Microbial Community Composition as Affected by Dryland Cropping Systems and Tillage in a Semiarid Sandy Soil


Abstract
:1. Introduction
2. Material and Methods
2.1. Cropping Systems and Tillage Treatments
2.2. Projected General Management and Soil Sampling
2.3. Selected Soil Properties
2.4. Microbial Community according to FAME Profiling
2.5. Pyrosequencing
2.6. Statistical Analysis

3. Results
3.1. Selected Soil Properties

{kind=link}
{kind=link}
{kind=link}
| Soil Properties | Srg-Ct | Ct-Rye-Srg | Srf-Rye | ANOVA | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| No-till | Till | No-till | Till | No-till | Till | Rotation | Tillage | Rot. × Till | |||
| Total C (g C kg-1 soil) | 5.47 (0.23) | 5.53 (0.23) | 6.10 (0.25) | 6.17 (0.30) | 7.27 (0.29) | 8.17 (0.71) | < 0.001 | n.s. | n.s. | ||
| Total N (g N kg-1 soil) | 0.50 (0.01) | 0.54 (0.04) | 0.58 (0.03) | 0.58 (0.02) | 0.70 (0.03) | 0.78 (0.05) | < 0.001 | 0.05 | n.s. | ||
| Organic C (g C kg-1 soil) | 4.93 (0.47) | 5.13 (0.20) | 4.97 (0.24) | 5.93 (0.37) | 6.70 (0.20) | 7.67 (0.79) | 0.001 | 0.1 | n.s. | ||
| Soil pH | 7.14 (0.02) | 7.10 (0.03) | 7.04 (0.06) | 7.02 (0.06) | 7.05 (0.02) | 7.14 (0.03) | n.s. | n.s. | n.s. | ||
| Microbial Biomass (MB) | |||||||||||
| MBC (mg C kg-1 soil) | 50.45 (3.95) | 50.76 (0.39) | 51.17 (4.66) | 49.77 (6.32) | 74.67 (1.27) | 80.38 (3.38) | < 0.001 | n.s. | n.s. | ||
| MBN (mg C kg-1 soil) | 2.52 (0.44) | 2.95 (0.18) | 3.45 (0.64) | 2.53 (0.34) | 3.73 (0.19) | 3.40 (0.22) | 0.1 | n.s. | n.s. | ||
| Enzyme Activities (EAs) | |||||||||||
| (mg PN kg-1 soil h-1) | |||||||||||
| β-Glucosidase activity | 64.34 (5.61) | 66.27 (6.06) | 94.40 (3.56) | 68.90 (5.21) | 141.92 (8.86) | 157.03 (12.70) | < 0.001 | n.s. | 0.05 | ||
| α-Galactosidase activity | 4.09 (0.38) | 4.22 (0.38) | 5.13 (0.07) | 6.50 (1.01) | 11.42 (0.73) | 13.04 (1.33) | < 0.001 | 0.1 | n.s. | ||
| β-Glucosaminidase activity | 6.59 (0.55) | 7.44 (0.55) | 8.03 (0.58) | 8.85 (0.08) | 16.22 (2.56) | 24.30 (4.09) | < 0.001 | 0.1 | n.s. | ||
| Alkaline Phosphatase act. | 75.93 (5.17) | 75.66 (5.26) | 76.95 (3.50) | 75.98 (7.95) | 123.73 (10.11) | 145.88 (4.09) | < 0.001 | n.s. | n.s. | ||
| Phosphodiesterase activity | 27.52 (3.00) | 28.79 (4.05) | 31.68 (6.94) | 38.34 (6.97) | 75.35 (9.17) | 81.52 (8.81) | < 0.001 | n.s. | n.s. | ||
| Arylsulfatase activity | 1.42 (0.71) | 2.13 (0.55) | 2.71 (0.90) | 2.13 (0.58) | 7.86 (0.67) | 9.68 (1.79) | < 0.001 | n.s. | n.s. | ||
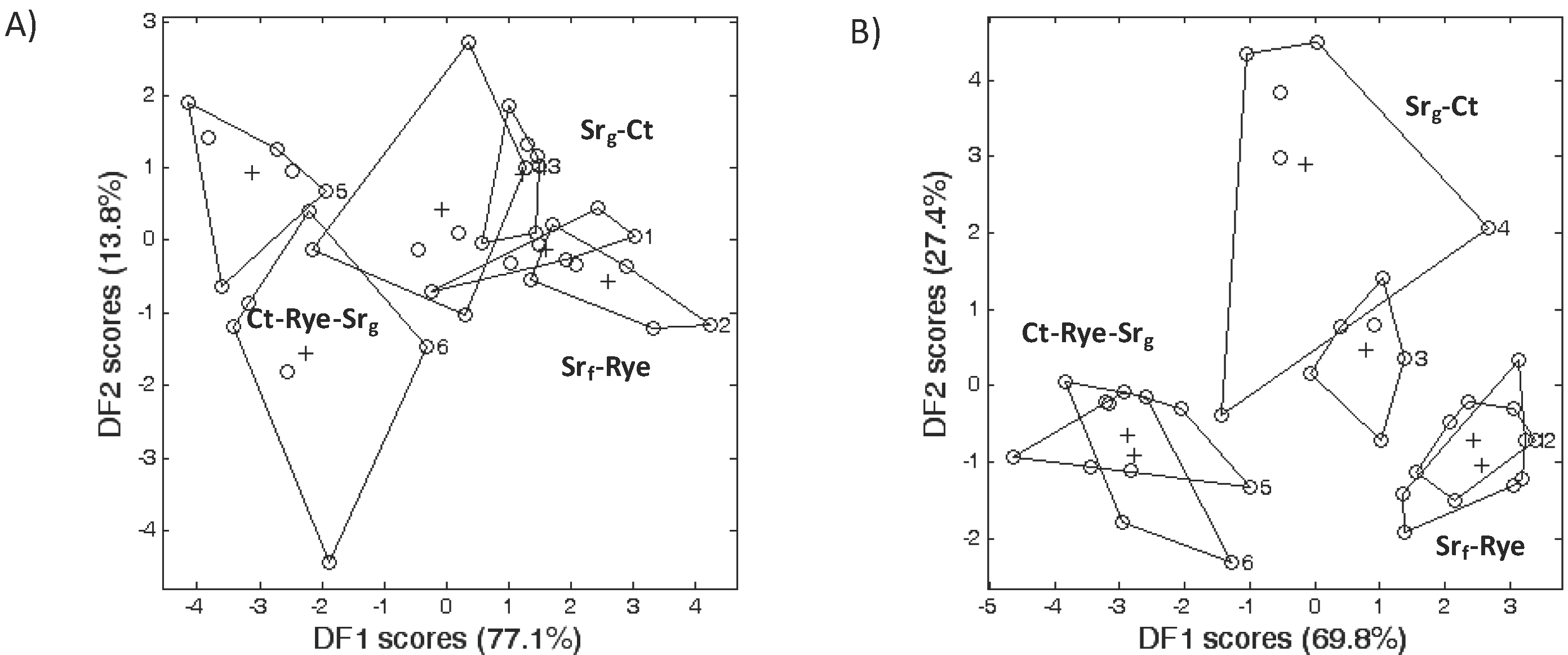
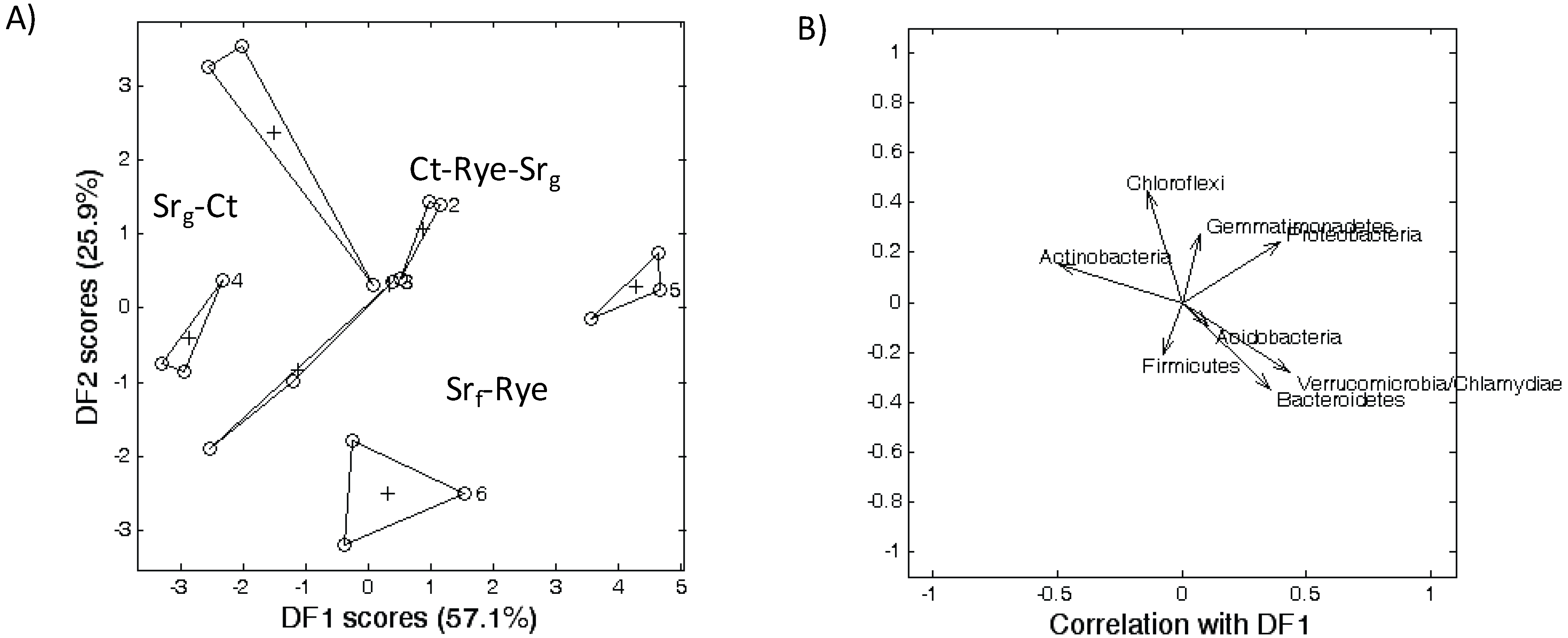
3.2. Microbial Community Structure
| FAME results | MIDI-FAME | EL-FAME | MIDI vs. EL | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Srg-Ct | Ct-Rye-Srg | Srf-Rye | ANOVA | Srg-Ct | Ct-Rye-Srg | Srf-Rye | ANOVA | ANOVA | |
| Bacteria (B) | ________ nmol g-1 soil _________ | ________ nmol g-1 soil _________ | |||||||
| Gram+ | |||||||||
| a15:0 | 1.52b | 1.73b | 2.36a | 0.005 | 0.13b | 0.28b | 0.51a | 0.002 | <0.001 |
| i15:0 | 2.21b | 2.62b | 3.39a | 0.007 | 0.23b | 0.45b | 0.79a | 0.008 | <0.001 |
| a17:0 | 0.52b | 0.62a | 0.67a | 0.023 | 0.19b | 0.26b | 0.42a | 0.001 | <0.001 |
| i17:0 | 0.51a | 0.62a | 0.63a | 0.020 | 0.15b | 0.22b | 0.31a | 0.005 | <0.001 |
| Gram- | |||||||||
| cy17:0 | 0.10 | n.d. | n.d. | n/a | 0.02b | 0.06b | 0.17a | 0.038 | n.s. |
| cy19:0 | n.d. | n.d. | n.d. | n/a | 0.21b | 0.19b | 0.31a | 0.026 | <0.001 |
| i13:0 3OH | 0.18b | 0.22b | 0.31a | 0.006 | n.d. | n.d. | n.d. | n/a | n/a |
| i17:0 3OH | 0.39b | 0.47b | 3.39a | 0.017 | n.d. | n.d. | n.d. | n/a | n/a |
| Acinomycetes | |||||||||
| 10Me16:0 | 1.13a | 1.44a | 1.27a | n.s. | 0.37b | 0.49ab | 0.72a | 0.017 | <0.001 |
| 10Me17:0 | 0.25a | 0.32a | 0.27a | n.s. | 0.03b | 0.08b | 0.12a | 0.047 | <0.001 |
| 10Me18:0 | 0.07a | 0.16a | 0.15a | n.s. | 0.08b | 0.14b | 0.25a | 0.012 | 0.03 |
| Fungi (F) | |||||||||
| 16:1ω5c | 6.39b | 3.64b | 23.10a | 0.040 | 0.18b | 0.08b | 2.11a | <0.001 | <0.001 |
| 18:1ω9c | 3.61b | 4.34b | 5.96a | <0.001 | 0.71b | 0.81b | 2.03a | 0.001 | <0.001 |
| 18:2ω6c | 2.82b | 2.85b | 4.66a | 0.002 | 0.31b | 0.34b | 1.02a | 0.106 | <0.001 |
| 18:3ω6c | 0.54b | 0.54b | 0.89a | 0.009 | 0.03b | 0.04b | 0.12a | 0.009 | <0.001 |
| Total (nmol g-1 soil) | 18.35b | 17.43b | 41.22a | 2.60b | 3.41b | 8.81a | |||
| Sum F indicators | 13.41b | 11.37b | 34.55a | <0.001 | 1.22b | 1.27b | 5.29a | 0.001 | <0.001 |
| Sum B indicators | 4.94b | 6.06ab | 6.67a | 0.036 | 1.39b | 2.13b | 3.52a | 0.004 | <0.001 |
| F:B ratios | 2.71b | 1.88b | 5.18a | 0.88b | 0.60b | 1.51a | |||

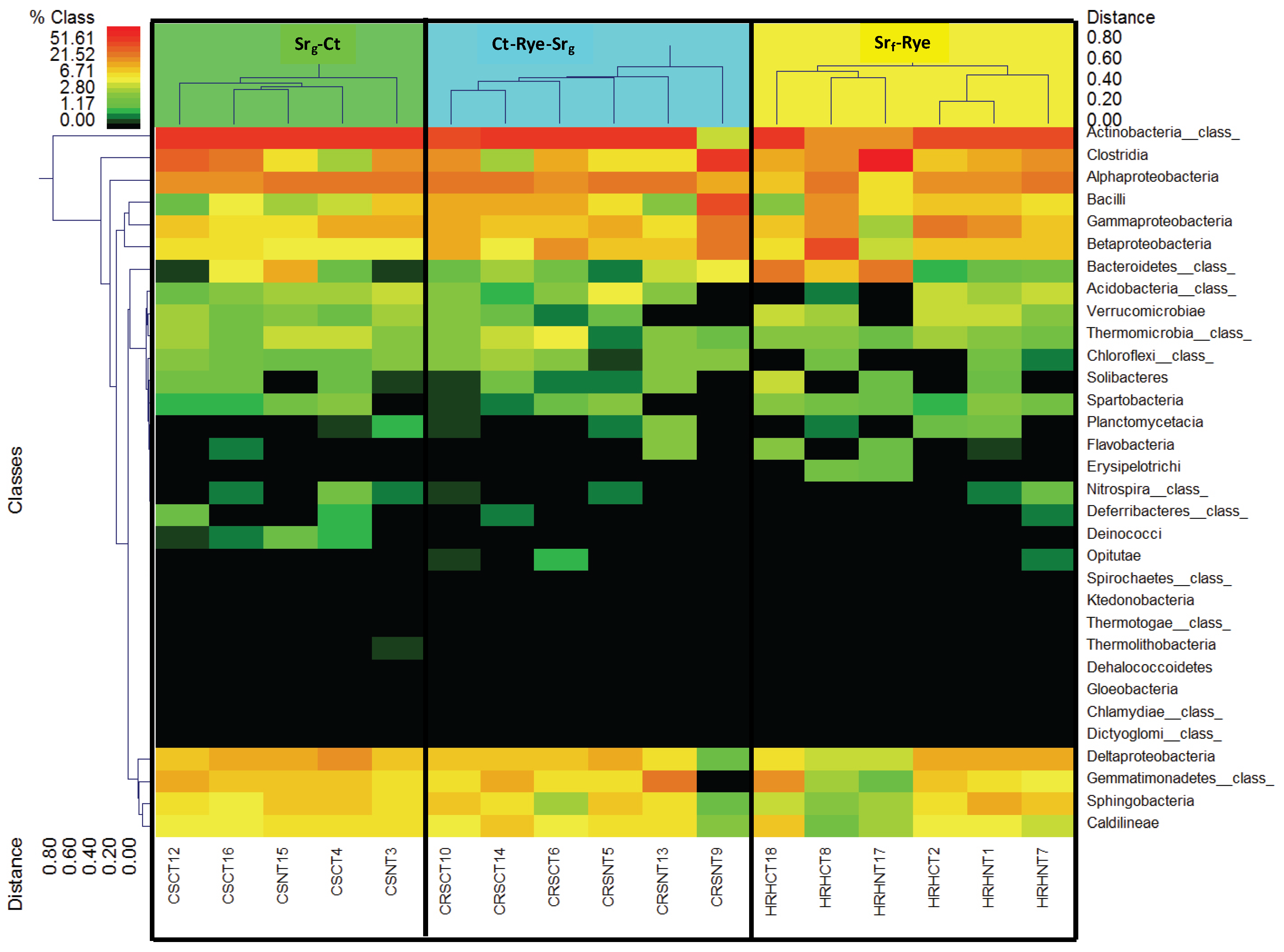
3.3. Bacterial Phyla Distribution in This Soil

3.4. Bacterial Phyla Distribution as Affected by the Cropping System and Tillage Management
| Relative abundance | Srg-Ct | Ct-Rye-Srg | Srf-Rye | ANOVA results | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| bacteria (%) | No-till | Till | No-till | Till | No-till | Till | Rotation | Tillage | Rot × Till | ||
| Proteobacteria | 23.52 (2.75) | 29.72 (7.74) | 34.22 (1.24) | 32.18 (2.76) | 28.13 (9.73) | 37.55 (8.34) | n.s. | n.s. | 0.053 | ||
| Actinobacteria | 33.63 (0.53) | 32.48 (3.47) | 22.84 (2.77) | 30.71 (10.13) | 20.44 (6.16) | 21.18 (5.58) | 0.010 | n.s. | 0.005 | ||
| Firmicutes | 23.62 (4.45) | 13.16 (12.86) | 22.44 (2.98) | 14.63 (15.53) | 27.91 (3.53) | 14.18 (14.68) | n.s. | 0.027 | n.s. | ||
| Chloroflexi | 4.88 (0.46) | 6.12 (2.46) | 5.55 (1.13) | 7.38 (1.18) | 4.05 (0.96) | 5.09 (0.86) | 0.018 | 0.071 | 0.010 | ||
| Bacteroidetes | 6.16 (0.76) | 5.75 (3.88) | 5.77 (1.15) | 5.27 (0.98) | 10.86 (4.23) | 10.30 (3.13) | 0.019 | n.s. | n.s. | ||
| Gemmatimonadetes | 4.08 (0.25) | 6.48 (0.89) | 5.60 (1.29) | 5.59 (3.67) | 2.94 (2.14) | 5.64 (1.11) | 0.001 | 0.029 | n.s. | ||
| Verrucomicrobia | |||||||||||
| /Chlamydiae | 1.51 (0.17) | 2.15 (0.83) | 0.74 (0.24) | 1.80 (0.74) | 2.55 (0.33) | 3.44 (0.92) | n.s. | 0.039 | 0.001 | ||
| Acidobacteria | 1.57 (0.26) | 2.34 (0.84) | 2.00 (0.04) | 1.63 (1.04) | 1.90 (0.76) | 1.99 (0.55) | n.s. | n.s. | n.s. | ||
| Diversity | Srg-Ct | Ct-Rye-Srg | Srf-Rye | |||
|---|---|---|---|---|---|---|
| Index | No-till | Till | No-till | Till | No-till | Till |
| OTU 3% | 223.67 | 333.67 | 161.33 | 261.67 | 166.01 | 145.00 |
| ACE 3% | 610.60 | 968.79 | 433.42 | 680.13 | 396.04 | 493.25 |
| Chao1 3% | 568.99 | 873.60 | 392.26 | 597.19 | 340.08 | 401.58 |
| 95% COI for Chao1 | 1,049.71–88.27 | 983.72–762.88 | 836.98–52.46 | 796.16–398.22 | 512.66–167.50 | 736.70–66.46 |

4. Discussion
4.1. Soil Microbial Communities As Affected by the Cropping Systems
4.2. Soil Microbial Communities As Affected by Tillage
4.3. Bacterial Distribution in this Soil
5. Conclusions
Acknowledgements
References
- Bailey, V.L.; Smith, J.L., Jr.; Bolton, H. Fungal-to-bacterial ratios in soils investigated for enhanced C sequestration. Soil Biol. Biochem. 2002, 34, 997–1008. [Google Scholar] [CrossRef]
- Acosta-Martinez, V.; Bell, C.E.; Morris, B.E.L.; Zak, J.; Allen, V.G. Long-term soil microbial community and enzyme activity responses to an integrated cropping-livestock system in a semi-arid region. Agric. Ecosys. Environ. 2010, 137, 231–240. [Google Scholar] [CrossRef]
- Acosta-Martinez, V.; Rowland, D.; Sorensen, R.B.; Yeater, K.M. Microbial community structure and functionality under peanut-based cropping systems in a sandy soil. Biol. Fertil. Soils 2008, 44, 681–692. [Google Scholar] [CrossRef]
- Zelles, L. Phospholipid fatty acid profiles in selected members of soil microbial communities. Chemosphere 1997, 35, 275–294. [Google Scholar] [CrossRef] [PubMed]
- Schutter, M.E.; Dick, R.P. Comparison of fatty acid methyl ester (FAME) methods for characterizing microbial communities. Soil Sci. Soc. Am. J. 2000, 64, 1659–1668. [Google Scholar] [CrossRef]
- Acosta-Martinez, V.; Upchurch, D.R.; Schubert, A.M.; Porter, D.; Wheeler, T. Early impacts of cotton and peanut cropping systems on selected soil chemical, physical, microbiological and biochemical properties. Biol. Fertil. Soils 2004, 40, 44–54. [Google Scholar] [CrossRef]
- Zak, J.C.; Visser, S. An appraisal of soil fungal biodiversity: the crossroads between taxonomic and functional biodiversity. Biodiv. Conser. 1996, 5, 169–183. [Google Scholar] [CrossRef]
- Zogg, G.P.; Zak, D.R.; Ringelberg, D.B.; MacDonald, N.W.; Pregitzer, K.S.; White, D.C. Compositional and functional shifts in microbial communities due to soil warming. Soil Sci. Soc. Am. J. 1997, 61, 475–481. [Google Scholar] [CrossRef]
- Ringelberg, D.B.; Stair, J.O.; Almeida, J.; Norby, R.J.; O’Neill, E.G.; White, D.S. Consequences of rising atmospheric carbon dioxide levels for the belowground microbiota associated with white oak. J. Environ. Qual. 1997, 26, 495–503. [Google Scholar] [CrossRef]
- Olsson, P.A. Signature fatty acids provide tools for determination of the distribution and interactions of mycorrhizal fungi in soil. FEMS Micro. Ecol. 1999, 29, 303–310. [Google Scholar] [CrossRef]
- Madan, R.; Pankhurst, C.; Hawke, B.; Smith, S. Use of fatty acids for identification of AM fungi and estimation of AM spores in soil. Soil Biol. Biochem. 2002, 34, 125–128. [Google Scholar] [CrossRef]
- Pinkhart, H.C.; Ringelberg, D.B.; Piceno, Y.M.; Macnaughton, S.J.; White, D.C. Biochemical approaches to biomass measurements and community structure analysis. In Manual of Environmental Microbiology; Hurst, C.J., Crawford, R.L., Knudsen, G.R., McInerney, M.J., Stentzenbach, L.D., Eds.; ASM Press: Washington, DC, USA, 2002; pp. 101–113. [Google Scholar]
- Ruess, L.; Häggblomb, M.M.; García Zapatac, E.J.; Dightonc, J. Fatty acids of fungi and nematodes-possible biomarkers in the soil food chain? Soil Biol. Biochem. 2002, 34, 745–756. [Google Scholar]
- Bardgett, R.D.; Hobbs, P.J.; Frostegard, A. Changes in soil fungal: bacterial biomass ratios following reductions in the intensity of management of an upland grassland. Biol. Fertil. Soils 1996, 22, 261–264. [Google Scholar] [CrossRef]
- Frostegard, A.; Baath, E. The use of phospholipid fatty acid analysis to estimate bacterial and fungal biomass in soil. Biol. Fertil. Soils 1996, 22, 59–65. [Google Scholar] [CrossRef]
- Rateledge, C.; Wilkinson, S.G. Microbial Lipids; Academic: London, UK, 1998. [Google Scholar]
- Fulthorpe, R.R.; Roesch, L.F.W.; Riva, A.; Triplett, E.W. Distantly sampled soils carry few species in common. ISME J. 2008, 2, 901–910. [Google Scholar] [CrossRef] [PubMed]
- Huse, S.M.; Huber, J.A.; Morrison, H.G. Accuracy and quality of massively parallel DNA pyrosequencing. Genom. Biol. 2007, 8, R143. [Google Scholar]
- Liu, Z.; Lozupone, C.; Hamady, M. Short pyrosequencing reads suffice for accurate microbial community analysis. Nucl. Acids Res. 2007, 35, e120. [Google Scholar] [CrossRef] [PubMed]
- Roesch, L.F.; Fulthrope, R.R.; Riva, A.; Casella, G.; Hadwin, A.K.M.; Kent, A.D.; Daroub, S.M.; Camargo, F.A.O.; Farmerie, W.G.; Triplett, E.W. Pyrosequencing enumerates and contrasts soil microbial diversity. ISME J. 2007, 1, 283–290. [Google Scholar] [PubMed]
- Cardenas, E.; Tiedje, J.M. New tools for discovering and characterizing microbial diversity. Curr. Opin.Biotech. 2008, 19, 544–549. [Google Scholar] [CrossRef] [PubMed]
- Acosta-Martínez, V.; Dowd, S.E.; Sun, Y.; Allen, V.G. Tag-encoded pyrosequencing analysis of bacterial diversity in a single soil type as affected by management and land use. Soil Biol. Biochem. 2008, 40, 2762–2770. [Google Scholar] [CrossRef]
- Acosta-Martínez, V.; Dowd, S.E.; Sun, Y.; Wester, D.; Allen, V.G. Pyrosequencing analysis for characterization of soil bacterial populations as affected by an integrated livestock-cotton production system. Appl. Soil Ecol. 2010, 45, 13–25. [Google Scholar] [CrossRef]
- Dowd, S.E.; Sun, Y.; Secor, P.R.; Rhoads, D.D.; Wolcott, B.M.; James, G.A.; Wolcott, R.D. Survey of bacterial diversity in chronic wounds using pyrosequencing, DGGE, and full ribosome shotgun sequencing. BMC Micrbiol. 2008, 8, 43. [Google Scholar] [CrossRef]
- Dowd, S.E.; Sun, Y.; Wolcott, R.D.; Carroll, J.A. Bacterial tag-encoded FLX amplicon pyrosequencing (bTEFAP) for microbiome studies: bacterial diversity in the ileum of newly weaned Salmonella-infected pigs. Foodbor. Path. Dis. 2008, 5, 459–472. [Google Scholar] [CrossRef]
- McGreehan, S.L.; Naylor, D.V. Automated instrumental analysis of carbon and nitrogen in plant and soil samples. Comm. Soil Sci. Plant Anal. 1988, 19, 493–505. [Google Scholar] [CrossRef]
- LECO Corporation; St. Joseph, MI. Dumas Method. Organic Application Note FP 2000. Nitrogen in Soil. 1988. Form No. 203-821-005. [Google Scholar]
- Brookes, P.C.; Landman, A.; Pruden, G.; Jenkinson, D.S. Chloroform fumigation and the release of soil nitrogen: A rapid direct extraction method to measure microbial biomass nitrogen in soil. Soil Biol. Biochem. 1985, 17, 837–842. [Google Scholar] [CrossRef]
- Vance, E.D.; Brookes, P.C.; Jenkinson, D.S. An extraction method for measuring microbial biomass C. Soil Biol. Biochem. 1987, 19, 703–707. [Google Scholar] [CrossRef]
- Wu, J.; Joegensen, R.G.; Pommerening, B.; Chaussod, R.; Brookes, P.C. Measurement of soil microbial biomass C by fumigation: a automated procedure. Soil Biol. Biochem. 1990, 22, 1167–1169. [Google Scholar]
- Jenkinson, D.S. Determination of microbial biomass carbon and nitrogen in soil. In Advances in Nitrogen Cycling in Agricultural Ecosystems; Wilson, J.R., Ed.; CAB Int.: Walling-ford, UK and Marcel Dekker, New York, NY, USA, 1988; pp. 368–386. [Google Scholar]
- Tabatabai, M.A. Soil enzymes. In Methods of Soil Analysis: Microbiological and Biochemical Properties; Weaver, R.W., Angle, J.S., Bottomley, P.S., Eds.; SSSA Book Series No. 5; Soil Science Society of America: Madison, WI, USA, 1994; pp. 775–833. [Google Scholar]
- Parham, J.A.; Deng, S.P. Detection, quantification and characterization of β-glucosaminidase activity in soil. Soil Biol. Biochem. 2000, 32, 1183–1190. [Google Scholar] [CrossRef]
- Dowd, S.E.; Zaragoza, J.; Rodriguez, J.R.; Oliver, M.J.; Payton, P.R. Windows.NET network distributed basic local alignment search toolkit (W.ND-BLAST). BMC Bioinform. 2005, 6, 93. [Google Scholar]
- Altschul, S.F.; Gish, W.; Miller, W. Basic local alignment search tool. J. Mol.Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef] [PubMed]
- Maidak, B.L.; Cole, J.R.; Lilburn, T.G.; Parker Jr., C.T.; Saxman, P.R.; Farris, R.J.; Garrity, G.M.; Olsen, G.J.; Schmidt, T.M.; Tiedje, J.M. The RDP-II (Ribosomal Database Project). Nucl. Acids Res. 2001, 29, 173–174. [Google Scholar] [CrossRef] [PubMed]
- Cole, J.R.; Chai, B.; Farris, R.J.; Wang, Q.; Kulam-Syed-Mohideen, A.S.; McGarrell, D.M.; Bandela, A.M.; Cardenas, E.; Garrity, G.M.; Tiedje, J.M. The ribosomal database project (RDP-II): introducing myRDP space and quality controlled public data. Nucl. Acids Res. 2007, 35, 169–172. [Google Scholar] [CrossRef]
- Edgar, R.C. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucl. Acids Res. 2004, 32, 792–1797. [Google Scholar]
- Felsenstein, J.P. Phylogeny Inference Packageversion 3.2. Cladistics1989; 5, 164–166.
- Felsenstein, J.P. Phylogeny Inference Package, version 3.6. Distributed by the author. Department of Genome Sciences, University of Washington: Seattle, WA, USA, 2005.
- Schloss, P.D.; Handelsman, J. Introducing DOTUR, a computer program for defining operational taxonomic units and estimating species richness. Appl. Environ. Microbiol. 2005, 71, 1501–1506. [Google Scholar] [CrossRef] [PubMed]
- Seber, G.A.F.; Wild, C.J. Nonlinear Regression; John Wiley: New York, NY, USA, 1989. [Google Scholar]
- Chao, A.; Bunge, J. Estimating the number of species in a stochastic abundance model. Biometrics 2002, 58, 531–539. [Google Scholar] [CrossRef] [PubMed]
- Acosta-Martinez, V.; Lascano, R.; Booker, J.D.; Zobeck, T.M.; Upchurch, D.R. Dryland Cropping Systems Influence Microbial Biomass and Enzyme Activities in a Sandy Soil in a Semiarid Region. Submitted to. Biol. Fertil. Soils. 2010, (in press). [Google Scholar]
- Liebig, M.; Carpenter-Boggs, L.; Johnson, J.M.F.; Wright, S.; Barbour, N. Cropping system effects on the soil biological characteristics in the Great Plains. Renew. Agric. Food Sys. 2006, 21, 36–48. [Google Scholar] [CrossRef]
- Lacey, J. Actinomycetes in soils, composts and fodders. In Actinomycetales: Characteristics and Practical Importance; Sykes, G., Skinner, F.A., Eds.; Academic Press: New York, NY, USA, 1973; pp. 231–251. [Google Scholar]
- Fierer, N.; Bradford, M.A.; Jackson, R.B. Toward an ecological classification of soil bacteria. Ecology 2007, 88, 1354–1364. [Google Scholar] [CrossRef] [PubMed]
- Kersters, K.; De Vos, P.; Gillis, M.; Swings, J.; Vandamme, P.; Stackebrandt, E. Introduction to the proteobacteria. In The Prokaryotes, 3rd ed.; Dwarkin, M., Falkow, S., Rosenberg, E., Schleifer, K.H., Stackebrandt, E., Eds.; Springer: New York, NY, USA, 2006; volume 5, pp. 3–37. [Google Scholar]
- Weiss, P.; Schweitzer, B.; Amann, R.; Simon, M. Identification in situ and dynamics of bacteria on limnetic organic aggregates (Lake Snow). Appl. Environ. Microbiol. 1996, 62, 1998–2005. [Google Scholar] [PubMed]
- Abell, G.C.J.; Bowman, J.P. Colonization and community dynamics of class Flavobacteria on diatom detritus in experimental mesocosms based on Southern Ocean seawater. FEMS Microbiol. Ecol. 2005, 53, 379–391. [Google Scholar] [CrossRef] [PubMed]
- Jangid, K.; Williams, M.A.; Franzluebbers, A.J.; Sanderlin, J.S.; Reeves, J.H.; Jenkins, M.B.; Endale, D.M.; Coleman, D.C.; Whitman, W.B. Relative impacts of land-use, management intensity and fertilization upon soil microbial community structure in agricultural systems. Soil Biol. Biochem. 2008, 40, 2843–2853. [Google Scholar] [CrossRef]
- Roldan, A.; Salinas-Garcia, J.R.; Alguacil, M.M.; Diaz, E.; Caravaca, F. Soil enzyme activities suggest advantages of conservation tillage practices in sorghum cultivation under subtropical conditions. Geoderma 2005, 129, 178–185. [Google Scholar] [CrossRef]
- Kennedy, A.C.; Schillinger, W.F. Soil quality and water intake in traditional-till vs. no-till Paired farms in Washington's Palouse region. Soil Sci.Soc. Am. J. 2006, 70, 940–949. [Google Scholar]
- Acosta-Martínez, V.; Mikha, M.; Vigil, M.F. Microbial communities and enzyme activities in soils Ander alternative crop rotations compared to wheat-fallow for the Central Great Plains. Appl. Soil Ecol. 2007, 37, 41–52. [Google Scholar] [CrossRef]
- Upchurch, R.; Chiu, C.Y.; Everett, K.; Dyszynski, G.; Coleman, D.C.; Whitman, W.B. Differences in the composition and diversity of bacterial communities from agricultural and forest soils. Soil Biol. Biochem. 2008, 40, 1294–1305. [Google Scholar]
- Janssen, P.H. Identifying the dominant soil bacteria taxa in libraries of 16S rRNA and 16S rRNA genes. Appl. Environ. Microbiol. 2006, 72, 1719–1728. [Google Scholar] [CrossRef] [PubMed]
- Dinamarca, M.A.; Cereceda-Balic, F.; Fadic, X.; Seeger, M. Analysis of striazine-degrading microbial communities in soils using most-probable-number enumeration and tetrazolium salt detection. Intern. Microbiol. 2007, 10, 209–215. [Google Scholar]
- Liebner, S.; Harder, J.; Wagner, D. Bacterial diversity and community structurein polygonal tundra soils from Samoylov Island, Lena Delta, Siberia. Int. Microbiol. 2008, 11, 195–202. [Google Scholar] [PubMed]
- Allen, V.G.; Brown, C.P.; Kellison, R.; Segarra, E.; Green, C.J.; Wheeler, T.A.; Dotray, P.A.; Conkwright, J.C.; Green, C.J.; Acosta-Martinez, V. Integrating cotton and beef production to reduce water withdrawal from the Ogallala aquifer in the Southern High Plains. Agron. J. 2005, 97, 556–567. [Google Scholar] [CrossRef]
- Allen, V.G.; Brown, C.P.; Segarra, E.; Green, C.J.; Wheeler, T.A.; Acosta-Martinez, V.; Zobeck, T.M. In search of sustainable agricultural systems for the Llano Estacado of the U.S. Southern High Plains. Agr. Ecosyst. Environ. 2008, 124, 3–12. [Google Scholar] [CrossRef]
- Larkin, R.P. Characterization of soil microbial communities under different potato cropping systems by microbial population dynamics, substrate utilization, and fatty acid profile. Soil Biol. Biochem. 2003, 35, 1451–1466. [Google Scholar] [CrossRef]
- Larkin, R.P.; Honeycutt, W. Effects of different 3-year cropping systems on soil microbial communities and Rhizoctonia diseases of potato. Phytopathology 2006, 96, 68–79. [Google Scholar] [CrossRef] [PubMed]
- Spain, A.M.; Krumholz, L.R.; Elshahed, M.S. Abundance, composition, diversity and novelty of soil proteobacteria. ISME J. 2009, 3, 992–1000. [Google Scholar] [CrossRef] [PubMed]
- Lauber, C.L.; Hamady, M.; Knight, R.; Fierer, N. Pyrosequencing-based assessment of soil pH as a predictor of soil bacterial community structure at the continental scale. Appl. Environ. Microbiol. 2009, 15, 5111–5120. [Google Scholar] [CrossRef]
© 2010 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Acosta-Martínez, V.; Dowd, S.E.; Bell, C.W.; Lascano, R.; Booker, J.D.; Zobeck, T.M.; Upchurch, D.R. Microbial Community Composition as Affected by Dryland Cropping Systems and Tillage in a Semiarid Sandy Soil. Diversity 2010, 2, 910-931. https://doi.org/10.3390/d2060910
Acosta-Martínez V, Dowd SE, Bell CW, Lascano R, Booker JD, Zobeck TM, Upchurch DR. Microbial Community Composition as Affected by Dryland Cropping Systems and Tillage in a Semiarid Sandy Soil. Diversity. 2010; 2(6):910-931. https://doi.org/10.3390/d2060910
Chicago/Turabian StyleAcosta-Martínez, Verónica, Scot E. Dowd, Colin W. Bell, Robert Lascano, Jill D. Booker, Ted M. Zobeck, and Dan R. Upchurch. 2010. "Microbial Community Composition as Affected by Dryland Cropping Systems and Tillage in a Semiarid Sandy Soil" Diversity 2, no. 6: 910-931. https://doi.org/10.3390/d2060910
APA StyleAcosta-Martínez, V., Dowd, S. E., Bell, C. W., Lascano, R., Booker, J. D., Zobeck, T. M., & Upchurch, D. R. (2010). Microbial Community Composition as Affected by Dryland Cropping Systems and Tillage in a Semiarid Sandy Soil. Diversity, 2(6), 910-931. https://doi.org/10.3390/d2060910