Y.; Raven, P.H.; Hong, D.Y. Flora of China, Volume 25 (Orchidaceae); Science Press: Beijing, China; Missouri Botanical Garden Press: St. Louis, MO, USA, 2009; pp. 33–44. [Google Scholar]
- Chen, S.C.; Liu, F.Y. Notes on some species of Paphiopedilum from Yunnan. Acta Bot. Yunnanica 1982, 4, 163–167. [Google Scholar]
- Cites, Appendices I, II and III. Available online: http://www.cites.org/eng/app/appendices.php (accessed on 22 June 2021).
- Han, J.Y.; Xiao, H.F.; Gao, J.Y. Seasonal dynamics of mycorrhizal fungi in Paphiopedilum spicerianum (Rchb. f) Pfitzer—A critically endangered orchid from China. Glob. Ecol. Conserv. 2016, 6, 327–338. [Google Scholar] [CrossRef]
- Parthibhan, S.; Ramasubbu, R. Mycorrhizal and endophytic fungal association in Paphiopedilum druryi (Bedd.) Stein—A strict endemic and critically endangered orchid of the Western Ghats. Ecol. Genet. Genom. 2020, 16, 100059. [Google Scholar] [CrossRef]
- Liu, Z.J.; Chen, X.Q.; Chen, L.J.; Lei, S.P. The Genus Paphiopedilum in China; Science Press: Beijing, China, 2009. [Google Scholar]
- Wang, X.G.; Yan, H.X.; Li, X.L.; He, J.Z.; Zhou, Z.G. Identification and Growth Promoting Analysis of Mycorrhizal Fungi from Paphiopedilum hirsutissimun (Orchidaceae). Southwest China J. Agric. Sci. 2021, 34, 119–125. [Google Scholar] [CrossRef]
- Khamchatra, N.; Dixon, K.W.; Tantiwiwat, S.; Piapukiew, J. Symbiotic seed germination of an endangered epiphytic slipper orchid, Paphiopedilum villosum (Lindl.) Stein. from Thailand. S. Afr. J. Bot. 2016, 104, 76–81. [Google Scholar] [CrossRef]
- Sutthinon, P.; Rungwattana, K.; Suwanphakdee, C.; Himaman, W.; Lueangjaroenkit, P. Endophytic Fungi from Root of Three Lady’s Slipper Orchids (Paphiopedilum spp.) in Southern Thailand. Chiang Mai J. Sci. 2021, 48, 853–866. [Google Scholar]
- Yang, W.-K.; Li, T.-Q.; Wu, S.-M.; Finnegan, P.M.; Gao, J.-Y. Ex situ seed baiting to isolate germination-enhancing fungi for assisted colonization in Paphiopedilum spicerianum, a critically endangered orchid in China. Glob. Ecol. Conserv. 2020, 23, e01147. [Google Scholar] [CrossRef]
- Yi, S. Mycorrhizal Fungi and Symbiotic Seed Germination of Four Paphiopedilum Species; Beijing Forestry University: Beijing, China, 2017; pp. 19–29. [Google Scholar]
- Zhu, X.-M.; Hu, H.; Li, S.-Y.; Yan, N. Interaction between Endophytic Fungi and Seedlings of Two Species of Paphiopedilum during Symbiotic Culture. Plant Divers. Resour. 2012, 34, 171–178. [Google Scholar] [CrossRef]
- Yuan, L.; Yang, Z.L.; Li, S.-Y.; Hu, H.; Huang, J.-L. Mycorrhizal specificity, preference, and plasticity of six slipper orchids from South Western China. Mycorrhiza 2010, 20, 559–568. [Google Scholar] [CrossRef]
- Meng, Y.-Y.; Zhang, W.-L.; Selosse, M.-A.; Gao, J.-Y. Are fungi from adult orchid roots the best symbionts at germination? A case study. Mycorrhiza 2019, 29, 541–547. [Google Scholar] [CrossRef]
- Rasmussen, H.N. Recent developments in the study of orchid mycorrhiza. Plant Soil 2002, 244, 149–163. [Google Scholar] [CrossRef]
- Rasmussen, H.N.; Dixon, K.; Jersakova, J.; Těšitelová, T. Germination and seedling establishment in orchids: A complex of requirements. Ann. Bot. 2015, 116, 391–402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bidartondo, M.I.; Read, D.J. Fungal specificity bottlenecks during orchid germination and development. Mol. Ecol. 2008, 17, 3707–3716. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Phillips, R.D.; Barrett, M.D.; Dixon, K.W.; Hopper, S.D. Do mycorrhizal symbioses cause rarity in orchids? J. Ecol. 2011, 99, 858–869. [Google Scholar] [CrossRef]
- Pecoraro, L.; Wang, X.; Venturella, G.; Gao, W.; Wen, T.; Gafforov, Y.; Gupta, V.K. Molecular evidence supports simultaneous association of the achlorophyllous orchid Chamaegastrodia inverta with ectomycorrhizal Ceratobasidiaceae and Russulaceae. BMC Microbiol. 2020, 20, 236. [Google Scholar] [CrossRef]
- Meng, Y.-Y.; Shao, S.-C.; Liu, S.-J.; Gao, J.-Y. Do the fungi associated with roots of adult plants support seed germination? A case study on Dendrobium exile (Orchidaceae). Glob. Ecol. Conserv. 2019, 17, e00582. [Google Scholar] [CrossRef]
- McCormick, M.K.; Whigham, D.F.; O’Neill, J. Mycorrhizal diversity in photosynthetic terrestrial orchids. New Phytol. 2004, 163, 425–438. [Google Scholar] [CrossRef]
- Jacquemyn, H.; Brys, R.; Cammue, B.P.A.; Honnay, O.; Lievens, B. Mycorrhizal associations and reproductive isolation in three closely related Orchis species. Ann. Bot. 2011, 107, 347–356. [Google Scholar] [CrossRef] [Green Version]
- Tsavkelova, E. Bacteria Associated with Orchid Roots. In Bacteria in Agrobiology: Plant Growth Responses; Springer Science & Business Media: Berlin, Germany, 2011; pp. 221–258. [Google Scholar]
- Kaur, J.; Sharma, J. Orchid Root Associated Bacteria: Linchpins or Accessories? Front. Plant Sci. 2021, 12, 661966. [Google Scholar] [CrossRef]
- Tsavkelova, E.A.; Egorova, M.A.; Leontieva, M.R.; Malakho, S.G.; Kolomeitseva, G.L.; Netrusov, A.I. Dendrobium nobile Lindl. seed germination in co-cultures with diverse associated bacteria. Plant Growth Regul. 2016, 80, 79–91. [Google Scholar] [CrossRef]
- Zhang, P.; Zhong, Y.-F.; Song, X.-Q.; Wang, X.-M.; Wang, J. Interaction Effect of Symbiotic Microorganisms on Seedling Growth of Dendrobium catenatum Lindley (Orchidaceae). Plant Sci. J. 2013, 31, 73–79. [Google Scholar] [CrossRef]
- Frey-Klett, P.; Garbaye, J. Mycorrhiza helper bacteria: A promising model for the genomic analysis of fungal-bacterial interactions. New Phytol. 2005, 168, 4–8. [Google Scholar] [CrossRef] [PubMed]
- Novotná, A.; Suárez, J.P. Molecular detection of bacteria associated with Serendipita sp., a mycorrhizal fungus from the orchid Stanhopea connata Klotzsch in southern Ecuador. Bot. Lett. 2018, 165, 307–313. [Google Scholar] [CrossRef]
- Bulgarelli, D.; Garrido-Oter, R.; Münch, P.C.; Weiman, A.; Dröge, J.; Pan, Y.; McHardy, A.C.; Schulze-Lefert, P. Structure and Function of the Bacterial Root Microbiota in Wild and Domesticated Barley. Cell Host Microbe 2015, 17, 392–403. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Waud, M.; Busschaert, P.; Ruyters, S.; Jacquemyn, H.; Lievens, B. Impact of primer choice on characterization of orchid mycorrhizal communities using 454 pyrosequencing. Mol. Ecol. Resour. 2014, 14, 679–699. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [Green Version]
- Schloss, P.D.; Westcott, S.L.; Ryabin, T.; Hall, J.R.; Hartmann, M.; Hollister, E.B.; Lesniewski, R.A.; Oakley, B.B.; Parks, D.H.; Robinson, C.J.; et al. Introducing mothur: Open-Source, Platform-Independent, Community-Supported Software for Describing and Comparing Microbial Communities. Appl. Environ. Microbiol. 2009, 75, 7537–7541. [Google Scholar] [CrossRef] [Green Version]
- Oksanen, J.; Kindt, R.; Legendre, P.; O’Hara, B.; Stevens, M.H.H.; Oksanen, M.J.; Suggests, M. The Vegan Package. Commun. Ecol. Package 2007, 10, 631–637. [Google Scholar]
- Wickham, H. Ggplot2: Elegant Graphics for Data Analysis; Springer: New York, NY, USA, 2016. [Google Scholar]
- Bulgarelli, D.; Rott, M.; Schlaeppi, K.; Van Themaat, E.V.L.; Ahmadinejad, N.; Assenza, F.; Rauf, P.; Huettel, B.; Reinhardt, R.; Schmelzer, E.; et al. Revealing structure and assembly cues for Arabidopsis root-inhabiting bacterial microbiota. Nature 2012, 488, 91–95. [Google Scholar] [CrossRef]
- Lundberg, D.S.; Lebeis, S.L.; Paredes, S.H.; Yourstone, S.; Gehring, J.; Malfatti, S.; Tremblay, J.; Engelbrektson, A.; Kunin, V.; Del Rio, T.G.; et al. Defining the core Arabidopsis thaliana root microbiome. Nature 2012, 488, 86–90. [Google Scholar] [CrossRef] [Green Version]
- Csardi, G.; Nepusz, T. The igraph software package for complex network research. Int. J. Complex Syst. 2006, 1695, 1–9. [Google Scholar]
- Katoh, K.; Standley, D.M. MAFFT Multiple Sequence Alignment Software Version 7: Improvements in Performance and Usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular Evolutionary Genetics Analysis across Computing Platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef]
- Kim, P.-J.; Price, N.D. Genetic Co-Occurrence Network across Sequenced Microbes. PLOS Comput. Biol. 2011, 7, e1002340. [Google Scholar] [CrossRef] [PubMed]
- Rasche, F.; Velvis, H.; Zachow, C.; Berg, G.; Van Elsas, J.D.; Sessitsch, A. Impact of transgenic potatoes expressing anti-bacterial agents on bacterial endophytes is comparable with the effects of plant genotype, soil type and pathogen infection. J. Appl. Ecol. 2006, 43, 555–566. [Google Scholar] [CrossRef]
- Xiong, C.; Zhu, Y.G.; Wang, J.T.; Singh, B.; Han, L.L.; Shen, J.; Li, P.; Wang, G.; Wu, C.; Ge, A.; et al. Host selection shapes crop microbiome assembly and network complexity. New Phytol. 2021, 229, 1091–1104. [Google Scholar] [CrossRef] [PubMed]
- Comeau, D.; Novinscak, A.; Joly, D.L.; Filion, M. Spatio-Temporal and Cultivar-Dependent Variations in the Cannabis Microbiome. Front. Microbiol. 2020, 24, 491. [Google Scholar] [CrossRef] [Green Version]
- Wei, G.; Ning, K.; Zhang, G.; Yu, H.; Yang, S.; Dai, F.; Dong, L.; Chen, S. Compartment Niche Shapes the Assembly and Network of Cannabis sativa-Associated Microbiome. Front. Microbiol. 2021, 12, 714993. [Google Scholar] [CrossRef]
- Chen, L.; Wang, Y.-C.; Qin, L.-Y.; He, H.-Y.; Yu, X.-L.; Yang, M.-Z.; Zhang, H.-B. Dynamics of fungal communities during Gastrodia elata growth. BMC Microbiol. 2019, 19, 158. [Google Scholar] [CrossRef]
- Tsavkelova, E.A.; Cherdyntseva, T.A.; Botina, S.G.; Netrusov, A.I. Bacteria associated with orchid roots and microbial production of auxin. Microbiol. Res. 2007, 162, 69–76. [Google Scholar] [CrossRef]
- Faria, D.C.; Dias, A.C.F.; Melo, I.S.; Costa, F.E.D.C. Endophytic bacteria isolated from orchid and their potential to promote plant growth. World J. Microbiol. Biotechnol. 2013, 29, 217–221. [Google Scholar] [CrossRef] [PubMed]
- Da Silva, J.A.T.; Tsavkelova, E.A.; Zeng, S.; Ng, T.B.; Parthibhan, S.; Dobránszki, J.; Cardoso, J.C.; Rao, M.V. Symbiotic in vitro seed propagation of Dendrobium: Fungal and bacterial partners and their influence on plant growth and development. Planta 2015, 242, 1–22. [Google Scholar] [CrossRef] [PubMed]
- Altinkaynak, H.; Ozkoc, I. Isolation and molecular characterization of plant growth promoting bacteria from the rhizosphere of orchids in Turkey. Rhizosphere 2020, 16, 100280. [Google Scholar] [CrossRef]
- Pei, C.; Mi, C.; Sun, L.; Liu, W.; Li, O.; Hu, X. Diversity of endophytic bacteria of Dendrobium officinale based on culture-dependent and culture-independent methods. Biotechnol. Biotechnol. Equip. 2017, 31, 112–119. [Google Scholar] [CrossRef] [Green Version]
- Alibrandi, P.; Schnell, S.; Perotto, S.; Cardinale, M. Diversity and Structure of the Endophytic Bacterial Communities Associated With Three Terrestrial Orchid Species as Revealed by 16S rRNA Gene Metabarcoding. Front. Microbiol. 2020, 11, 604964. [Google Scholar] [CrossRef]
- Lin, S.-Y.; Hameed, A.; Liu, Y.-C.; Wen, C.-Z.; Lai, W.-A.; Hsu, Y.-H.; Young, C.-C. Bacillus lycopersici sp. nov., isolated from a tomato plant (Solanum lycopersicum L.). Int. J. Syst. Evol. Microbiol. 2015, 65, 2085–2090. [Google Scholar] [CrossRef]
- Ali, A.; Shahzad, R.; Khan, A.L.; Halo, B.A.; Al-Yahyai, R.; Al-Harrasi, A.; Al-Rawahi, A.; Lee, I.-J. Endophytic bacterial diversity of Avicennia marina helps to confer resistance against salinity stress in Solanum lycopersicum. J. Plant Interact. 2017, 12, 312–322. [Google Scholar] [CrossRef] [Green Version]
- Huang, X.P.; Monk, C. Purification and characterization of a cellulase (CMCase) from a newly isolated thermophilic aerobic bacterium Caldibacillus cellulovorans gen. nov., sp. nov. World J. Microbiol. Biotechnol. 2004, 20, 85–92. [Google Scholar] [CrossRef]
- Spaepen, S.; Vanderleyden, J. Auxin and Plant-Microbe Interactions. Cold Spring Harb. Perspect. Biol. 2011, 3, a001438. [Google Scholar] [CrossRef] [Green Version]
- Urdaci, M.C.; Stal, L.J.; Marchand, M. Occurrence of nitrogen fixation among Vibrio spp. Arch. Microbiol. 1988, 150, 224–229. [Google Scholar] [CrossRef]
- Bakker, P.A.H.M.; Ran, L.X. Mercado-Blanco, J. Rhizobacterial salicylate production provokes headaches! Plant Soil 2014, 382, 1–16. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Yam, T.W.; Meng, Q.; Zhu, J.; Zhang, P.; Wu, H.; Wang, J.; Zhao, Y.; Song, X. The dual inoculation of endophytic fungi and bacteria promotes seedlings growth in Dendrobium catenatum (Orchidaceae) under in vitro culture conditions. Plant Cell, Tissue Organ Cult. (PCTOC) 2016, 126, 523–531. [Google Scholar] [CrossRef]
- Aspray, T.J.; Jones, E.E.; Davies, M.W.; Shipman, M.; Bending, G.D. Increased hyphal branching and growth of ectomycorrhizal fungus Lactarius rufus by the helper bacterium Paenibacillus sp. Mycorrhiza 2013, 23, 403–410. [Google Scholar] [CrossRef] [PubMed]
- LabbE, J.L.; Weston, D.; Edunkirk, N.; Pelletier, D.A.; Tuskan, G.A. Newly identified helper bacteria stimulate ectomycorrhizal formation in Populus. Front. Plant Sci. 2014, 5, 579. [Google Scholar] [CrossRef] [PubMed]
- Frey-Klett, P.; Garbaye, J.; Tarkka, M. The mycorrhiza helper bacteria revisited. New Phytol. 2007, 176, 22–36. [Google Scholar] [CrossRef]
- Yang, S.; Zhang, X.; Cao, Z.; Zhao, K.; Wang, S.; Chen, M.; Hu, X. Growth-promoting Sphingomonas paucimobilis ZJSH1 associated with Dendrobium officinale through phytohormone production and nitrogen fixation. Microb. Biotechnol. 2014, 7, 611–620. [Google Scholar] [CrossRef]
- Pan, L.; Chen, J.; Ren, S.; Shen, H.; Rong, B.; Liu, W.; Yang, Z. Complete genome sequence of Mycobacterium Mya-zh01, an endophytic bacterium, promotes plant growth and seed germination isolated from flower stalk of Doritaenopsis. Arch. Microbiol. 2020, 202, 1965–1976. [Google Scholar] [CrossRef]
- Jiang, J.; Zhang, K.; Cheng, S.; Nie, Q.; Zhou, S.-X.; Chen, Q.; Zhou, J.; Zhen, X.; Li, X.T.; Zhen, T.W.; et al. Fusarium oxysporum KB-3 from Bletilla striata: An orchid mycorrhizal fungus. Mycorrhiza 2019, 29, 531–540. [Google Scholar] [CrossRef]
- Sisti, L.S.; Flores-Borges, D.N.A.; De Andrade, S.A.L.; Koehler, S.; Bonatelli, M.L.; Mayer, J.L.S. The Role of Non-Mycorrhizal Fungi in Germination of the Mycoheterotrophic Orchid Pogoniopsis schenckii Cogn. Front. Plant Sci. 2019, 10, 1589. [Google Scholar] [CrossRef]
- Jacquemyn, H.; Brys, R.; Waud, M.; Busschaert, P.; Lievens, B. Mycorrhizal networks and coexistence in species-rich orchid communities. New Phytol. 2015, 206, 1127–1134. [Google Scholar] [CrossRef]
- Rafter, M.; Yokoya, K.; Schofield, E.J.; Zettler, L.W.; Sarasan, V. Non-specific symbiotic germination of Cynorkis purpurea (Thouars) Kraezl., a habitat-specific terrestrial orchid from the Central Highlands of Madagascar. Mycorrhiza 2016, 26, 541–552. [Google Scholar] [CrossRef] [PubMed]
- Martos, F.; Munoz, F.; Pailler, T.; Kottke, I.; Gonneau, C.; Selosse, M.-A. The role of epiphytism in architecture and evolutionary constraint within mycorrhizal networks of tropical orchids. Mol. Ecol. 2012, 21, 5098–5109. [Google Scholar] [CrossRef] [PubMed]
- Yagame, T.; Ogura-Tsujita, Y.; Kinoshita, A.; Iwase, K.; Yukawa, T. Fungal partner shifts during the evolution of mycoheterotrophy in Neottia. Am. J. Bot. 2016, 103, 1630–1641. [Google Scholar] [CrossRef]
- Huang, H.; Zi, X.-M.; Lin, H.; Gao, J.-Y. Host-specificity of symbiotic mycorrhizal fungi for enhancing seed germination, protocorm formation and seedling development of over-collected medicinal orchid, Dendrobium devonianum. J. Microbiol. 2018, 56, 42–48. [Google Scholar] [CrossRef] [PubMed]
- Xing, X.; Ma, X.; Men, J.; Chen, Y.; Guo, S. Phylogenetic constrains on mycorrhizal specificity in eight Dendrobium (Orchidaceae) species. Sci. China Life Sci. 2017, 60, 536–544. [Google Scholar] [CrossRef] [PubMed]
- Smith, S.E.; Read, D.J. Mycorrhizal Symbiosis; Academic Press: San Diego, CA, USA, 1997. [Google Scholar]
- Gebauer, G.; Preiss, K.; Gebauer, A.C. Partial mycoheterotrophy is more widespread among orchids than previously assumed. New Phytol. 2016, 211, 11–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| OTU | Accession Number | Taxonomic Affiliation | Closest Match in GenBank (Accession Number) | Identity | Total Score | Host | Site | Development Stage | ||
|---|---|---|---|---|---|---|---|---|---|---|
| Phase S | Phase M | Phase L | ||||||||
| OTU1 | OL891761 | Tulasnellaceae | GQ241861 | 99.77% | 804 | Paphiopedilum armeniacum | Kunming greenhouse | 0.0027 | 0.3424 | 0.6549 |
| OTU3 | OL891762 | Tulasnellaceae | FJ786674 | 99.77% | 800 | Paphiopedilum armeniacum | Baoshan wild | 0 | 0.3875 | 0.6125 |
| OTU11 | OL891763 | Tulasnellaceae | GQ241789 | 97.78% | 699 | Paphiopedilum micranthum | Xingyi greenhouse | 0 | 0 | 1 |
| OTU33 | OL891764 | Tulasnellaceae | GQ241786 | 98.51% | 713 | Paphiopedilum micranthum | Xingyi greenhouse | 0 | 0.4662 | 0.5338 |
| OTU67 | OL891765 | Tulasnellaceae | AB369932 | 97.62% | 795 | Cypripedium macranthos var. rebunense | Japan | 0 | 0.9936 | 0.0064 |
| OTU74 | OL891766 | Tulasnellaceae | KP053823 | 98.61% | 763 | Liparis viridiflora | Yunnan | 0 | 1 | 0 |
| OTU178 | OL891767 | Tulasnellaceae | AJ313458 | 97.73% | 756 | Spathaglotis plicata | Singapore | 0 | 0 | 1 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cao, X.; Wang, X.; Wang, T.; Chen, Y.; Yao, N. Dynamic Shifts in the Root Microbiota of Cultivated Paphiopedilum armeniacum during Different Stages of Growth. Diversity 2022, 14, 321. https://doi.org/10.3390/d14050321
Cao X, Wang X, Wang T, Chen Y, Yao N. Dynamic Shifts in the Root Microbiota of Cultivated Paphiopedilum armeniacum during Different Stages of Growth. Diversity. 2022; 14(5):321. https://doi.org/10.3390/d14050321
Chicago/Turabian StyleCao, Xiaolu, Xiaojing Wang, Tao Wang, Yan Chen, and Na Yao. 2022. "Dynamic Shifts in the Root Microbiota of Cultivated Paphiopedilum armeniacum during Different Stages of Growth" Diversity 14, no. 5: 321. https://doi.org/10.3390/d14050321
APA StyleCao, X., Wang, X., Wang, T., Chen, Y., & Yao, N. (2022). Dynamic Shifts in the Root Microbiota of Cultivated Paphiopedilum armeniacum during Different Stages of Growth. Diversity, 14(5), 321. https://doi.org/10.3390/d14050321