
Thrips Microbiome Study in Commercial Avocado (Persea americana Mill.) from Northwest Colombian Andes (Antioquia, Colombia) Shows the Presence of Wolbachia, Ehrlichia, Enterobacter

 , , , and
, , , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
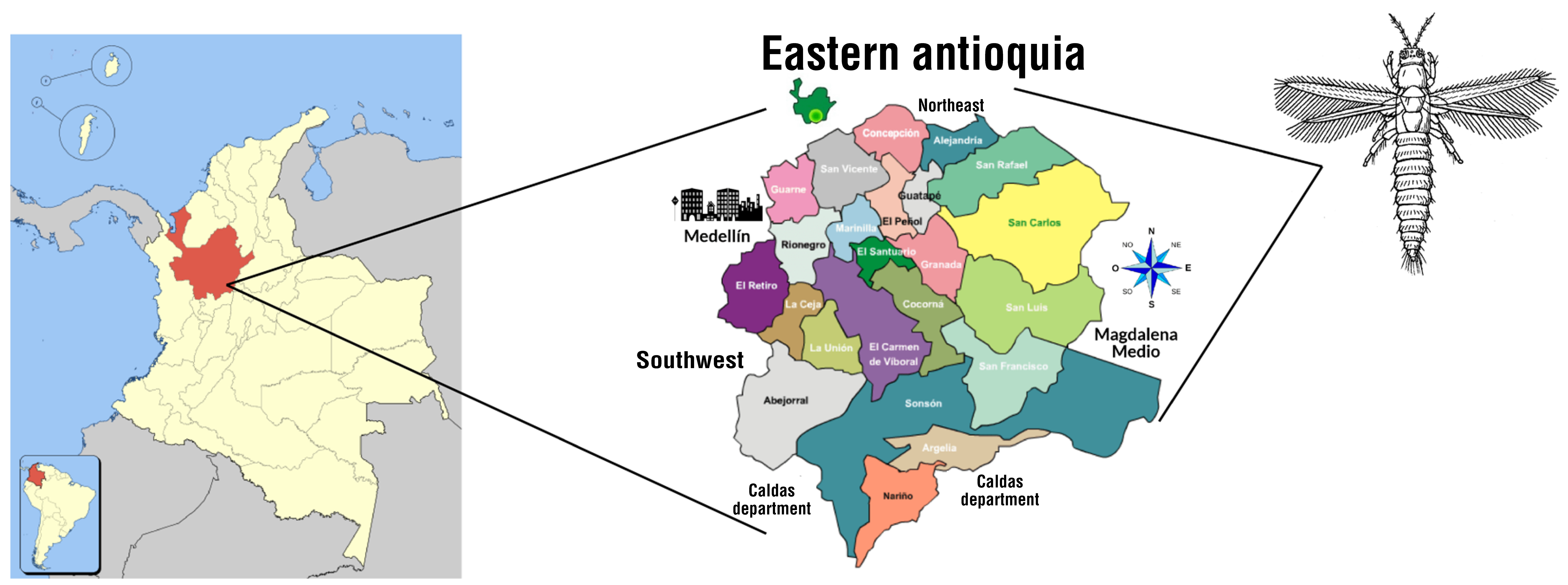
2.1. Insect Collection
2.2. Culture-Dependent Assays
2.2.1. Isolation and Purification of Bacteria from Thrips
2.2.2. Extraction of Genomic DNA and PCR of the Transcribed Internal Spacer-ITS, 16S rRNA and gyrB Genes
2.3. Culture-Independent Assays
2.3.1. Temperature Gradient Gel Electrophoresis (TGGE)
2.3.2. Sequencing and Phylogenetic Analysis
2.3.3. Analysis of the Bacterial Community by PCR-TGGE
2.3.4. 16S Amplicon Next Generation Sequencing (NGS)
3. Results
3.1. Bacterial Diversity through Culture-Dependent Assays
3.1.1. Isolation and Culture of Microbial Isolates
3.1.2. Identification of Bacterial Isolates Using 16S rDNA and gyrB Sequencing
3.1.3. Phylogenetic Analysis with Sequences of the 16S rRNA and gyrB Genes
3.2. Bacterial Diversity through Culture-Independent Methods
3.2.1. PCR-TGGE Analysis
3.2.2. Phylogenetic Affiliation of the Sequences Obtained from the TGGE Bands
3.2.3. Quality Analysis of 16S rDNA Sequence and Bacterial Identity Assignment by High-Throughput Sequencing-NGS
3.2.4. Microbiota Composition
3.2.5. Bacterial Diversity
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Salazar-López, N.J.; Domínguez-Avila, J.A.; Yahia, E.M.; Belmonte-Herrera, B.H.; Wall-Medrano, A.; Montalvo-González, E.; González-Aguilar, G.A. Avocado fruit and by-products as potential sources of bioactive compounds. Food Res. Int. 2020, 138, 109774. [Google Scholar] [CrossRef] [PubMed]
- Cambero-campos, J.; Johansen-Naime, R.; García-Martínez, O.; Cantu-Sifuentes, M.; Cerna-Chavez, E.; Renata-Salazar, A. Especies Depredadoras de Trips (Thysanoptera) asociadas a huertas de aguacate en Nayarit, México. Acta Zoológica Mex. 2011, 27, 115–121. [Google Scholar] [CrossRef]
- Monje, B.; Delgadillo, D.; Gómez, J.; Varón, E. Manejo de Neohydatothrips signifer Priesner (Thysanoptera: Thripidae) en maracuyá (Passiflora edulis f. flavicarpa Degener) en el departamento del Huila (Colombia). Cienc. Y Tecnol. Agropecu. 2012, 13, 21–30. [Google Scholar] [CrossRef] [Green Version]
- Mound, L.; Hoddle, M. The Scirtothrips perseae species-group (Thysanoptera), with one new species from avocado, Persea americana. Zootaxa 2016, 4079, 388–392. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mound, L.A.; Marullo, R. The thrips of central and south America: An introduction (Insecta: Thysanoptera). Florida Entomol. 1996, 79, 270–271. [Google Scholar] [CrossRef]
- Douglas, A.E. Multiorganismal insects: Diversity and function of resident microorganisms. Annu. Rev. Entomol. 2015, 60, 17–34. [Google Scholar] [CrossRef] [Green Version]
- Dickey, A.M.; Trease, A.J.; Jara-Cavieres, A.; Kumar, V.; Christenson, M.K.; Potluri, L.P.; Morgan, J.K.; Shatters, R.G., Jr.; McKenzie, C.L.; Davis, P.H.; et al. Estimating bacterial diversity in Scirtothrips dorsalis (Thysanoptera: Thripidae) via next generation sequencing. Florida Entomol. 2014, 97, 362–366. [Google Scholar] [CrossRef]
- Douglas, A.E. The microbial dimension in insect nutritional ecology. Funct. Ecol. 2009, 23, 38–47. [Google Scholar] [CrossRef]
- Engel, P.; Moran, N.A. The gut microbiota of insects—Diversity in structure and function. FEMS Microbiol. Rev. 2013, 37, 699–735. [Google Scholar] [CrossRef]
- Duron, O.; Hurst, G.D. Arthropods and inherited bacteria: From counting the symbionts to understanding how symbionts count. BMC Biol. 2013, 11, 45. [Google Scholar] [CrossRef] [Green Version]
- Gawande, S.J.; Anandhan, S.; Ingle, A.; Roylawar, P.; Khandagale, K.; Gawai, T.; Jacobson, A.; Asokan, R.; Singh, M. Microbiome profiling of the onion thrips, Thrips tabaci Lindeman (Thysanoptera: Thripidae). PLoS ONE 2019, 14, e0223281. [Google Scholar] [CrossRef] [PubMed]
- Liberti, J.; Engel, P. The gut microbiota—Brain axis of insects. Curr. Opin. Insect Sci. 2020, 39, 6–13. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.; Zhang, H. The Impact of environmental heterogeneity and life stage on the hindgut microbiota of Holotrichia Parallela larvae (Coleoptera: Scarabaeidae). PLoS ONE 2013, 8, e57169. [Google Scholar] [CrossRef]
- Chanbusarakum, L.; Ullman, D. Characterization of bacterial symbionts in Frankliniella occidentalis (Pergande), Western flower thrips. J. Invertebr. Pathol. 2008, 99, 318–325. [Google Scholar] [CrossRef]
- de Vries, E.J.; Jacobs, G.; Sabelis, M.W.; Menken, S.B.J.; Breeuwer, J.A.J. Diet-dependent effects of gut bacteria on their insect host: The symbiosis of Erwinia sp. and western flower thrips. Proc. R. Soc. Lond. B 2004, 271, 2171–2178. [Google Scholar] [CrossRef] [Green Version]
- De Vries, E.; Breeuwer, J.; Jacobs, G.; Mollema, C. The association of Western flower thrips, Frankliniella occidentalis, with a near Erwinia species gut bacteria: Transient or permanent? J. Invertebr. Pathol. 2001, 77, 120–128. [Google Scholar] [CrossRef]
- de Vries, E.J.; Jacobs, G.; Breeuwer, J.A.J. Growth and transmission of gut bacteria in the western flower thrips, Frankliniella occidentalis. J. Invertebr. Pathol. 2001, 77, 129–137. [Google Scholar] [CrossRef] [PubMed]
- Facey, P.D.; Méric, G.; Hitchings, M.D.; Pachebat, J.A.; Hegarty, M.J.; Chen, X.; Morgan, L.V.A.; Hoeppner, J.E.; Whitten, M.M.A.; Kirk, W.D.J.; et al. Draft genomes, phylogenetic reconstruction, and comparative genomics of two novel cohabiting bacterial symbionts isolated from Frankliniella occidentalis. Genome Biol. Evol. 2015, 7, 2188–2202. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Vries, E.J.; Van Der Wurff, A.G.; Jacobs, G.; Breeuwer, J. Onion thrips, Thrips tabaci, have gut bacteria that are closely related to the symbionts of the western flower thrips, Frankliniella occidentalis. J. Insect Sci. 2008, 8, 23. [Google Scholar] [CrossRef] [Green Version]
- Dutta, B.; Barman, A.K.; Srinivasan, R.; Avci, U.; Ullman, D.; Langston, D.B., Jr.; Gitaitis, R. Transmission of Pantoea ananatis and Pantoea agglomerans causal agents of center rot of onion (Allium cepa L.) by onion thrips (Thrips tabaci Lindeman). Phytopathology 2014, 104, 812–819. [Google Scholar] [CrossRef] [Green Version]
- Arakaki, N.; Miyoshi, T.; Noda, H. Wolbachia-mediated parthenogenesis in the predatory thrips Franklinothrips vespiformis (Thysanoptera: Insecta). Proc. R. Soc. B Biol. Sci. 2001, 268, 1011–1016. [Google Scholar] [CrossRef] [Green Version]
- Weeks, A.R.; Velten, R.; Stouthamer, R. Incidence of a new sex-ratio-distorting endosymbiotic bacterium among arthropods. Proc. R. Soc. B Biol. Sci. 2003, 270, 1857–1865. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumm, S.; Moritz, G. First detection of Wolbachia in arrhenotokous populations of thrips species (Thysanoptera: Thripidae and Phlaeothripidae) and its role in reproduction. Environ. Entomol. 2008, 37, 1422–1428. [Google Scholar] [CrossRef] [PubMed]
- Saurav, G.; Daimei, G.; Singh, V.R.; Popli, S.; Rajagopal, R. Detection and localization of Wolbachia in Thrips palmi Karny (Thysanoptera: Thripidae). Indian J. Microbiol. 2016, 56, 167–171. [Google Scholar] [CrossRef] [Green Version]
- Kaczmarczyk, A.; Kucharczyk, H.; Kucharczyk, M.; Kapusta, P.; Sell, J.; Zielińska, S. First insight into microbiome profile of fungivorous thrips Hoplothrips carpathicus (Insecta: Thysanoptera) at different developmental stages: Molecular evidence of Wolbachia endosymbiosis. Sci. Rep. 2018, 8, 14376. [Google Scholar] [CrossRef] [PubMed]
- Cao, Y.; Fanning, S.; Proos, S.; Jordan, K.; Srikumar, S. A review on the applications of next generation sequencing technologies as applied to food-related microbiome studies. Front. Microbiol. 2017, 8, 1829. [Google Scholar] [CrossRef]
- Castañeda-Monsalve, V.A.; Junca, H.; García-Bonilla, E.; Montoya-Campuzano, O.I.; Moreno-Herrera, C.X. Characterization of the gastrointestinal bacterial microbiome of farmed juvenile and adult white Cachama (Piaractus brachypomus). Aquaculture 2019, 512, 734325. [Google Scholar] [CrossRef]
- Martin, J.E. comp. Part 1. Collecting, preparing, and preserving insects, mites and spiders. In The Insects and Arachnids of Canada; Agriculture Canada: Ottawa, ON, Canada, 1976; pp. 9–175. ISBN 0660016508. [Google Scholar]
- Pham, V.H.T.; Kim, J. Improvement for isolation of soil bacteria by using common culture media. J. Pure Appl. Microbiol. 2016, 10, 49. [Google Scholar]
- Silva-Bedoya, L.M.; Sánchez-Pinzón, M.S.; Cadavid-Restrepo, G.E.; Moreno-Herrera, C.X. Bacterial community analysis of an industrial wastewater treatment plant in Colombia with screening for lipid-degrading microorganisms. Microbiol. Res. 2016, 192, 313–325. [Google Scholar] [CrossRef]
- Hammer, Ø.; Harper, D.A.T.; Ryan, P.D. PAST: Paleontological statistics software package for education and data analysis. Palaeontol. Electron. 2001, 4, 9. [Google Scholar]
- Sanders, E.R.; Miller, J.H. I, Microbiologist: A Discovery-Based Course in Microbial Ecology and Molecular Evolution; ASM Press: Los Angeles, CA, USA, 2010; ISBN 978-1-55581-470-0. [Google Scholar]
- Jensen, M.A.; Webster, J.A.; Straus, N. Rapid identification of bacteria on the basis of polymerase chain reaction-amplified ribosomal DNA spacer polymorphisms. Appl. Environ. Microbiol. 1993, 59, 945–952. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moreno, C.; Romero, J.; Espejo, R.T. Polymorphism in repeated 16S PRNA genes is a common property of type strains and environmental isolates of the genus Vibrio. Microbiology 2002, 148, 1233–1239. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yim, O.; Ramdeen, K.T. Hierarchical cluster analysis: Comparison of three linkage measures and application to psychological data. Quant. Methods Psychol. 2015, 11, 8–21. [Google Scholar] [CrossRef] [Green Version]
- Yamamoto, S.; Harayama, S. PCR Amplification and direct sequencing of gyrB genes with universal primers and their application to the detection and taxonomic analysis of pseudomonas putida strains. Appl. Environ. Microbiol. 1995, 61, 1104–1109. [Google Scholar] [CrossRef] [Green Version]
- Yin, H.; Cao, L.; Qiu, G.; Wang, D.; Kellogg, L.; Zhou, J.; Liu, X.; Dai, Z.; Ding, J.; Liu, X. Molecular diversity of 16S rRNA and gyrB genes in copper mines. Arch. Microbiol. 2008, 189, 101–110. [Google Scholar] [CrossRef]
- Goldenberger, D.; Perschil, I.; Ritzler, M.; Altwegg, M. A Simple “Universal” DNA extraction procedure using SDS and proteinase K is compatible with direct PCR AMPLIFICATION. PCR Methods Appl. 1995, 4, 368–370. [Google Scholar] [CrossRef] [Green Version]
- Moreno, C.X.; Moy, F.; Daniels, T.J.; Godfrey, H.P.; Cabello, F.C. Molecular analysis of microbial communities identified in different developmental stages of Ixodes scapularis ticks from Westchester and Dutchess Counties, New York. Environ. Microbiol. 2006, 8, 761–772. [Google Scholar] [CrossRef]
- Altschul, S.F.; Madden, T.L.; Schäffer, A.A.; Zhang, J.; Zhang, Z.; Miller, W.; Lipman, D.J. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Oxford Univ. Press 1997, 25, 3389–3402. [Google Scholar] [CrossRef] [Green Version]
- Cole, J.R.; Wang, Q.; Cardenas, E.; Fish, J.; Chai, B.; Farris, R.J.; Kulam-Syed-Mohideen, A.S.; McGarrell, D.M.; Marsh, T.; Garrity, G.M.; et al. The Ribosomal Database Project: Improved alignments and new tools for rRNA analysis. Nucleic Acids Res. 2009, 37, 141–145. [Google Scholar] [CrossRef] [Green Version]
- Edgar, R.C. MUSCLE: A multiple sequence alignment method with reduced time and space complexity. BMC Bioinform. 2004, 5, 113. [Google Scholar] [CrossRef] [Green Version]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular evolutionary genetics analysis across computing platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef] [PubMed]
- Ronquist, F.; Teslenko, M.; Van Der Mark, P.; Ayres, D.L.; Darling, A.; Höhna, S.; Larget, B.; Liu, L.; Suchard, M.A.; Huelsenbeck, J.P. Mrbayes 3.2: Efficient bayesian phylogenetic inference and model choice across a large model space. Syst. Biol. 2012, 61, 539–542. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vivero, R.J.; Gil Jaramillo, N.; Cadavid-Restrepo, G.; Uribe Soto, S.; Moreno Herrera, C.X. Structural differences in gut bacteria communities in developmental stages of natural populations of Lutzomyia evansi from Colombia’s Caribbean coast. Parasit. Vectors 2016, 9, 496. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Camarinha-Silva, A.; Jáuregui, R.; Chaves-Moreno, D.; Oxley, A.P.A.; Schaumburg, F.; Becker, K.; Wos-Oxley, M.L.; Pieper, D.H. Comparing the anterior nare bacterial community of two discrete human populations using Illumina amplicon sequencing. Environ. Microbiol. 2014, 16, 2939–2952. [Google Scholar] [CrossRef] [PubMed]
- Parada, A.E.; Needham, D.M.; Fuhrman, J.A. Every base matters: Assessing small subunit rRNA primers for marine microbiomes with mock communities, time series and global field samples. Environ. Microbiol. 2016, 18, 1403–1414. [Google Scholar] [CrossRef] [PubMed]
- Apprill, A.; Mcnally, S.; Parsons, R.; Weber, L. Minor revision to V4 region SSU rRNA 806R gene primer greatly increases detection of SAR11 bacterioplankton. Aquat. Microb. Ecol. 2015, 75, 129–137. [Google Scholar] [CrossRef] [Green Version]
- Caporaso, J.G.; Kuczynski, J.; Stombaugh, J.; Bittinger, K.; Bushman, F.; Costello, E.; Fierer, N.; Gonzalez, A.; Goodrich, J.; Gordon, J.; et al. QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 2010, 7, 335–336. [Google Scholar] [CrossRef] [Green Version]
- Wang, Q.; Garrity, G.M.; Tiedje, J.M.; Cole, J.R. Naïve Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl. Environ. Microbiol. 2007, 73, 5261–5267. [Google Scholar] [CrossRef] [Green Version]
- Pérez-Jaramillo, J.E.; Carrión, V.J.; Bosse, M.; Ferrão, L.F.V.; De Hollander, M.; Garcia, A.A.F.; Ramírez, C.A.; Mendes, R.; Raaijmakers, J.M. Linking rhizosphere microbiome composition of wild and domesticated Phaseolus vulgaris to genotypic and root phenotypic traits. ISME J. 2017, 11, 2244–2257. [Google Scholar] [CrossRef] [Green Version]
- McMurdie, P.J.; Holmes, S. Phyloseq: An R Package for Reproducible Interactive Analysis and Graphics of Microbiome Census Data. PLoS ONE 2013, 8, e61217. [Google Scholar] [CrossRef] [Green Version]
- Oksanen, J.; Blanchet, F.G.; Kindt, R.; Legendre, P.; Minchin, P.R.; O’Hara, R.B.; Simpson, G.L.; Solymos, P.; Stevens, M.H.M.; Wagner, H. Vegan: Community Ecology Package. Available online: http://vegan.r-forge.r-project.org/ (accessed on 12 February 2020).
- De Vries, E.; Van de Wetering, F.; Van der Hoek, M.; Jacobs, G.; Breeuwer, J. Symbiotic bacteria (Erwinia sp.) in the gut of Frankliniella occidentalis (Thysanoptera: Thripidae) do not affect its ability to transmit Tospovirus. Eur. J. Entomol. 2012, 109, 261–266. [Google Scholar] [CrossRef] [Green Version]
- Yun, J.; Roh, W.; Whon, W.; Jung, M.; Kim, M.; Park, D.; Yoon, C.; Nam, Y.-D.; Kim, Y.-J.; Choi, J.-H.; et al. Insect gut bacterial diversity determined by environmental habitat, diet, developmental stage, and phylogeny of host. Appl. Environ. Microbiol. 2014, 80, 5254–5264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walterson, A.M.; Stavrinides, J. Pantoea: Insights into a highly versatile and diverse genus within the Enterobacteriaceae. FEMS Microbiol. Rev. 2015, 39, 968–984. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pileggi, M.; Alvim, S.; Pileggi, V.; Olchanheski, R.; Garbugio, P.; Munoz, A.M.; Koskinen, W.C.; Barber, B.; Sadowsky, M. Isolation of mesotrione—Degrading bacteria from aquatic environments in Brazil. Chemosphere 2012, 86, 1127–1132. [Google Scholar] [CrossRef] [Green Version]
- Walterson, A.M.; Smith, D.D.N.; Stavrinides, J. Identification of a Pantoea biosynthetic cluster that directs the synthesis of an antimicrobial natural product. PLoS ONE 2014, 9, e96208. [Google Scholar] [CrossRef] [Green Version]
- Kumar, D.; Kim, J. Sphingomonas olei sp. nov., with the ability to degrade aliphatic hydrocarbons, isolated from oil-contaminated soil. Int. J. Syst. Evol. Microbiol. 2017, 67, 2731–2738. [Google Scholar] [CrossRef]
- An, R.; Sreevatsan, S.; Grewal, P.S. Moraxella osloensis gene expression in the slug host Deroceras reticulatum. BMC Microbiol. 2008, 8, 19. [Google Scholar] [CrossRef] [Green Version]
- Kaibara, F.; Liyama, K.; Chieda, Y.; Lee, J.M.; Kusakabe, T.; Yasunaga-aoki, C.; Shimizu, S. Construction of serralysin-like metalloprotease-deficient mutants of Serratia liquefaciens and their virulence in the silkworm, Bombyx mori. J. Insect Biotechnol. Sericologynsect Biotechnol. Sericology 2012, 81, 55–61. [Google Scholar] [CrossRef]
- Wang, L.; Lee, F.; Tai, C.; Kasai, H. Comparison of gyrB gene sequences, 16S rRNA gene sequences and DNA–DNA hybridization in the Bacillus subtilis group. Int. J. Syst. Evol. Microbiol. 2007, 57, 1846–1850. [Google Scholar] [CrossRef] [Green Version]
- Shafi, J.; Tian, H.; Ji, M. Bacillus species as versatile weapons for plant pathogens: A review. Biotechnol. Biotechnol. Equip. 2017, 31, 446–459. [Google Scholar] [CrossRef] [Green Version]
- Cawoy, H.; Bettiol, W.; Fickers, P.; Ongena, M. Bacillus-based biological control of plant diseases. In Pesticides in the Modern World—Pests Control and Pesticides Exposure; Stoytcheva, M., Ed.; InTech: Rijeka, Croatia, 2011; pp. 273–302. ISBN 978-953-307-459-7. [Google Scholar]
- Choudhary, D.K.; Johri, B.N. Interactions of Bacillus spp. and plants—With special reference to induced systemic resistance (ISR). Microbiol. Res. 2009, 164, 493–513. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, D.T.; Spooner-hart, R.N.; Riegler, M. Polyploidy versus endosymbionts in obligately thelytokous thrips. BMC Evol. Biol. 2015, 15, 23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Ramírez-Ahuja, M.L.; Gómez-Govea, M.A.; Lugo-Trampe, A.; Borrego-Soto, G.; Delgado-Enciso, I.; Ponce-Garcia, G.; Martínez-Fierro, M.L.; Ramírez-Valles, E.G.; Treviño, V.; Flores-Suarezs, A.E.; et al. Microbiota of Telenomus tridentatus (Platygastroidea: Scelionidae ): An unwanted parasitoid. Appl. Entomol. 2019, 143, 834–841. [Google Scholar] [CrossRef]
- Shelomi, M. Journal of Asia-Paci fi c Entomology Bacterial and eukaryote microbiomes of mosquito habitats in dengue- endemic southern Taiwan. J. Asia. Pac. Entomol. 2019, 22, 471–480. [Google Scholar] [CrossRef]
- Barak, H.; Kumar, P.; Zaritsky, A.; Mendel, Z.; Ment, D.; Kushmaro, A.; Ben-dov, E. Diversity of Bacterial Biota in Capnodis tenebrionis (Coleoptera: Buprestidae) Larvae. Pathogens 2019, 8, 4. [Google Scholar] [CrossRef] [Green Version]
- Chua, K.; Song, S.; Yong, H.; See-too, W.-S.; Yin, W.; Chan, K.-G. Microbial community composition reveals spatial variation and distinctive core microbiome of the weaver ant Oecophylla smaragdina in Malaysia. Sci. Rep. 2018, 8, 10777. [Google Scholar] [CrossRef] [Green Version]
- Powell, C.M.; Lopez Montiel, A.; Beddingfield, B.; Hanson, J.D.; Bextine, B.R. Comparison of bacterial communities of flower thrips (Frankliniella tritici) and potato psyllid (Bactericera cockerelli). Southwest. Entomol. 2015, 40, 765–773. [Google Scholar] [CrossRef]
- Ambika, S.; Rajagopal, R. Lumen anatomy and localization of Wolbachia sp. in the thrips, Plicothrips apicalis (Bagnall). Curr. Sci. 2018, 115, 1297–1304. [Google Scholar] [CrossRef]
- Augustinos, A.A.; Kyritsis, G.A.; Papadopoulos, N.T.; Abd-Alla, A.M.M.; Cáceres, C.; Bourtzis, K. Exploitation of the medfly gut microbiota for the enhancement of sterile insect technique: Use of Enterobacter sp. in larval diet-based probiotic applications. PLoS ONE 2015, 10, e0136459. [Google Scholar] [CrossRef] [Green Version]
- Kyritsis, G.A.; Augustinos, A.A.; Ntougias, S.; Papadopoulos, N.T.; Bourtzis, K.; Cáceres, C. Enterobacter sp. AA26 gut symbiont as a protein source for Mediterranean fruit fly mass-rearing and sterile insect technique applications. BMC Microbiol. 2019, 19, 288. [Google Scholar] [CrossRef] [Green Version]
- Caragata, E.P.; Rocha, M.N.; Pereira, T.N.; Mansur, S.B.; Heverton, L.; Id, C.D.; Id, L.A.M. Pathogen blocking in Wolbachia infected Aedes aegypti is not affected by Zika and dengue virus co-infection. PLoS Negl. Trop. Dis. 2019, 13, e0007443. [Google Scholar] [CrossRef] [PubMed]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cano-Calle, D.; Montoya-Porras, L.M.; Ochoa-Giraldo, S.; Junca, H.; Garcia-Bonilla, E.; Saldamando-Benjumea, C.; Moreno-Herrera, C.X.; Arango-Isaza, R.E. Thrips Microbiome Study in Commercial Avocado (Persea americana Mill.) from Northwest Colombian Andes (Antioquia, Colombia) Shows the Presence of Wolbachia, Ehrlichia, Enterobacter. Diversity 2022, 14, 540. https://doi.org/10.3390/d14070540
Cano-Calle D, Montoya-Porras LM, Ochoa-Giraldo S, Junca H, Garcia-Bonilla E, Saldamando-Benjumea C, Moreno-Herrera CX, Arango-Isaza RE. Thrips Microbiome Study in Commercial Avocado (Persea americana Mill.) from Northwest Colombian Andes (Antioquia, Colombia) Shows the Presence of Wolbachia, Ehrlichia, Enterobacter. Diversity. 2022; 14(7):540. https://doi.org/10.3390/d14070540
Chicago/Turabian StyleCano-Calle, Daniela, Luisa Maria Montoya-Porras, Sebastian Ochoa-Giraldo, Howard Junca, Erika Garcia-Bonilla, Clara Saldamando-Benjumea, Claudia Ximena Moreno-Herrera, and Rafael E. Arango-Isaza. 2022. "Thrips Microbiome Study in Commercial Avocado (Persea americana Mill.) from Northwest Colombian Andes (Antioquia, Colombia) Shows the Presence of Wolbachia, Ehrlichia, Enterobacter" Diversity 14, no. 7: 540. https://doi.org/10.3390/d14070540