Abstract
Pinus squamata is a rare and endangered tree endemic to northeastern Yunnan Province, China, and it is listed as a Plant Species with Extremely Small Populations (PSESP) in China for requiring urgent conservation. Furthermore, the actions of ex situ conservation and reintroduction based on artificial propagation have been carried out since some 15 years ago. The rhizosphere microbiome plays an important role in soil quality and plant health. However, how the fungal communities of the rhizosphere differ between wild, ex situ, and reintroduced examples of Pinus squamata remains unclear. Illumina sequencing of the internal transcribed spacer 2 (ITS2) region was used to investigate fungal communities in the P. squamata rhizosphere soil. Rhizospheric fungal community composition, structure, diversity, and ecological function in the soil surrounding wild, ex situ, and reintroduced P. squamata individuals were elucidated. The ex situ site Kunming (EK) had the highest fungal community richness and diversity. The samples collected from six different sites were well separated (R = 0.95, p = 0.001), suggesting significant differences between the sites. Soil total potassium (TK), available phosphorus (AP), and pH were the main factors driving fungal community (0.01 < p ≤ 0.05). Prediction of fungal functional guild in the P. squamata rhizosphere demonstrated that the fungi could be classified as ectomycorrhizal, endophyte, and plant pathogenic fungi. Our research will provide a basis to guide the further selection of conservation sites for P. squamata based on fungal diversity and offer guidance on the antagonistic fungi and plant pathogenic fungi that may be of relevance to the conservation of this rare plant.
1. Introduction
The plant microbiome consists of microorganisms present in the rhizosphere (the soil-root interface), phyllosphere (the plant aerial surfaces), and endosphere (the internal tissues). The microbiome is strongly influenced by the plant genome and may be considered as a second genome or pan-genome [1,2]. Rhizosphere microorganisms include phages, bacteria, archaea, and fungi [3]. The rhizosphere microbiome consists of micro-organisms that can be beneficial to plant growth and health, including the nitrogen-fixing bacteria, mycorrhizal fungi, plant growth promoting rhizobacteria (PGPR), biocontrol microorganisms, mycoparasitic fungi, and protozoa, as well as micro-organisms that can be detrimental, including the pathogenic fungi, oomycetes, bacteria, and nematodes [4,5]. Fungi are of major importance in plant ecology, decomposing organic matter and cycling nutrients across trophic levels [6]. Soil fungi can be classified into three functional groups, including the biological controllers, the ecosystem regulators, and the species participating in organic matter decomposition and compound transformations [7]. Soil microbial characteristics reflect changes in soil quality, and thus, can also be used as bio-indicators of soil health [8]. The soil microbiome is of critical importance for the conservation of soil health [9]. Therefore, studying the interactions between the soil environment, the host plants, and the rhizospheric microorganisms has become a hot topic in microbial molecular ecology research.
The Plant Species with Extremely Small Populations (PSESP) conservation concept was developed in China in 2005. Small remaining populations, a restricted and narrow habitat, severe anthropogenic disturbance, and a high risk of extinction are the four prominent characteristics of plants defined as PSESPs [10,11]. National- and provincial-level programs and projects rescuing PSESPs and supporting their conservation have been issued and implemented [12], and the PSESP concept and resulting conservation action have attracted widespread attention in the field of conservation biology [13,14]. Pinus squamata was listed as a national first-level key protected wild plant in China in both 1999 and 2021 [15,16], and was classified as Critically Endangered (CR) by the International Union for Conservation of Nature and Natural Resources (IUCN) in 2001 [17]. P. squamata is currently listed as 1 of 62 species of PSESPs in Yunnan Province, and also as 1 of the 20 key PSESPs of Yunnan. Moreover, the species was included as 1 of 120 nationally important PSESPs in China in 2011 [11].
Pinus squamata was described as a new species in 1992 [18], and has important research value in the elucidation of the evolution of the genus Pinus. Only 34 P. squamata individuals are known from the wild, and the species is found only in the Yunnan Yaoshan National Nature Reserve, with an area of only two square kilometers in Qiaojia County, Yunnan. The species is therefore known locally as the Qiaojia five-needle pine. Various aspects of P. squamata biology have been investigated, including floristic geography [19], endangerment reason [20], genetic structure [21,22], disease control [23], tissue culture [24], and ecological stoichiometric characteristics [25]. However, the microbial communities associated with the rhizospheric soil of P. squamata remain unclear.
The threats to endangered plants include internal factors such as restricted heritability and fertility, and external factors including environmental, human, and other interferences. Rhizosphere microorganisms, as external factors, also play an extremely important role in the growth and development of plants [26]. In this study, we investigated communities of rhizospheric microorganisms in the soil surrounding the roots of endangered P. squamata individuals, as well as the causes underlying observed patterns in fungal diversity. We collected the rhizosphere soil from wild, ex situ, and reintroduced P. squamata individuals at different sites. We investigated the community structure and diversity of the rhizosphere fungi and determined the soil physicochemical properties. We also analyzed the relationships between the fungal communities and soil physicochemical factors and conducted a functional prediction study. We hypothesized that the rhizosphere fungi will have different effects on the wild, ex situ, and reintroduced individuals of P. squamata, and that wild individuals will have the highest fungal diversity. We also predict that the soil physical and chemical properties will affect the communities of rhizospheric fungi. It is our aim that this study will suggest possible conservation strategies for P. squamata and provide guidance for the selection of future conservation sites for this critically endangered plant based on fungal diversity. We hope that our research can also help to protect P. squamata from soil-borne fungal pathogens.
2. Materials and Methods
2.1. Samples Collection
Samples of P. squamata rhizosphere soil were collected from different protected sites, including from individuals growing in wild (W), Ex-introduction (Ex), and Re-introduction (Re) sites in Yunnan Province in June 2020. Samples from the wild site were collected in Qiaojia County, Zhaotong City (WQ); ex situ sites included Caojian Forestry Farm, Dali Bai Autonomous Prefecture (EC), Yipinglang Forestry Station, Chuxiong Yi Autonomous Prefecture (EY), Kunming Botanical Garden (EK), and Qiaojia County (EQ). The re-introduction site was in Qiaojia County (RQ). The selected areas had no connection between the wild, ex situ, and reintroduced P. squamata individuals, and the populations were independent. The diameter at breast height (DBH) and height of selected P. squamata individuals were measured, and three individuals with the similar DBH were randomly selected from each of the six sites, representing three replicates from each site.
A total of 18 rhizospheric and bulk soil samples were collected, including from the wild Qiaojia site (26°52′03.96″ N, 103°00′42.44″ E, 2206 m), ex situ Caojian site (25°45′32.73″ N, 99°06′53.94″ E, 2502 m), ex situ Yipinglang site (25°08′08″ N, 101°54′01″ E, 1893 m), ex situ Kunming site (25°08′40.13″ N, 102°44′28.96″ E, 1990 m), ex situ Qiaojia site (27°0′30.34″ N, 102°57′ 25.47″ E, 1876.62 m), and the reintroduced Qiaojia site (26°52′00″ N, 103°00′39.9″ E, 2133 m). To collect the samples, we first removed the surface litter with a small shovel, then we gently followed a tree root to its tip, and dug out the root in four directions at a depth of 0–20 cm from the surface. Any loosely bound soil was gently shaken from the root and was used as “bulk soil”. Then, we collected the soil within 1–10 mm of the root surface in 4 directions according to the shaking-off method described previously [26]. Samples were packed into sterilized bags and thoroughly mixed as the rhizospheric soil from each individual. Samples were stored in liquid nitrogen during transportation and were taken immediately back to the laboratory. If the soil was too sticky, we used tweezers on ice to remove impurities such as plant dead branches, roots, and crushed stones, and to mix the samples well together. Samples were then passed through a 0.355 mm mesh sieve, put into 5 mL cryopreservation tubes and stored in a −80 °C freezer.
2.2. Measurement of Soil Physical and Chemical Properties
Bulk soil samples were ground and air-dried, and stored in self-sealing bag to determine soil physical and chemical properties. The soil physicochemical parameters were determined using the National Standards of the People’s Republic of China [27]: soil pH (NY/T1377-2007): Glass electrode method, soil organic matter (NY/T1121.6-2006): Sulfuric acid-potassium dichromate volumetric method, soil total nitrogen (NY/T1121.24-2012): Kjeldahl method, soil total phosphorus (NY/T88-1988): Alkaline solubility-molybdenum antimony anticolorimetric method, soil total potassium (NY/T87-1988): Atomic absorption spectrophotometry, soil available phosphorus (NY/T1121.7-2014): Spectrophotometry, soil available nitrogen (LY/T1228-2015): Alkalinolytic diffusion, and soil available potassium (NY/T889-2004): Atomic absorption spectrophotometry. Determination of soil physical and chemical properties was conducted by the Yunnan Sanbiao Agriculture and Forestry Technology Co., Ltd. in Kunming, China.
2.3. DNA Extraction, PCR Amplification, and High Throughput Sequencing
Total soil microbial total DNA was extracted directly using the Power Soil DNA Isolation Kit (MoBio Laboratories, San Diego, CA, USA). DNA purity and concentration were assessed using a NanoDrop 2000 spectrometer (Thermo Fisher Scientific, Wilmington, DE, USA). DNA integrity was measured using 1% agarose gel electrophoresis. The fungal universal primer pairs ITS3F (GCATCGATGAAGAACGCAGC)_ITS4R (TCCTCCGCTTATTGATATGC) were used to amplify the Internal Transcribed Spacer 2 (ITS2) region [28].
PCR amplification was conducted using TransGen AP221-02: TransStart FastPfu DNA Polymerase (TransGen Biotech, Beijing, China) and was performed in a GeneAmp 9700 thermal cycler (Applied Biosystems, Foster City, CA, USA). The reaction mixture included 4 µL of 5×FastPfu buffer, 2 µL of 2.5 mM dNTPs, 0.4 µL of FastPfu Polymerase, 0.8 µL of the forward primer (5 µM), 0.8 µL of the reverse primer (5 µM), 0.2 µL of BSA, and 10 ng of template DNA, and double distilled water (ddH2O) was added to a final volume of 20 µL. Thermal cycling conditions were as follows: 3 min for denaturing at 95 °C, 35 cycles (30 s at 95 °C; 30 s for annealing at 55 °C; and 45 s at 72 °C), 10 min for a final extension at 72 °C, and the reactions were then kept at 10 °C until halted. PCR amplification was detected using 2% agarose gel electrophoresis, and the target fragments were cut out and recovered.
Next, the products were purified, quantified, and homogenized to form a sequencing library, and the constructed library was checked for quality. Finally, qualified libraries were subjected to bidirectional high-throughput sequencing using Illumina MiSeq PE300 (Illumina, San Diego, CA, USA). The sequencing of all samples in this study was performed by the Shanghai Majorbio Bio-pharm Technology Co., Ltd. (Shanghai, China) The National Microbiology Data Center (NMDC, https://nmdc.cn/, accessed on 19 June 2023) allows a huge amount of microbiological data to be organized and integrated in an effective way and shared in an open manner [29]. Data were deposited in the NMDC under BioProject ID NMDC10018294 and accession numbers NMDC40029871–NMDC40029888 (https://nmdc.cn/resource/genomics/sample/detail/NMDC40029871-NMDC40029888), and accessed on 19 June 2023.
2.4. Data Analysis
FLASH (version 1.2.11) software was used to join the reads from each sample and obtain high-quality clean reads [30]. The QIIME (version 1.9.1) and Fastp (version 0.19.6) programs were then used to filter the clean tags and obtain effective tags. The sequences were then clustered into operational taxonomic units (OTUs) with UPARSE (version 7.0.1090) based on a 97% nucleotide similarity threshold [31]. Taxonomic assignments were performed using the RDP classifier algorithm, and the fungal ITS database in UNITE (version 8.0) [32]. OTUs aligned to the chloroplast and mitochondrion sequences were removed from the dataset.
Alpha diversity metrics reflecting the richness and diversity of the communities were calculated using Mothur (version v.1.35.1). Beta diversity was also estimated as a representation of the compositional differences between communities. Principal coordinates analysis (PCoA) using the Bray–Curtis distance metric was used to evaluate similarities across community structures. Analysis of similarities (ANOSIM) was used to test whether the differences between groups were significantly greater than the within group differences. Common and unique taxonomic communities among the different groups were visualized with a Venn diagram. Significant differences in relative abundance were tested with Kruskal–Wallis rank sum tests. Linear discriminant analysis Effect Size (LEfSe) measurements analysis was conducted to identify taxonomic biomarkers for different groups. The relationships between soil physicochemical parameters and fungal community were analyzed using Redundancy analysis (RDA) and correlation heatmap analysis. The functional prediction and classification of the fungal communities were performed according to the FUNGuild database [33].
Statistical analyses were conducted using SPSS 22.0 software (SPSS Inc., Chicago, IL, USA) and the results are shown as means ± SD (standard deviations), with a p < 0.05 considered to be statistically significant. Majorbio Cloud is a one-stop, comprehensive bioinformatic platform for multi-omics analyses [34]. Bioinformatics analyses were performed using the online Majorbio Cloud Platform (www.majorbio.com, accessed on 19 June 2023).
3. Results
3.1. Sequencing Quality
The 18 rhizosphere soil samples were subjected to sequencing of the whole community ITS on an Illumina MiSeq PE 300 platform. After filtering out low-quality reads, ITS2 sequencing obtained 1,197,640 clean reads and acquired 322 bp average length of sequence, respectively (Supplementary Table S1). With the increase in the number of sequencing reads, the rarefaction curves eventually became flat (Supplementary Figure S1), indicating that the sequencing depth of all samples are reasonable, and that the samples can truly and comprehensively reflect the structure and composition of the rhizospheric fungal community of P. squamata at different sites. In addition, the Good’s coverage rate of samples was higher than 99.00% (Table 1), indicating that the integrity of the sequencing data are high and therefore the data are reliable, with the probability of undetected fungal microbiota sequences in each sample being extremely low.

Table 1.
Alpha diversity values of P. squamata rhizosphere fungi at different conserving sites.
3.2. Alpha Diversity
The alpha diversity of rhizosphere fungi at the Operational Taxonomic Units (OTU) level were demonstrated (Table 1). The sobs, ace, and chao1 indices reflect community richness, and the simpson and shannon indices represent community diversity. The smaller the simpson index, and the larger the shannon index, the higher the community diversity. The EK site had the highest sobs, ace, and chao1 indices, which were 799.00, 979.90, and 975.72, respectively, and these values were significantly different from those at the other 5 sites (p < 0.05). The EY site had the lowest values of these three indices, with values of 209.00, 271.02, and 267.06, respectively. Thus, the EK site had the highest fungal community richness, while EY site had the lowest fungal community richness. The shannon index of the EK site was the largest at 4.30, and was significantly different from those of the EY, WQ, and RQ sites (p < 0.05). The simpson index of the EK site was the smallest at 0.0614, and was significantly different from that at the RQ site (p < 0.05). Combined with shannon and simpson indices, this demonstrates that the EK site had high fungal community diversity, while the RQ site had low fungal community diversity.
3.3. Fungal Communities in Rhizosphere Soil of P. squamata
3.3.1. Community Structure and Composition
The fungal communities of the 18 P. squamata rhizosphere soils contained a total of 18 phyla, 58 classes, 154 orders, 343 families, 715 genera, 1164 species, and 2775 OTUs. At the phylum level, five phyla were detected in all of the six sites, including Ascomycota, Basidiomycota, unclassified_k_Fungi, Rozellomycota, and Mortierellomycota. Phyla with a relative abundance of less than 0.01 (1%) in all samples were considered to be “others” (Figure 1A). The relative abundance of Ascomycota (55.67–85.40%), and Basidiomycota (5.44–36.15%) together made up between 87.64% (RQ) and 99.00% (EY) of the total taxa. At the genus level, genera with a relative abundance of less than 0.05 (5%) in all samples were categorized as “others” (Figure 1B). Suillus (15.17%) was abundant at the EC site, while Cenococcum (30.41%) and Oidiodendron (10.26%) were dominant at the EY site, and Fusarium (10.81%) and Chrysosporium (10.78%) were abundant at the EK site. Rhizopogon (14.23%) was dominant at the WQ site, and Rhizopogon (21.73%) and Tuber (13.77%) were abundant at the RQ sites, Penicillium (32.52%) was dominant at the EQ sites.
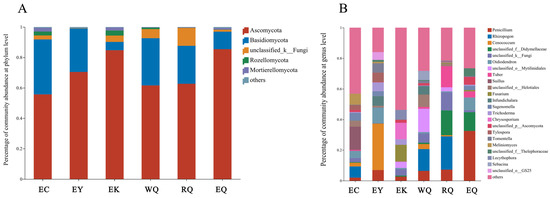
Figure 1.
Composition of fungal communities in P. squamata rhizosphere soil at the phylum (A) and genus (B) levels. WQ wild Qiaojia, RQ reintroduced Qiaojia, EQ ex situ Qiaojia, EC ex situ Caojian, EY ex situ Yipinglang, and EK ex situ Kunming (n = 3).
3.3.2. Unique and Common Communities
Unique and common communities at the different sites were analyzed using Venn diagrams (Figure 2). At the phylum level, two (11.11%) phyla, Entorrhizomycota (87.80%), and Monoblepharomycota (12.20%) were unique to the EC site. A single phylum, Calcarisporiellomycota (5.56%) was specific to the EY site, and two (11.11%) phyla were unique to the EK site. Seven (38.89%) of the eighteen phyla were common to all of the six sites (Figure 2A). These seven shared phyla were the Ascomycota (70.13%), Basidiomycota (22.97%), Rozellomycota (1.18%), Mortierellomycota (1.02%), and others (0.35%).
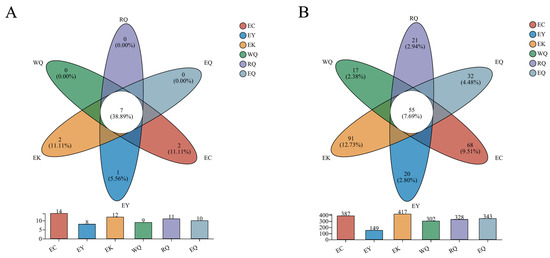
Figure 2.
Venn diagrams of fungal communities in P. squamata rhizosphere soil at different sites at the phylum (A) and genus (B) levels. Note: The bar plots represent the number of phyla and genera found at each site. The intersections of the Venn diagram represent the number of phyla and genera common to different sites. The outer areas of the diagram mean the number of phyla and genera unique to each different sites. WQ wild Qiaojia, RQ reintroduced Qiaojia, EQ ex situ Qiaojia, EC ex situ Caojian, EY ex situ Yipinglang, and EK ex situ Kunming (n = 3).
At the genus level, the number of genera unique to each site were: EC (68), EY (20), EK (91), WQ (17), RQ (21), and EQ (32). In total, 55 (7.69%) of the 715 genera were shared in six different sites (Figure 2B). The EK and EC sites each had more specific genera than they had shared genera. The genera common to all six sites were Penicillium (21.16%), Oidiodendron (9.42%), Fusarium (4.48%), Infundichalara (4.36%), Sagenomella (4.14%), Trichoderma (4.03%), Saitozyma (2.98%), Mortierella (2.24%), Cladosporium (1.25%), Coniochaeta (1.06%), Chaetosphaeria (1.03%), and others (11.06%).
3.4. Beta Diversity
Analysis of similarities (ANOSIM) is used to test whether the differences between groups is significantly greater than that within groups. The R value can range between −1 and 1, with an R > 0, indicating that the differences between groups is greater than the difference within the group. A p < 0.05 signifies significant differences between groups. The samples collected from six different sites were well separated (R = 0.95, p = 0.001), suggesting significant differences between the sites. Variation in rhizospheric fungal communities is mainly correlated with spatial location: the trees from the three sampling locations that were close to each other are clearly clustered. We also ran a principal coordinates analysis (PCoA) of fungal communities in rhizosphere soil of P. squamata at the OTU level (Figure 3). PC1 explained 17.02% of the variation in the data, and PC2 illustrated 14.44%, together accounting for 31.46% of the observed differences in fungal communities between sites. The EC and EY sites were clearly separated from other sites on PC1 dimension, while the PC2 dimension clearly separated the EC and EK sites from the other sites.
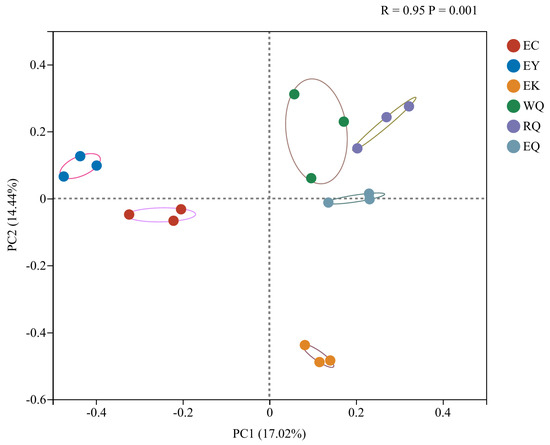
Figure 3.
Principal coordinate analysis of fungal communities in the rhizosphere soil of P. squamata at different sites at the OTU level. WQ wild Qiaojia, RQ reintroduced Qiaojia, EQ ex situ Qiaojia, EC ex situ Caojian, EY ex situ Yipinglang, and EK ex situ Kunming (n = 3).
3.5. Difference Analysis of Fungal Communities
Linear discriminant analysis Effect Size (LEfSe) analysis was used to identify taxonomic groups associated with the different sites. Thus, identification of fungal biomarkers to distinguishing different conservation sites was conducted at the genus-to-genus levels based on the all-against-all strategy. Only taxa above the linear discriminant analysis (LDA) significance threshold of >4.0 are presented for the six soil groups (Figure 4). At the genus level, Sagenomella, Cadophora, Truncatella, and Pseudeurotium were enriched at EC, Cenococcum, Tomentella, and Infundichalara were abundant at EY, and Fusarium, Coniochaeta, and Pyrenochaetopsis were enriched at EK. Meanwhile, Stilbella and Pseudogymnoascus were abundant at WQ, Rhizopogon and Tuber were enriched at RQ.
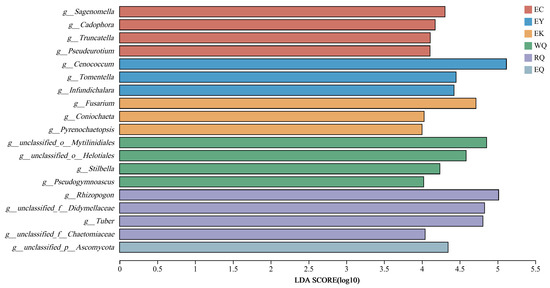
Figure 4.
Linear discriminant analysis of fungal communities in the rhizosphere soil of P. squamata Note: the larger the LDA score, the greater the influence of species abundance on the differential effect. WQ wild Qiaojia, RQ reintroduced Qiaojia, EQ ex situ Qiaojia, EC ex situ Caojian, EY ex situ Yipinglang, and EK ex situ Kunming (n = 3).
Significant difference between groups among the top 10 phyla and genera were analyzed using Kruskal–Wallis rank sum tests (Supplementary Figure S2). At the phylum level, the abundance of the Rozellomycota and Olpidiomycota were found to be different between different sites (0.01 < p ≤ 0.05). Furthermore, at the genus level, Rhizopogon, Cenococcum, Tuber, and Suillus were significantly different between the six different sites (0.01 < p ≤ 0.05).
3.6. Soil Physicochemical Properties and Their Relationships with Fungal Communities
3.6.1. Soil Physical and Chemical Properties
Soil physical and chemical properties at different conservation sites of P. squamata were shown in Table 2. Soil pH was <7 at the WQ, RQ, EC, and EY sites, and was significantly different from the pH of the soil at the EK and EQ sites (p < 0.05). Soil organic matter (OM), total nitrogen (TN), total phosphorus (TP), available nitrogen (AN), and available phosphorus (AP) levels at the EY site were the lowest, reaching 30.23 g/kg, 0.76 g/kg, 0.25 g/kg, 68.31 mg/kg, and 6.93 mg/kg, respectively. The OM content at the EY site was significantly different from that at the EC and EK sites (p < 0.05), TN and AN levels at the EY site were significantly different from those at the EQ, EC, and EK sites (p < 0.05). The TP content at the EY site were significantly different from those at other sites (p < 0.05), and the AP content of the soil at the EY site was significantly different from that at the RQ, EQ, and EK sites (p < 0.05). Soil total potassium (TK) at the EK site was the lowest, reaching 7.27 g/kg, but available potassium (AK) at the EK site was the highest, reaching 174.50 mg/kg, with that at the WQ site being the lowest, at 99.77 mg/kg. The TK content of the EK site was significantly different from that at other sites, with the exception of EQ site (p < 0.05). The AK content of the soil at the WQ site was significantly different from that at EQ and EK sites (p < 0.05).
Table 2.
Soil physical and chemical properties of P. squamata rhizosphere at different conservation sites.
3.6.2. Redundancy Analysis
A redundancy analysis (RDA) was conducted to evaluate the relative contribution of different soil physicochemical factors on variation in the observed fungal communities in the rhizosphere soil of P. squamata. At the phylum level, the interpretation rate of RDA1 was 51.84%, and the RDA2 was 5.13%, together accounting for 56.97% of the variation (Figure 5). Of the eight soil physicochemical parameters, soil TK, AP, and pH affected community structures (0.01 < p ≤ 0.05), while the other five parameters did not (p > 0.05) (Supplementary Table S2). TK, AP, and pH were therefore the main factors driving fungal community structure.
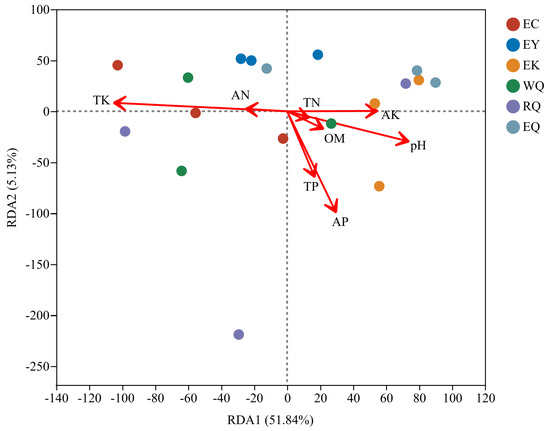
Figure 5.
Redundancy analysis of fungal communities and soil physicochemical properties of P. squamata rhizosphere soil at different conservation sites at the phylum level. Note: the length of the environmental factor arrow can represent the degree of influence of the environmental factor on the species. WQ wild Qiaojia, RQ reintroduced Qiaojia, EQ ex situ Qiaojia, EC ex situ Caojian, EY ex situ Yipinglang, and EK ex situ Kunming (n = 3).
3.6.3. Heatmap of the Correlation between Fungal Community and Soil Physicochemical Properties
Correlations between soil physicochemical parameters and the relative abundances of the top 10 most abundant fungal phyla were visualized using heatmaps at the phylum level. TK and other factors clustered into two branches (Figure 6). TK was extremely significantly positively correlated with Basidiomycota, positively correlated with Mucoromycota. TK was also negatively correlated with Chytridiomycota and extremely significantly negatively correlated with Ascomycota. AP was significantly positively correlated with unclassified_k_Fungi and Chytridiomycota, and negatively correlated with Mucoromycota. pH was significantly positively correlated with Mortierellomycota, and positively correlated with Glomeromycota and Ascomycota. pH was also negatively correlated with Basidiomycota. AK was significantly positively correlated with Olpidiomycota, and positively correlated with Mortierellomycota and Glomeromycota, while it was negatively correlated with Basidiomycota. AN was positively correlated with Rozellomycota. OM and TN were significantly positively correlated with Rozellomycota, and positively correlated with Olpidiomycota.
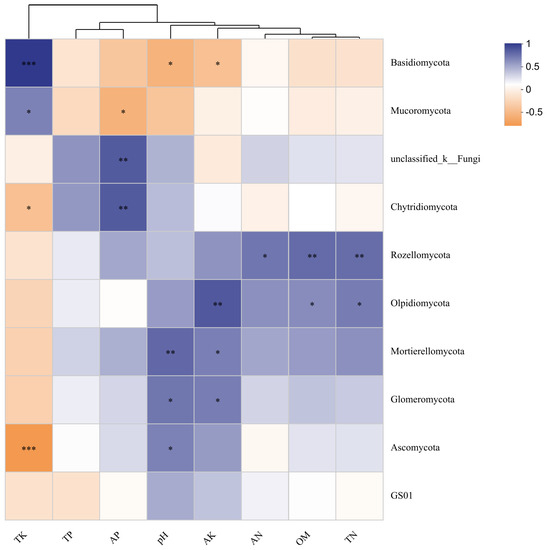
Figure 6.
Heatmap of the correlation between fungal community and soil physicochemical properties of P. squamata rhizosphere soil at different conservation sites at the phylum level. Note: * 0.01 < p ≤ 0.05, ** 0.001 < p ≤ 0.01, *** p ≤ 0.001. Red means a positive correlation, blue means a negative correlation. Darker colors indicate stronger correlations.
3.7. Functional Prediction in Rhizosphere Fungal Community of P. squamata
FUNGuild functional classification prediction in the rhizosphere soil of P. squamata was conducted. Any classes with a relative abundance < 0.01 were merged into “others” (Supplementary Figure S3). Trophic mode is mainly based on saprotrophic fungi in the P. squamata rhizosphere. Fungal functional guild classification prediction in the P. squamata rhizosphere soil suggested that the most common guilds were likely to be ectomycorrhizal, and endophyte fungi, as well as plant pathogens. The relative abundance of ectomycorrhizal fungi at the conservation sites were: 32.51% (EC), 49.20% (EY), 0.16% (EK), 24.27% (WQ), 37.32% (RQ), and 9.02% (EQ). The relative abundances of plant pathogens in P. squamata rhizosphere soil at the different sites are 0.42% (EC), 0.13% (EY), 2.85% (EK), 0.30% (WQ), 0.42% (RQ), and 1.30% (EQ), indicating that there were large numbers of antagonistic microorganisms in the EY and WQ sites.
4. Discussion
4.1. Fungal Diversity of P. squamata Rhizosphere Soil
Microbial community richness and diversity can reflect the complexity of the microbial communities. Upon pathogen or insect attack, plants are able to recruit protective microorganisms, and enhance the activity of these microbes to suppress pathogens in the rhizosphere [4]. In our study, the EK site had the highest fungal community richness and diversity. Because of the stronger stability of the community structure in EK, this ex situ Kunming site may therefore be the most suitable of all the sites tested for the ex situ conservation of P. squamata. The wild Qiaojia (WQ) and ex situ Yipinglang (EY) sites had the lowest community richness of all the tested sites, while the ex situ Yipinglang (EY) and reintroduced Qiaojia (RQ) sites had lowest fungal community diversity. These less stable fungal community structures may be related to the older ages of the sampled trees in WQ, EY, and RQ sites. Previous research into the effects of rhizospheric microorganisms on seven wild and two reintroduced populations of Scutellaria tsinyunensis, suggested that the population reintroduced into Helong garden (B42) had higher fungal richness and diversity than the other tested sites, and it showed more trend in the process of field reintroduction, more beneficial to S. tsinyunensis growth in the field [26]. Our study demonstrated that at Qiaojia site, the fungal community richness at the wild Qiaojia site (WQ) was lower than at the reintroduced (RQ) and ex situ Qiaojia (EQ) sites, and that the fungal community diversity at the ex situ Qiaojia (EQ) site was higher than that at the wild (WQ) and reintroduced Qiaojia (RQ) sites. Similarly, Shen et al. (2020) found that the richness of the fungal communities was lower in the wild Magnolia sinica rhizosphere than in that of the reintroduced M. sinica rhizosphere, but that the diversity of fungal communities in the wild plants was higher than that in the rhizosphere of reintroduced plants. Moreover, the DBH and height of the wild plants was significantly larger than that of the reintroduced plants [35]. Our results are consistent with those from the M. sinica study in the richness and diversity of soil fungal communities in wild and reintroduced plant individuals.
4.2. Community Structure of P. squamata Rhizosphere Fungi
In our study, the Ascomycota and Basidiomycota were the dominant phyla in the fungal communities of P. squamata rhizosphere soil at different conservation sites. Previous research compared the diversity and structure of the rhizosphere fungal community between the straight and twisted trunk types of Pinus yunnanensis. The fungal communities’ composition in the rhizosphere of straight and twisted bark varieties included Basidiomycota (72.96 and 68.35%, respectively) and Ascomycota (26.12 and 29.76%, respectively), with an overall relative abundance of more than 98% in both groups [36]. Many fungi convert dead organic matter into biomass, carbon dioxide, and organic acids, can effectively decompose organic matter and soil components, and thereby, regulate the balance of carbon and nutrients [7].
Ascomycota play an essential role in decomposing plant residues and degrading OM in the soil [37]. In this study, the relative abundance of Ascomycota was higher in the reintroduced Qiaojia site (RQ) than the wild Qiaojia (WQ). The relative abundance of Ascomycota at Qiaojia was higher in the ex situ site (EQ) than the reintroduced (RQ) and wild (WQ) sites. However, the OM content at the ex situ Caojian (EC) and ex situ Kunming (EK) sites were significantly higher than that in the RQ, EY, suggesting that the large number of Ascomycota at EY promoted the decomposition of OM, at EC and EK, they reduced the decomposition of OM. A previous study investigating rhizosphere soils surrounding wild and reintroduced Magnolia sinica individuals showed that the relative abundance of Ascomycota was greater in the reintroduced rhizospheric soil than in the wild rhizospheric soil [35]. Our findings were consistent with these results.
Basidiomycota can efficiently decompose lignocellulose, fallen, and litter [38]. In our study, the relative abundance of Basidiomycota was higher in WQ than RQ. This is consistent with our observations that when we collected the samples, we found that there were a large number of fallen leaves and a large amount of litter where the wild plants were growing, whereas there was only a small amount of litter associated with the reintroduced P. squamata. Of the four ex situ sites, the relative abundance of Basidiomycota was highest at EC. Furthermore, the relative abundance of Basidiomycota at Qiaojia was higher in the wild (WQ) individuals than those in reintroduction (RQ) and ex situ (EQ) conservation programs. Previous results have demonstrated that the relative abundance of Basidiomycota in the wild plant rhizosphere is significantly greater than that in the rhizosphere surrounding reintroduced plants [35]. Our findings are consistent with these results.
Some strains of Mortierella belong to the plant growth-promoting fungi (PGPF) and are valuable decomposers. Key characteristics, including the ability to survive under very unfavorable environmental conditions and the utilization of carbon sources contained in polymers like cellulose, hemicellulose, and chitin, mean that these fungi are efficient as the agricultural inoculants [39]. In this study, the relative abundances of Mortierella in the EC and EK sites were higher than that at EQ. Trichoderma are plant symbionts, and also behave as a low cost, effective and ecofriendly biocontrol agents [40]. In our study, the relative abundance of Trichoderma was higher at the WQ site than at RQ. Of the four ex situ sites, the relative abundance of Trichoderma was higher in EY than EK, EQ, and EC, indicating that there were a large number of antagonistic fungi in the EY site, that could potentially be screened for use in biocontrol programs. Our results suggested that the relative abundance of Mortierella in the reintroduced plant rhizosphere was higher than that in the wild plant rhizosphere, but that the relative abundance of Trichoderma in the wild plant rhizosphere was 1.56 times higher than that in the reintroduced plant rhizosphere [35]. The low relative abundances of Mortierella and Trichoderma in wild and reintroduced Qiaojia soils may be a biotic factor contributing to the scarcity of P. squamata.
Penicillium is a diverse genus occurring worldwide and its species play important roles as decomposers of organic materials [41]. In our study, the relative abundance of Penicillium was higher in RQ than WQ. Of the four ex situ sites, the relative abundance of Penicillium was highest at EQ, indicating that there were a large number of plant pathogens fungi at EQ. Furthermore, the genus Fusarium contains many important plant pathogens, and mycotoxin producers, as well as opportunistic human pathogens [42]. In this study, the relative abundance of Fusarium was higher at the EK site than EQ and RQ, suggesting that there may be many plant pathogenic fungi at EK. The presence of these Fusarium species may represent a potential risk for P. squamata conservation at EK.
4.3. Relationships between Fungal Community and Soil Physicochemical Properties
Abiotic stresses and soil nutrient limitations are major environmental conditions that reduce plant growth, productivity, and quality [43]. In this study, we determined eight soil physical and chemical parameters at different P. squamata conservation sites. The tested parameters included pH, OM, TN, TP, TK, AN, AP, AK. Soil pH at the EK and EQ sites was found to be >7, indicating alkaline soils, which is not conducive to the growth of P. squamata. The biogeochemistry of soil organic matter is a highly complex and dynamic soil property and is of vital importance for the health and ecological functioning of ecosystems [44]. Nitrogen (N), phosphorus (P), and potassium (K) are essential macronutrients for plant growth and development [45]. Soil organic matter (OM), total nitrogen (TN), total phosphorus (TP), available nitrogen (AN), and available phosphorus (AP) contents were all the lowest at the EY site, indicating that this site had the lowest soil fertility and nutrients.
The diversity and activity of soil fungi is regulated by various biotic (plants and other organisms) and abiotic (soil pH, moisture, salinity, structure, and temperature) factors [7]. In our study, of the eight soil physicochemical parameters, soil TK, AP, and pH affected community structures significantly (0.01 < p ≤ 0.05), while the other five parameters did not (p > 0.05). TK, AP, and pH were thus the main factors driving fungal community structure. Soil pH, AK, TN, TP, and TK have also been found to be important factors influencing the fungal communities associated with the rhizosphere soil surrounding wild and reintroduced M. sinica individuals [35]. Moreover, available K was found to be correlated with different fungal communities (p = 0.01) in the soils of twisted and straight-trunk individuals of P. yunnanensis [36]. Our results are therefore similar to those of previous studies.
4.4. Functional Prediction of P. squamata Rhizosphere Fungal Communities
According to the means by which they obtain nutrition, fungi can be divided into three trophic modes: pathotrophs, saprotrophs, and symbiotrophs [33]. In our study, the dominant trophic mode in the rhizosphere of P. squamata was saprotroph. This group of fungi obtains most of its nutrients by decomposing litter; therefore, the litter itself is important and can influence the functional diversity of the P. squamata rhizosphere fungal communities. This is similar to results from studies in wild and reintroduced M. sinica rhizospheres, where most of the fungi in the wild rhizospheric soils around wild M. sinica individuals were predicted to be saprotrophs, followed by pathotrophs, with only a small proportion of symbiotrophs. However, although most fungi in the rhizospheric soil surrounding the reintroduced plants were saprotrophic, the second largest predicted trophic mode was symbiotroph [35], suggesting that the trophic modes of fungi in the rhizospheric soils of P. squamata and M. sinica are different. We found a large number of antagonistic microorganisms in the EY and WQ sites. Our results are consistent with the results from previous studies suggesting that the relative abundance of plant pathogens in the rhizosphere of wild M. sinica individuals was significantly lower than that in rhizosphere of the reintroduced plants [35].
5. Conclusions
The present study explored the diversity, composition, and function of rhizosphere soil fungal communities in conservation sites where P. squamata is grown in the wild, or in ex situ, and reintroduced conservation programs. The ex situ Kunming (EK) site had the highest fungal community richness and diversity, and may therefore be most suitable for the ex situ conservation of P. squamata. The low relative abundance of Mortierella and Trichoderma in wild and reintroduced Qiaojia soils may be a biotic factor contributing to the threats facing P. squamata, and sites chosen for the conservation of P. squamata should be selected by comparison of fungal diversity. Nevertheless, we acknowledge that this study has limitations. This study only explored the impacts of the rhizosphere on P. squamata at different conservation sits. Furthermore, we investigated only the fungal communities of the P. squamata rhizosphere, although rhizosphere microbes may include further microbial communities, including prokaryotes (bacteria and archaea), as well as protists and viruses. Moreover, the effects of different plant compartments (root endosphere and phyllosphere) and non-rhizosphere soil on the growth of P. squamata should be fully investigated. In the future, culture-dependent methods or a combination of multi-omics methods should also be considered to assess the patterns in microbial diversity in the P. squamata rhizosphere.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/d15070868/s1, Figure S1: Rarefaction curves of fungal community in rhizosphere of P. squamata; Figure S2: Difference analysis of fungal communities in rhizosphere of P. squamata; Figure S3: FUNGuild Function prediction in rhizosphere of P. squamata; Table S1: Sample sequencing information of fungi in rhizosphere of P. squamata; Table S2: Network nodes of fungi in rhizosphere of P. squamata; Table S3: Network centrality of fungi in rhizosphere of P. squamata; Table S4: Relationships between soil physicochemical parameters and fungal community variation at phylum level in rhizosphere of P. squamata.
Author Contributions
All authors contributed to the conception and design of the study. Material preparation, data collection, and analysis were performed by F.L. The first draft of the manuscript was written by F.L. and W.S. commented on subsequent versions of the manuscript. All authors have read and agreed to the published version of the manuscript.
Funding
This work was equally supported by the Science and Technology Basic Resources Investigation Program of China (2017FY100100), and the Second Tibetan Plateau Scientific Expedition and Research Program (2019QZKK0502).
Institutional Review Board Statement
Not applicable.
Data Availability Statement
The datasets generated during the current study are available in the National Microbiology Data Center (NMDC), under BioProject ID NMDC10018294 and accession numbers https://nmdc.cn/resource/genomics/sample/detail/NMDC40029871-NMDC40029888, accessed on 19 June 2023.
Acknowledgments
We would like to thank Shuai Chang and Pin Zhang for help with sampling.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Turner, T.R.; James, E.K.; Poole, P.S. The plant microbiome. Genome Biol. 2013, 14, 209. [Google Scholar] [CrossRef] [PubMed]
- Philippot, L.; Raaijmakers, J.M.; Lemanceau, P.; van der Putten, W.H. Going back to the roots: The microbial ecology of the rhizosphere. Nat. Rev. Microbiol. 2013, 11, 789–799. [Google Scholar] [CrossRef] [PubMed]
- Buee, M.; De Boer, W.; Martin, F.; van Overbeek, L.; Jurkevitch, E. The rhizosphere zoo: An overview of plant-associated communities of microorganisms, including phages, bacteria, archaea, and fungi, and of some of their structuring factors. Plant Soil 2009, 321, 189–212. [Google Scholar] [CrossRef]
- Berendsen, R.L.; Pieterse, C.M.J.; Bakker, P.A.H.M. The rhizosphere microbiome and plant health. Trends Plant Sci. 2012, 17, 478–486. [Google Scholar] [CrossRef]
- Mendes, R.; Garbeva, P.; Raaijmakers, J.M. The rhizosphere microbiome: Significance of plant beneficial, plant pathogenic, and human pathogenic microorganisms. FEMS Microbiol. Rev. 2013, 37, 634–663. [Google Scholar] [CrossRef]
- Nilsson, R.H.; Anslan, S.; Bahram, M.; Wurzbacher, C.; Baldrian, P.; Tedersoo, L. Mycobiome diversity: High-throughput sequencing and identification of fungi. Nat. Rev. Microbiol. 2019, 17, 95–109. [Google Scholar] [CrossRef]
- Frac, M.; Hannula, S.E.; Belka, M.; Jedryczk, M. Fungal biodiversity and their role in soil health. Front. Microbiol. 2018, 9, 707. [Google Scholar] [CrossRef]
- Zhou, L.X.; Ding, M.M. Soil microbial characteristics as bioindicators of soil health. Biodivers. Sci. 2007, 15, 162–171. [Google Scholar] [CrossRef]
- Zhu, Y.G.; Peng, J.J.; Wei, Z.; Shen, Q.R.; Zhang, F.S. Linking the soil microbiome to soil health. Sci. Sin. Vitae 2021, 51, 1–11. [Google Scholar] [CrossRef]
- Sun, W.B.; Han, C.Y. Researches and conservation for plant species with extremely small populations (PSESP). Biodivers. Sci. 2015, 23, 426–429. [Google Scholar] [CrossRef]
- Sun, W.B.; Yang, J.; Dao, Z.L. Study and Conservation of Plant Species with Extremely Small Populations (PSESP) in Yunnan Province; Science Press: Beijing, China, 2019. [Google Scholar]
- Yang, J.; Cai, L.; Liu, D.T.; Chen, G.; Gratzfeld, J.; Sun, W.B. China’s conservation program on Plant Species with Extremely Small Populations (PSESP): Progress and perspectives. Biol. Conserv. 2020, 244, 108535. [Google Scholar] [CrossRef]
- Cogoni, D.; Fenu, G.; Dessi, C.; Deidda, A.; Giotta, C.; Piccitto, M.; Bacchetta, G. Importance of plants with extremely small populations (PSESPs) in endemic-rich areas, elements often forgotten in conservation strategies. Plants 2021, 10, 1504. [Google Scholar] [CrossRef]
- Crane, P. Conserving our global botanical heritage: The PSESP plant conservation program. Plant Divers. 2020, 42, 319–322. [Google Scholar] [CrossRef]
- National Forestry Bureau and Agriculture Ministry of China. List of National Key Protected Wild Plants; National Forestry Bureau and Agriculture Ministry of China: Beijing, China, 1999.
- National Forestry Bureau and Agriculture Ministry of China. List of National Key Protected Wild Plants; National Forestry Bureau and Agriculture Ministry of China: Beijing, China, 2021.
- IUCN Species Survival Commission. IUCN Red List Categories and Criteria: Version 3.1; IUCN: Gland, Switzerland; Cambridge, UK, 2001. [Google Scholar]
- Li, X.W. A new series and a new species of Pinus from Yunnan. Acta Bot. Yunnanica 1992, 14, 259–260. [Google Scholar]
- Fan, G.S.; Li, X.W.; Deng, L.L. A study of floristic geography of Pinus squamata flora. J. Cent.-South For. Univ. 1996, 16, 23–27+33. [Google Scholar] [CrossRef]
- Lu, S.J.; Deng, L.L.; Li, X.W. A study on endangered causes of Pinus squamata. J. Northwest For. Univ. 1999, 14, 44–46. [Google Scholar]
- Zhang, Z.Y.; Chen, Y.Y.; Li, D.Z. Detection of low genetic variation in a critically endangered chinese pine, Pinus squamata, using RAPD and ISSR Markers. Biochem. Genet. 2005, 43, 239–249. [Google Scholar] [CrossRef]
- Ruan, Z.Y.; Wang, B.Y.; Ouyang, Z.Q.; Liao, L.B.; Su, T.W.; Qiao, L. Characterization of microsatellites in genome of Pinus squamata, a critically endangered species in the world. Bull. Bot. Res. 2016, 36, 775–781. [Google Scholar] [CrossRef]
- Wu, Y.; Su, T.W.; Wu, J.R. Investigation and prevention on major diseases and pests of Pinus squamata artificial breeding populations. For. Inventory Plan. 2016, 41, 73–77. [Google Scholar] [CrossRef]
- Ruan, Z.Y.; Qiao, L.; Su, T.W.; Bai, B.; Cao, Y.C. A study on tissue culture of Pinus squamata. J. West China For Sci. 2017, 46, 44–49. [Google Scholar] [CrossRef]
- Yang, X.; Wang, P.; Gao, D.W.; Gao, N.N.; Li, L.S.; Yang, X.L.; Zhong, Q.J. Ecological stoichiometry of form. Quercus pannosa and form. Pinus squamata in the Yaoshan nature reserve, Yunnan. Acta Ecol. Sin. 2019, 39, 4021–4028. [Google Scholar]
- Yu, F.Q. Effects of Rhizospheric Microorganisms on Populations of Scutellaria tsinyunensis. Master’s Thesis, Southwest University, Chongqing, China, 2018. [Google Scholar]
- National Agricultural Technology Extension and Service Center. Technical Specification for Soil Analysis; Chinese Agricultural Press: Beijing, China, 2006. [Google Scholar]
- Tedersoo, L.; Anslan, S.; Bahram, M.; Polme, S.; Riit, T.; Liiv, I.; Koljalg, U.; Kisand, V.; Nilsson, R.H.; Hildebrand, F.; et al. Shotgun metagenomes and multiple primer pair-barcode combinations of amplicons reveal biases in metabarcoding analyses of fungi. MycoKeys 2015, 10, 1–43. [Google Scholar] [CrossRef]
- Fan, G.M.; Sun, Q.L.; Shi, W.Y.; Qi, H.Y.; Sun, D.Z.; Li, F.H.; Pang, H.F.; Ma, J.C.; Wu, L.H. The services and applications of national microbiology data center. Acta Microbiol. Sin. 2021, 61, 3761–3773. [Google Scholar] [CrossRef]
- Magoc, T.; Salzberg, S.L. FLASH: Fast length adjustment of short reads to improve genome assemblies. Bioinformatics 2011, 27, 2957–2963. [Google Scholar] [CrossRef]
- Edgar, R.C. UPARSE: Highly accurate OTU sequences from microbial amplicon reads. Nat. Methods 2013, 10, 996–998. [Google Scholar] [CrossRef]
- Nilsson, R.H.; Larsson, K.H.; Taylor, A.F.S.; Bengtsson-Palme, J.; Jeppesen, T.S.; Schigel, D.; Kennedy, P.; Picard, K.; Glockner, F.O.; Tedersoo, L.; et al. The UNITE database for molecular identification of fungi: Handling dark taxa and parallel taxonomic classifications. Nucleic Acids Res. 2019, 47, D259–D264. [Google Scholar] [CrossRef]
- Nguyen, N.H.; Song, Z.W.; Bates, S.T.; Branco, S.; Tedersoo, L.; Menke, J.; Schilling, J.S.; Kennedy, P.G. FUNGuild: An open annotation tool for parsing fungal community datasets by ecological guild. Fungal Ecol. 2016, 20, 241–248. [Google Scholar] [CrossRef]
- Ren, Y.; Yu, G.; Shi, C.P.; Liu, L.M.; Guo, Q.; Han, C.; Zhang, D.; Zhang, L.; Liu, B.X.; Gao, H.; et al. Majorbio Cloud: A one-stop, comprehensive bioinformatic platform for multi-omics analyses. iMeta 2022, 1, e12. [Google Scholar] [CrossRef]
- Shen, Q.Q.; Yang, J.Y.; Su, D.F.; Li, Z.Y.; Xiao, W.; Wang, Y.X.; Cui, X.L. Comparative analysis of fungal diversity in rhizospheric soil from wild and reintroduced Magnolia sinica estimated via high-throughput sequencing. Plants 2020, 9, 600. [Google Scholar] [CrossRef]
- Li, P.L.; Zong, D.; Gan, P.H.; Li, H.L.; Wu, Z.Y.; Li, F.H.; Zhao, C.L.; Li, L.G.; He, C.Z. Comparison of the diversity and structure of the rhizosphere microbial community between the straight and twisted trunk types of Pinus yunnanensis. Front. Microbiol. 2023, 14, 1066805. [Google Scholar] [CrossRef]
- Egidi, E.; Delgado-Baquerizo, M.; Plett, J.M.; Wang, J.T.; Eldridge, D.J.; Bardgett, R.D.; Maestre, F.T.; Singh, B.K. A few Ascomycota taxa dominate soil fungal communities worldwide. Nat. Commun. 2019, 10, 2369. [Google Scholar] [CrossRef]
- He, M.Q.; Zhao, R.L.; Hyde, K.D.; Begerow, D.; Kemler, M.; Yurkov, A.; McKenzie, E.H.C.; Raspe, O.; Kakishima, M.; Sanchez-Ramirez, S.; et al. Notes, outline and divergence times of Basidiomycota. Fungal Divers. 2019, 99, 105–367. [Google Scholar] [CrossRef]
- Ozimek, E.; Hanaka, A. Mortierella Species as the Plant Growth-Promoting Fungi Present in the Agricultural Soils. Agriculture 2021, 11, 7. [Google Scholar] [CrossRef]
- Sood, M.; Kapoor, D.; Kumar, V.; Sheteiwy, M.S.; Ramakrishnan, M.; Landi, M.; Araniti, F.; Sharma, A. Trichoderma: The “Secrets” of a Multitalented Biocontrol Agent. Plants 2020, 9, 762. [Google Scholar] [CrossRef]
- Visagie, C.M.; Houbraken, J.; Frisvad, J.C.; Hong, S.B.; Klaassen, C.H.W.; Perrone, G.; Seifert, K.A.; Varga, J.; Yaguchi, T.; Samson, R.A. Identification and nomenclature of the genus Penicillium. Stud. Mycol. 2014, 78, 343–371. [Google Scholar] [CrossRef]
- Ma, L.J.; Geiser, D.M.; Proctor, R.H.; Rooney, A.P.; O’Donnell, K.; Trail, F.; Gardiner, D.M.; Manners, J.M.; Kazan, K. Fusarium Pathogenomics. Annu. Rev. Microbiol. 2013, 67, 399–416. [Google Scholar] [CrossRef]
- Gong, Z.Z.; Xiong, L.M.; Shi, H.Z.; Yang, S.H.; Herrera-Estrella, L.R.; Xu, G.H.; Chao, D.Y.; Li, J.R.; Wang, P.Y.; Qin, F.; et al. Plant abiotic stress response and nutrient use efficiency. Sci. China Life Sci. 2020, 63, 635–674. [Google Scholar] [CrossRef] [PubMed]
- Ondrasek, G.; Begic, H.B.; Zovko, M.; Filipovic, L.; Merino-Gergichevich, C.; Savic, R.; Rengel, Z. Biogeochemistry of soil organic matter in agroecosystems & environmental implications. Sci. Total Environ. 2019, 658, 1559–1573. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Chen, Y.F.; Wu, W.H. Potassium and phosphorus transport and signaling in plants. J. Integr. Plant Biol. 2021, 63, 34–52. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).